In Vitro and In Silico Analyses of Nicotine Release from a Gelisphere-Loaded Compressed Polymeric Matrix for Potential Parkinson’s Disease Interventions



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Formulation of the Reinforced Alginate-HEC Gelispheres
2.2.2. Formulation of the Gelisphere-Loaded External Polymeric Matrices
2.2.3. Evaluation of the Flowability and Friability of the Polymeric Material
2.2.4. Determination of the Brinell Hardness Number of the Compressed Polymeric Matrices
2.2.5. Evaluation of Conductivity Changes Following Polymeric Matrix Gelation
2.2.6. Evaluation of the Matrix Erosion upon Hydration
2.2.7. Evaluation of Surface Morphology of the Matrices
2.2.8. Textural Analysis of the Hydrated and Unhydrated Gelisphere-Loaded External Polymeric Matrices
2.2.9. In Vitro Drug Release Studies of the Gelisphere-Loaded External Polymeric Matrices
2.2.10. Static Lattice Atomistic Molecular Structural Simulations
3. Results and Discussion
3.1. Flowability and Friability of the Polymeric Matrices
3.2. Changes in the Brinell Hardness Number (BHN) of Compressed External Polymeric Matrices Following Hydration
3.3. Evaluation of Erosion of Compressed External Polymeric Matrix
3.4. Changes in Conductivity of Compressed External Polymeric Matrices Following Hydration
3.5. Evaluation of Porosity Changes upon Hydration
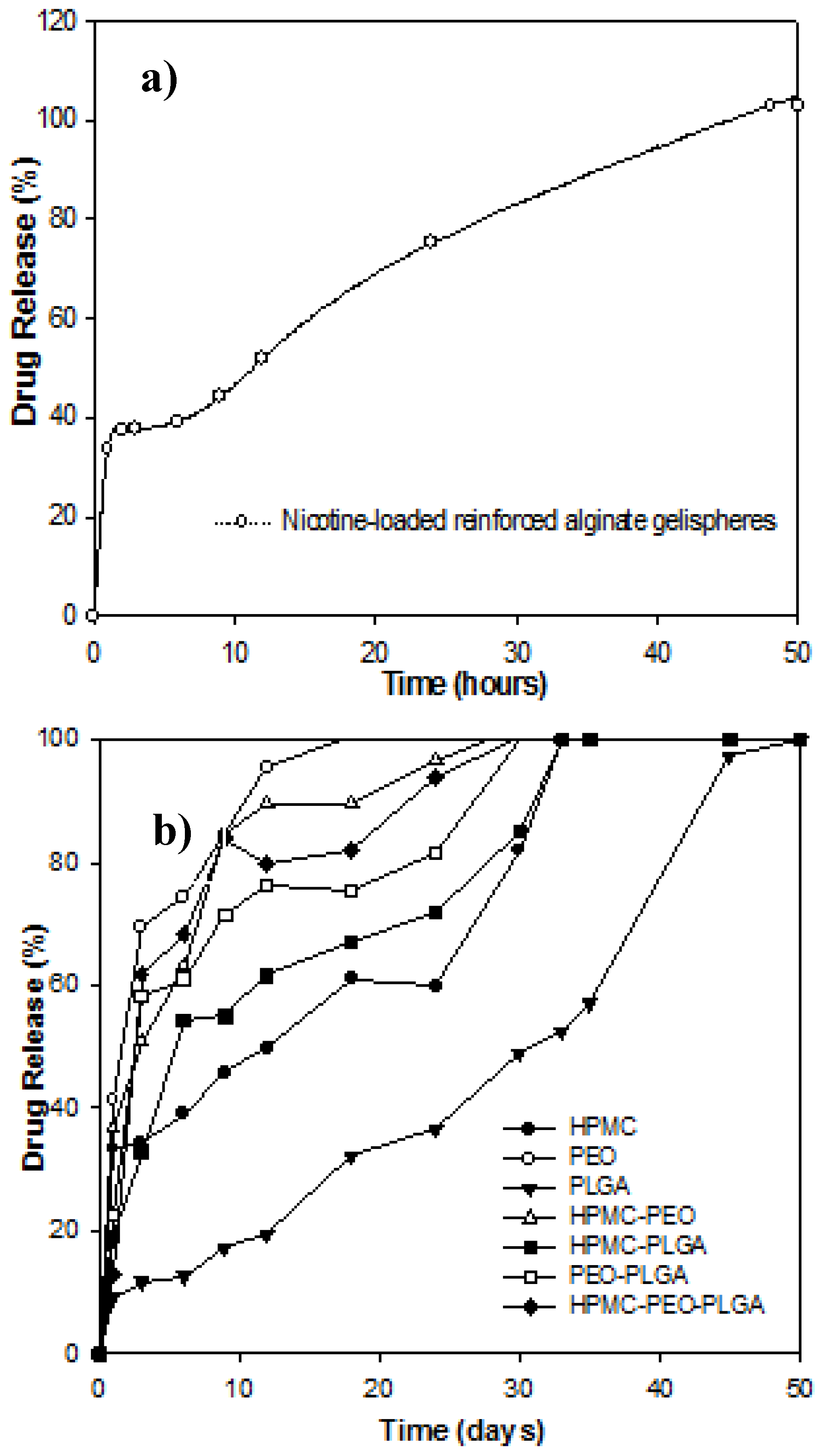
3.6. In Vitro Drug Release from Nicotine-Loaded Compressed Gelisphere Polymeric Matrices
3.7. Molecular Mechanics Energy Relationship (MMER) Analysis
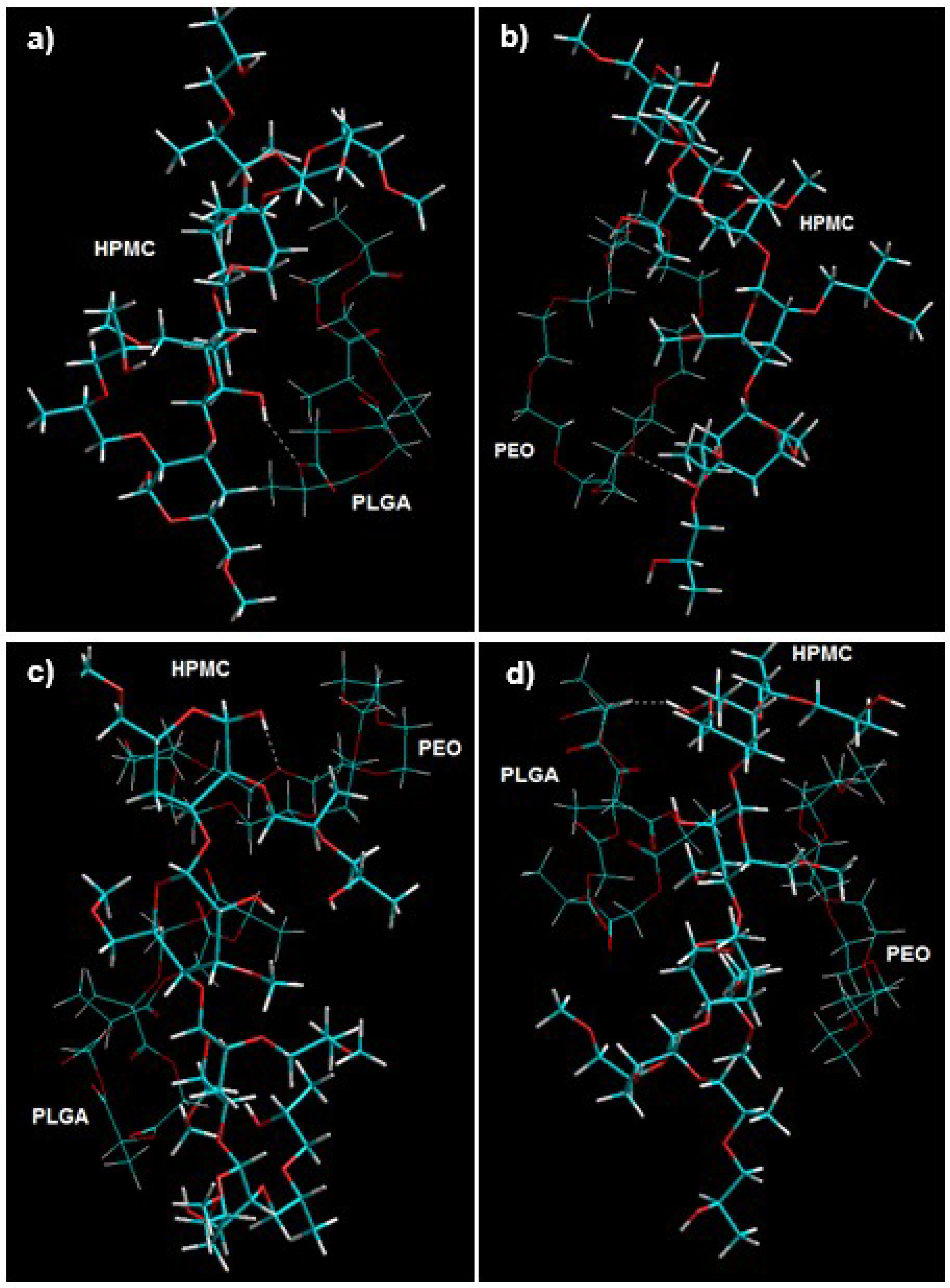
3.7.1. Energy Minimizations Involving Drug-Polymer Morphologies
3.7.2. Mechanistic Elucidation of Crosslinked Polymer Conformations
3.7.3. 3D Computational Modeling for Polymer–Polymer Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Moll, H. The treatment of post encephalitic Parkinsonism by nicotine. Br. Med. J. 1926, 1, 1079–1081. [Google Scholar] [CrossRef] [PubMed]
- Quik, M.; Zhang, D.; McGregor, M.; Bordia, T. Alpha7 nicotinic receptors as therapeutic targets for Parkinson’s disease. Biochem. Pharmacol. 2015, 97, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Barreto, G.E.; Iarkov, A.; Moran, V.E. Beneficial effects of nicotine, cotinine and its metabolites as potential agents for Parkinson’s disease. Front. Aging Neurosci. 2014, 6, 340. [Google Scholar] [CrossRef] [PubMed]
- Mouhape, C.; Costa, G.; Ferreira, M.; Abin-Carriquiry, J.A.; Dajas, F.; Prunell, G. Nicotine-induced neuroprotection in rotenone in vivo and in vitro models of Parkinson’s disease: Evidences for the involvement of the labile iron pool level as the underlying mechanism. Neurotox. Res. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Villafane, G.; Thiriez, C.; Audureau, E.; Straczek, C.; Kerschen, P.; Cormier-Dequaire, F.; Van Der Gucht, A.; Gurruchaga, J.-M.; Quéré-Carne, M.; Paul, M.; et al. High-dose transdermal nicotine in Parkinson’s disease patients: A randomized, open-label, blinded-endpoint evaluation phase 2 study. Eur. J. Neurol. 2018, 25, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, S.S.M.; Oliveira, F.S.; Gaitani, C.M. Microparticles as a strategy for low-molecular-weight heparin delivery. J. Pharm. Sci. 2011, 100, 1783–1792. [Google Scholar] [CrossRef] [PubMed]
- Pahwa, R.; Saini, N.; Kumar, V.; Kohli, K. Chitosan-based gastroretentive floating drug delivery technology: An updated review. Exp. Opin. Drug Deliv. 2012, 9, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable controlled-release polymers and polymeric nanoparticles: Mechanisms of controlling drug release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Chen, C.; Mantle, M.D.; Wolf, B.; Gladden, L.F.; Rajabi-Siahboomi, A.; Missaghi, S.; Mason, L.; Melia, C.D. The properties of HPMC:PEO extended release hydrophilic matrices and their response to ionic environments. Pharm. Res. 2017, 34, 941–956. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Fassihi, M.A.; Fassihi, R. Delivery considerations of highly viscous polymeric fluids mimicking concentrated biopharmaceuticals: Assessment of injectability via measurement of total work done “WT”. AAPS PharmSciTech 2018, 19, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Bhatia, M. Functionalization of chitosan/methylcellulose interpenetrating polymer network microspheres for gastroretentive application using central composite design. PDA J. Pharm. Sci. Technol. 2010, 64, 497–506. [Google Scholar] [PubMed]
- Colombo, P.; Bettini, R.; Peppas, N.A. Observation of swelling process and diffusion front position during swelling in hydroxypropyl methyl cellulose (HPMC) matrices containing a soluble drug. J. Control. Release 1999, 61, 83–91. [Google Scholar] [CrossRef]
- Jamzad, S.; Tutunji, L.; Fassihi, R. Analysis of macromolecular changes and drug release from hydrophilic matrix systems. Int. J. Pharm. 2005, 292, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Mansour, H.M.; Sohn, M.; Al-Ghananeem, A. Materials for pharmaceutical dosage forms: Molecular pharmaceutics and controlled release drug delivery aspects. Int. J. Mol. Sci. 2010, 11, 3298–3322. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, D.N.; Bhatia, A.; Kaur, R.; Sharma, R.; Kaur, G.; Dhawan, S. PLGA: A unique polymer for drug delivery. Ther. Deliv. 2015, 6, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Fournier, E.; Passirani, C.; Montero-Menei, C.N.; Benoit, J.P. Biocompatibility of implantable synthetic polymeric drug carriers: Focus on brain biocompatibility. Biomaterials 2003, 24, 3311–3331. [Google Scholar] [CrossRef]
- Choonara, Y.E.; Pillay, V.; Khan, R.A.; Singh, N.; Du Toit, L.C. Mechanistic evaluation of alginate-HEC gelisphere compacts for controlled intrastriatal nicotine release in Parkinson’s disease. J. Pharm. Sci. 2009, 98, 2059–2072. [Google Scholar] [CrossRef] [PubMed]
- Bawa, P.; Pillay, V.; Choonara, Y.E.; du Toit, L.C.; Ndesendo, V.M.K.; Kumar, P. A composite polyelectrolytic matrix for controlled oral drug delivery. AAPS PharmSciTech 2011, 12, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Choonara, Y.E.; Pillay, V. In silico analytico-mathematical interpretation of biopolymeric assemblies: Quantification of energy surfaces and molecular attributes via atomistic simulations. Bioeng. Transl. Med. 2018, 3, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Caccavo, D.; Cascone, S.; Lamberti, G.; Barba, A.A. Controlled drug release from hydrogel-based matrices: Experiments and modeling. Int. J. Pharm. 2015, 486, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Rafiei, P.; Haddadi, A. Pharmacokinetic Consequences of PLGA nanoparticles in docetaxel drug delivery. Pharm. Nanotechnol. 2017, 5, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Choonara, Y.E.; Toit, L.C.; Modi, G.; Naidoo, D.; Pillay, V. Novel high-viscosity polyacrylamidated chitosan for neural tissue engineering: Fabrication of anisotropic neurodurable scaffold via molecular disposition of persulfate-mediated polymer slicing and complexation. Int. J. Mol. Sci. 2012, 13, 13966–13984. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation Code | HPMC (mg) | PEO (mg) | PLGA (mg) | % WL4 | % WL20 |
|---|---|---|---|---|---|
| HPMC | 500 | - | - | 0.081 | 0.307 |
| PEO | - | 500 | - | 0.101 | 0.133 |
| PLGA | - | - | 500 | 0.000 | 0.026 |
| HPMC-PEO | 250 | 250 | - | 0.050 | 0.107 |
| HPMC-PLGA | 250 | - | 250 | 0.051 | 0.064 |
| PEO-PLGA | - | 250 | 250 | 0.038 | 0.102 |
| HPMC-PEO-PLGA | 166 | 166 | 166 | 0.025 | 0.051 |
| Structure | Molecular Attributes | ||
|---|---|---|---|
| Surface Area (grid) | Volume | Surface-To-Volume Ratio | |
| ALG | 828.99 | 1518.13 | 0.546 |
| NCT2 # | 603.16 | 1088.42 | 0.554 |
| ALG–NCT2 # | 1026.16 | 2273.41 | 0.451 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, P.; Choonara, Y.E.; Du Toit, L.C.; Singh, N.; Pillay, V. In Vitro and In Silico Analyses of Nicotine Release from a Gelisphere-Loaded Compressed Polymeric Matrix for Potential Parkinson’s Disease Interventions. Pharmaceutics 2018, 10, 233. https://doi.org/10.3390/pharmaceutics10040233
Kumar P, Choonara YE, Du Toit LC, Singh N, Pillay V. In Vitro and In Silico Analyses of Nicotine Release from a Gelisphere-Loaded Compressed Polymeric Matrix for Potential Parkinson’s Disease Interventions. Pharmaceutics. 2018; 10(4):233. https://doi.org/10.3390/pharmaceutics10040233
Chicago/Turabian StyleKumar, Pradeep, Yahya E. Choonara, Lisa C. Du Toit, Neha Singh, and Viness Pillay. 2018. "In Vitro and In Silico Analyses of Nicotine Release from a Gelisphere-Loaded Compressed Polymeric Matrix for Potential Parkinson’s Disease Interventions" Pharmaceutics 10, no. 4: 233. https://doi.org/10.3390/pharmaceutics10040233
APA StyleKumar, P., Choonara, Y. E., Du Toit, L. C., Singh, N., & Pillay, V. (2018). In Vitro and In Silico Analyses of Nicotine Release from a Gelisphere-Loaded Compressed Polymeric Matrix for Potential Parkinson’s Disease Interventions. Pharmaceutics, 10(4), 233. https://doi.org/10.3390/pharmaceutics10040233