Mechanism and Improved Dissolution of Glycyrrhetinic Acid Solid Dispersion by Alkalizers

Abstract
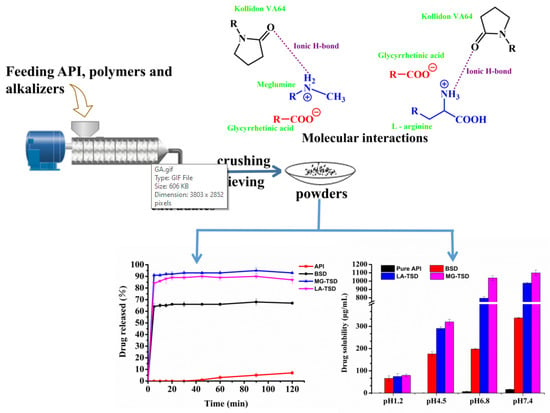
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Pre-Prescription Study
2.2.1. Solubility Study of the Alkalizers and Polymers
2.2.2. Preparation of Solid Dispersions
2.2.3. Dissolution Study of SDs
2.3. Characterization and Analysis of Optimal Formulation
2.3.1. Dissociation Constant (pKa)
2.3.2. Scanning Electron Microscopy (SEM)
2.3.3. Differential Scanning Calorimetry (DSC)
2.3.4. X-ray Powder Diffraction (XRPD)
2.3.5. Fourier Transform Infrared Spectroscopy (FTIR)
2.3.6. Raman Spectroscopy
2.3.7. X-ray Photoelectron Spectroscopy (XPS)
2.3.8. Molecular Modeling
2.3.9. Molecular Docking
2.3.10. Molecular Dynamic Simulation
2.3.11. Solubility and Dissolution of Optimal Formulation
3. Results and Discussion
3.1. Pre-Prescription Study
3.1.1. Solubility Screening
3.1.2. Dissolution Screening of Several Formulations with Different Drug/Alkalizer/Polymer Ratios
3.2. Characterization and Analysis of Optimal Formulation
3.2.1. Dissociation Constant (pKa)
3.2.2. SEM
3.2.3. DSC
3.2.4. XRPD
3.2.5. FTIR
3.2.6. Raman Spectroscopy
3.2.7. XPS
3.2.8. Molecular Docking
3.2.9. Molecular Dynamic Simulation
3.2.10. Solubility and Dissolution of Optimal Formulation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SEM | Scanning electron microscopy |
| DSC | Differential scanning calorimetry |
| XRPD | X-ray powder diffraction |
| FTIR | Fourier transform infrared spectroscopy |
| XPS | X-ray photoelectron spectroscopy |
| EC | Ethyl cellulose |
| HPMC | Hydroxypropyl methyl cellulose |
| HME | Hot melt extrusion |
| LA | L-arginine |
| MG | Meglumine |
| GA | Glycyrrhetinic |
| SD | Solid dispersion |
| BSD | Binary solid dispersion |
| TSD | Ternary solid dispersion |
| LA-TSD | L-arginine ternary solid dispersion |
| MG-TSD | Meglumine ternary solid dispersion |
| BPM | Binary physical mixture |
| LA-TPM | L-arginine physical mixture |
| MG-TPM | Meglumine physical mixture |
| LA-VA | L-arginine-Kollidon® VA64 extrudate |
| MG-VA | Meglumine-Kollidon® VA64 extrudate |
| GA-MG | Glycyrrhetinic-Meglumine extrudate |
References
- Liu, Y.; Zhao, J.; Wang, L.; Yan, B.; Gu, Y.; Chang, P.; Wang, Y. Nanocrystals Technology for Transdermal Delivery of Water-Insoluble Drugs. Curr. Drug Deliv. 2018, 15, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins: Basic science and product development. J. Pharm. Pharmacol. 2010, 62, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, K.; Wen, H.; Ouyang, D.; Li, X.; Gao, Y.; Pan, W.; Yang, X. Design and Evaluation of Hydrophilic Matrix System Containing Polyethylene Oxides for the Zero-Order Controlled Delivery of Water-Insoluble Drugs. AAPS PharmSciTech 2017, 18, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Korn, C.; Balbach, S. Compound selection for development—Is salt formation the ultimate answer? Experiences with an extended concept of the “100 mg approach”. Eur. J. Pharm. Sci. 2014, 57, 257–263. [Google Scholar] [CrossRef]
- Park, C.; Meghani, N.M.; Shin, Y.; Oh, E.; Park, J.B.; Cui, J.H.; Cao, Q.R.; Tran, T.T.; Tran, P.H.; Lee, B.J. Investigation of Crystallization and Salt Formation of Poorly Water-Soluble Telmisartan for Enhanced Solubility. Pharmaceutics 2019, 11, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigfridsson, K.; Ahlqvist, M.; Lindsjö, M.; Paulsson, S. Salt formation improved the properties of a candidate drug during early formulation development. Eur. J. Pharm. Sci. 2018, 120, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Lv, Y.; Li, T.L. Hybrid drug nanocrystals. Adv. Drug Deliv. Rev. 2019, 143, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Dibaei, M.; Rouini, M.-R.; Sheikholeslami, B.; Gholami, M.; Dinarvand, R. The effect of surface treatment on the brain delivery of curcumin nanosuspension: In vitro and in vivo studies. Int. J. Nanomed. 2019, 14, 5477–5490. [Google Scholar] [CrossRef] [Green Version]
- ElShagea, H.N.; ElKasabgy, N.A.; Fahmy, R.H.; Basalious, E.B. Freeze-Dried Self-Nanoemulsifying Self-Nanosuspension (SNESNS): A New Approach for the Preparation of a Highly Drug-Loaded Dosage Form. AAPS PharmSciTech 2019, 20, 258. [Google Scholar] [CrossRef]
- Dong, D.; Hsiao, C.H.; Giovanella, B.C.; Wang, Y.; Chow, D.S.; Li, Z. Sustained delivery of a camptothecin prodrug—CZ48 by nanosuspensions with improved pharmacokinetics and enhanced anticancer activity. Int. J. Nanomed. 2019, 14, 3799–3817. [Google Scholar] [CrossRef] [Green Version]
- Mohandoss, S.; Atchudan, R.; Immanuel Edison, T.N.J.; Palanisamy, S.; You, S.; Napoleon, A.A.; Shim, J.J.; Lee, Y.R. Enhanced solubility of guanosine by inclusion complexes with cyclodextrin derivatives: Preparation, characterization, and evaluation. Carbohydr. Polym. 2019, 224, 115166. [Google Scholar] [CrossRef] [PubMed]
- Tai, K.; Rappolt, M.; He, X.; Wei, Y.; Zhu, S.; Zhang, J.; Mao, L.; Gao, Y.; Yuan, F. Effect of β-sitosterol on the curcumin-loaded liposomes: Vesicle characteristics, physicochemical stability, in vitro release and bioavailability. Food Chem. 2019, 293, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.; Zalba, S.; Seynhaeve Ann, L.B.; Debets, R.; Ten Hagen, T.L.M. Liposomes targeted to MHC-restricted antigen improve drug delivery and antimelanoma response. Int. J. Nanomed. 2019, 14, 2069–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Katai, H.; Higashi, K.; Ueda, K.; Kawakami, K.; Moribe, K. Cryo-TEM and AFM Observation of the Time-Dependent Evolution of Amorphous Probucol Nanoparticles Formed by the Aqueous Dispersion of Ternary Solid Dispersions. Mol. Pharm. 2019, 16, 2184–2198. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Jin, L.; Liu, Q.; Xu, H.; Wu, H.; Zhang, X.; Mao, S. Exploration of supersaturable lacidipine ternary amorphous solid dispersion for enhanced dissolution and in vivo absorption. Eur. J. Pharm. Sci. 2019, 139, 105043. [Google Scholar] [CrossRef]
- Kambayashi, A.; Kiyota, T.; Fujiwara, M.; Dressman, J.B. PBPK modeling coupled with biorelevant dissolution to forecast the oral performance of amorphous solid dispersion formulations. Eur. J. Pharm. Sci. 2019, 135, 83–90. [Google Scholar] [CrossRef]
- Martínez, L.M.; Videa, M.; López Silva, T.; Castro, S.; Caballero, A.; Lara-Díaz, V.J.; Castorena-Torres, F. Two-phase amorphous-amorphous solid drug dispersion with enhanced stability, solubility and bioavailability resulting from ultrasonic dispersion of an immiscible system. Eur. J. Pharm. Biopharm. 2017, 119, 243–252. [Google Scholar] [CrossRef]
- Kwon, J.; Giri, B.R.; Song, E.; Castro, S.; Caballero, A.; Lara-Díaz, V.J.; Castorena-Torres, F. Spray-Dried Amorphous Solid Dispersions of Atorvastatin Calcium for Improved Supersaturation and Oral Bioavailability. Pharmaceutics 2019, 11, 461. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, A.; Kumar, N.S.K.; Suryanarayanan, R. Crosslinking: An avenue to develop stable amorphous solid dispersion with high drug loading and tailored physical stability. J. Control. Release 2019, 311, 212–224. [Google Scholar] [CrossRef]
- Ricarte, R.G.; Van Zee, N.J.; Li, Z.; Johnson, L.M.; Lodge, T.P.; Hillmyer, M.A. Recent Advances in Understanding the Micro- and Nanoscale Phenomena of Amorphous Solid Dispersions. Mol. Pharm. 2019, 16, 4089–4103. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Danda, L.J.A.; Batista, L.M.; Melo, V.C.S.; Soares Sobrinho, J.L.; Soares, M.F.R. Combining amorphous solid dispersions for improved kinetic solubility of posaconazole simultaneously released from soluble PVP/VA64 and an insoluble ammonio methacrylate copolymer. Eur. J. Pharm. Sci. 2019, 133, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Ben Osman, Y.; Liavitskaya, T.; Vyazovkin, S. Polyvinylpyrrolidone affects thermal stability of drugs in solid dispersions. Int. J. Pharm. 2018, 551, 111–120. [Google Scholar] [CrossRef] [PubMed]
- McFall, H.; Sarabu, S.; Shankar, V.; Bandari, S.; Murthy, S.N.; Kolter, K.; Langley, N.; Kim, D.W.; Repka, M.A. Formulation of aripiprazole-loaded pH-modulated solid dispersions via hot-melt extrusion technology: In vitro and in vivo studies. Int. J. Pharm. 2019, 554, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.D.; Tran, H.L.; Lee, B.J. Dissolution-modulating mechanism of alkalizers and polymers in a nanoemulsifying solid dispersion containing ionizable and poorly water-soluble drug. Eur. J. Pharm. Biopharm. 2009, 72, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Kwon, S.H.; Lee, S.E.; Jang, W.S.; Byeon, J.C.; Jeong, H.M.; Park, J.S. Use of acidifier and solubilizer in tadalafil solid dispersion to enhance the in vitro dissolution and oral bioavailability in rats. Int. J. Pharm. 2017, 526, 77–87. [Google Scholar] [CrossRef]
- Sun, W.; Pan, B. Effect of micro-environment modification and polymer type on the in-vitro dissolution behavior and in-vivo performance of amorphous solid dispersions. Eur. J. Pharm. Sci. 2017, 104, 240–254. [Google Scholar] [CrossRef]
- Armanini, D.; Fiore, C.; Mattarello, M.J.; Bielenberg, J.; Palermo, M. History of the Endocrine Effects of Licorice. Exp. Clin. Endocrinol. Diabetes 2002, 110, 257–261. [Google Scholar] [CrossRef]
- Wang, Y.; Du, H.; Zhai, G. Recent advances in active hepatic targeting drug delivery system. Curr. Drug Targets 2014, 15, 573–599. [Google Scholar] [CrossRef]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar] [CrossRef]
- Ribeiro, R.; Schmidt, C. Determination of acid dissociation constants (pKa) of cephalosporin antibiotics: Computational and experimental approaches. Chemosphere 2017, 169, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Völgyi, G.; Ruiz, R.; Box, K.; Comer, J.; Boschb, E.; Takács-Novák, K. Potentiometric and spectrophotometric pKa determination of water-insoluble compounds: Validation study in a new cosolvent system. Anal. Chim. Acta 2007, 583, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Shayesteh, H.; Radmehr, M.; Khajavi, F.; Mahjub, R. Application of chemometrics in determination of the acid dissociation constants (pKa) of several benzodiazepine derivatives as poorly soluble drugs in the presence of ionic surfactants. Eur. J. Pharm. Sci. 2015, 69, 44–50. [Google Scholar] [CrossRef]
- Taniguchi, C.; Kawabata, Y.; Wada, K.; Yamada, S.; Onoue, S. Microenvironmental pH-modification to improve dissolution behavior and oral absorption for drugs with pH-dependent solubility. Expert Opin. Drug Deliv. 2014, 11, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Bassi, P.; Kaur, G. pH modulation: A mechanism to obtain pH-independent drug release. Expert Opin. Drug Deliv. 2010, 7, 845–857. [Google Scholar] [CrossRef]
- Shi, N.Q.; Jin, Y.; Zhang, Y.; Che, X.X.; Xiao, X.; Cui, G.H.; Chen, Y.Z.; Feng, B.; Li, Z.Q.; Qi, X.R. The Influence of Cellulosic Polymer’s Variables on Dissolution/Solubility of Amorphous Felodipine and Crystallization Inhibition from a Supersaturated State. AAPS PharmSciTech 2018, 20, 12–26. [Google Scholar] [CrossRef]
- Xue, X.; Chen, G.; Xu, X.; Wang, J.; Wang, J.; Ren, L. A Combined Utilization of Plasdone-S630 and HPMCAS-HF in Ziprasidone Hydrochloride Solid Dispersion by Hot-Melt Extrusion to Enhance the Oral Bioavailability and No Food Effect. AAPS PharmSciTech 2019, 20, 37–49. [Google Scholar] [CrossRef]
- Skakle, J. Applications of X-ray powder diffraction in materials chemistry. Chem. Rec. 2005, 5, 252–262. [Google Scholar] [CrossRef]
- Leuner, C.; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Tang, X.C.; Pikal, M.J.; Taylor Lynne, S. A spectroscopic investigation of hydrogen bond patterns in crystalline and amorphous phases in dihydropyridine calcium channel blockers. Pharm. Res. 2002, 19, 477–483. [Google Scholar] [CrossRef]
- Hao, J.; Sun, Y.; Wang, Q.; Tong, X.; Zhang, H.; Zhang, Q. Effect and mechanism of penetration enhancement of organic base and alcohol on glycyrrhetinic acid in vitro. Int. J. Pharm. 2010, 399, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.; Tran, P.H.; Choi, H.G.; Han, H.K.; Lee, B.J. The roles of acidifiers in solid dispersions and physical mixtures. Int. J. Pharm. 2010, 384, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Samiei, N.; Mangas-Sanjuan, V.; González-Álvarez, I.; Foroutan, M.; Shafaati, A.; Zarghi, A.; Bermejo, M. Ion-pair strategy for enabling amifostine oral absorption: Rat in situ and in vivo experiments. Eur. J. Pharm. Sci. 2013, 49, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Elshaer, A.; Khan, S.; Perumal, D.; Hanson, P.; Mohammed, A.R. Use of Amino Acids as Counterions Improves the Solubility of the BCS II Model Drug, Indomethacin. Curr. Drug Deliv. 2011, 8, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ling, J.; Wei, C.; Zhang, H. Phase Behaviors and Self-Assembled Properties of Ion-paring Amphiphile Molecules Formed by Medium-chain Fatty Acids and L-Arginine Triggered by External Conditions. New J. Chem. 2017, 41, 14486–14497. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, L.; Zhang, F. Reactive Melt Extrusion to Improve the Dissolution Performance and Physical Stability of Naproxen Amorphous Solid Dispersions. Mol. Pharm. 2017, 14, 658–673. [Google Scholar] [CrossRef]
- Cheng, M.; Chen, H.; Wang, Y.; Xu, H.; He, B.; Han, J.; Zhang, Z. Optimized synthesis of glycyrrhetinic acid-modified chitosan 5-fluorouracil nanoparticles and their characteristics. Int. J. Nanomed. 2014, 9, 695–710. [Google Scholar]
- Qi, S.; Belton, P.; Nollenberger, K.; Gryczke, A.; Craig, D.Q. Compositional analysis of low quantities of phase separation in hot-melt-extruded solid dispersions: A combined atomic force microscopy, photothermal fourier-transform infrared microspectroscopy, and localised thermal analysis approach. Pharm. Res. 2011, 28, 2311–2326. [Google Scholar] [CrossRef]
- Konno, H.; Taylor, L.S. Influence of different polymers on the crystallization tendency of molecularly dispersed amorphous felodipine. J. Pharm. Sci. 2006, 95, 2692–2705. [Google Scholar] [CrossRef]
- Van Eerdenbrugh, B.; Taylor, L.S. Application of mid-IR spectroscopy for the characterization of pharmaceutical systems. Int. J. Pharm. 2011, 417, 3–16. [Google Scholar] [CrossRef]
- Taylor, L.S.; Langkilde, F.W.; Zografi, G. Fourier transform Raman spectroscopic study of the interaction of water vapor with amorphous polymers. J. Pharm. Sci. 2001, 90, 888–901. [Google Scholar] [CrossRef] [PubMed]
- Kestur, U.S.; Taylor, L.S. Role of polymer chemistry in influencing crystal growth rates from amorphous felodipine. CrystEngComm 2010, 12, 2390–2397. [Google Scholar] [CrossRef]
- Mesallati, H.; Umerska, A.; Paluch, K.J.; Tajber, L. Amorphous Polymeric Drug Salts as Ionic Solid Dispersion Forms of Ciprofloxacin. Mol. Pharm. 2017, 14, 2209–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Liu, C.; Yang, D.; Wan, X.; Shang, R.; Quan, P.; Fang, L. Molecular mechanism of ion-pair releasing from acrylic pressure sensitive adhesive containing carboxyl group: Roles of doubly ionic hydrogen bond in the controlled release process of bisoprolol ion-pair. J. Control. Release 2018, 289, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Lozoya-Agullo, I.; Planelles, M.; Merino-Sanjuán, M.; Bermejo, M.; Sarmento, B.; González-Álvarez, I.; González-Álvarez, M. Ion-pair approach coupled with nanoparticle formation to increase bioavailability of a low permeability charged drug. Int. J. Pharm. 2019, 557, 36–42. [Google Scholar] [CrossRef]
- Jiang, Q.; Wang, J.; Ma, P.; Liu, C.; Sun, M.; Sun, Y.; He, Z. Ion-pair formation combined with a penetration enhancer as a dual strategy to improve the transdermal delivery of meloxicam. Drug Deliv. Transl. Res. 2018, 8, 64–72. [Google Scholar] [CrossRef]
- Aloisio, C.; de Oliveira, A.G.; Longhi, M. Solubility and release modulation effect of sulfamerazine ternary complexes with cyclodextrins and meglumine. J. Pharm. Biomed. Anal. 2014, 100, 64–73. [Google Scholar] [CrossRef]
- Gupta, P.; Bansal, A.K. Molecular interactions in celecoxib-PVP-meglumine amorphous system. J. Pharm. Pharmacol. 2005, 57, 303–310. [Google Scholar] [CrossRef]
- Rojek, B.; Wesolowski, M. FTIR and TG analyses coupled with factor analysis in a compatibility study of acetazolamide with excipients. Spectrochim. Acta 2019, 208, 285–293. [Google Scholar] [CrossRef]
- Aliaga, A.E.; Garrido, C.; Leyton, P.; Diaz, G.; Gomez-Jeria, J.S.; Aguayo, T.; Clavijo, E.; Campos-Vallette, M.M.; Sanchez-Cortes, S. SERS and theoretical studies of arginine. Spectrochim. Acta 2010, 76, 458–463. [Google Scholar] [CrossRef]
- Zare-Zardini, H.; Amiri, A.; Shanbedi, M.; Taheri-Kafrani, A.; Kazi, S.N.; Chew, B.T.; Razmjou, A. In vitro and in vivo study of hazardous effects of Ag nanoparticles and Arginine-treated multi walled carbon nanotubes on blood cells: Application in hemodialysis membranes. J. Biomed. Mater. Res. 2015, 103, 2959–2965. [Google Scholar] [CrossRef] [PubMed]
- Luis Miguel, R.G.; Gonzalo, G.; Dalila, A.; Amaia, Q. Carbon-supported Pt-free catalysts with high specificity and activity toward the oxygen reduction reaction in acidic medium. Appl. Catal. B Environ. 2016, 184, 12–19. [Google Scholar]
- Wang, A.; Yu, W.; Huang, Z.; Zhou, F.; Song, J.; Song, Y.; Long, L.; Cifuentes, M.P.; Humphrey, M.G.; Zhang, L.; et al. Covalent functionalization of reduced graphene oxide with porphyrin by means of diazonium chemistry for nonlinear optical performance. Sci. Rep. 2016, 6, 23325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, J.S.; De Luca, A.C.; Pelendritis, M.; Terenghi, G.; Downes, S.; Schroeder, S.L.M. Quantitative analysis of complex amino acids and RGD peptides by X-ray photoelectron spectroscopy (XPS). Surf. Interface Anal. 2013, 45, 1238–1246. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.Y.; Ning, Y.S.; Yong, K.S.; Cai, Y.H.; Tang, H.H.; Shao, Y.X.; Alshahateet, S.F.; Sun, Y.M.; Xu, G.Q. Binding of glycine and L-cysteine on Si(111)-7 × 7. Langmuir 2007, 23, 6218–6226. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zemlyanov, D.; Chen, X.; Nie, H.; Su, Z.; Fang, K.; Yang, X.; Smith, D.; Byrn, S.; Lubach, J.W. Acid-Base Interactions of Polystyrene Sulfonic Acid in Amorphous Solid Dispersions Using a Combined UV/FTIR/XPS/ssNMR Study. Mol. Pharm. 2016, 13, 483–492. [Google Scholar] [CrossRef]
- Macaveiu, L.; Göbel, M.; Klapötke, T.M.; Murray, J.S.; Peter, P. The unique role of the nitro group in intramolecular interactions: Chloronitromethanes. Struct. Chem. 2010, 21, 139–146. [Google Scholar] [CrossRef]
- Zhu, Y.; Yan, J.; Liu, C.; Zhang, D. Modeling interactions between a β-O-4 type lignin model compound and 1-allyl-3-methylimidazolium chloride ionic liquid. Biopolymers 2017, 107, e23022. [Google Scholar] [CrossRef]
- Yang, D.; Wan, X.; Quan, P.; Liu, C.; Fang, L. The role of carboxyl group of pressure sensitive adhesive in controlled release of propranolol in transdermal patch: Quantitative determination of ionic interaction and molecular mechanism characterization. Eur. J. Pharm. Sci. 2018, 115, 330–338. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | GA (g) | pH Modifier | Kollidon® VA64 (g) | Ratio | Total (g) | |
|---|---|---|---|---|---|---|
| Mg(OH)2 (g) | Na2CO3 (g) | |||||
| F1 | 5 | - | - | 75 | 1:0:15 | 80 |
| F2 | 8 | - | - | 72 | 1:0:9 | 80 |
| F3 | 16 | - | - | 64 | 1:0:4 | 80 |
| F4 | 32 | - | - | 48 | 2:0:3 | 80 |
| F5 | 6 | 6 | - | 48 | 1:1:8 | 60 |
| F6 | 6 | 12 | - | 42 | 1:2:7 | 60 |
| F7 | 6 | 18 | - | 36 | 1:3:6 | 60 |
| F8 | 12 | 18 | - | 30 | 2:3:5 | 60 |
| F9 | 6 | - | 6 | 48 | 1:1:8 | 60 |
| F10 | 6 | - | 12 | 42 | 1:2:7 | 60 |
| F11 | 6 | - | 18 | 36 | 1:3:6 | 60 |
| F12 | 12 | - | 18 | 30 | 2:3:5 | 60 |
| Formulation | GA (g) | pH Modifier | Kollidon® VA64 (g) | Ratio | Total (g) | |
|---|---|---|---|---|---|---|
| Meglumine (g) | L-arginine (g) | |||||
| F13 | 6 | 6 | - | 48 | 1:1:8 | 60 |
| F14 | 6 | 12 | - | 42 | 1:2:7 | 60 |
| F15 | 6 | 18 | - | 36 | 1:3:6 | 60 |
| F16 | 12 | 18 | - | 30 | 2:3:5 | 60 |
| F17 | 6 | - | 6 | 48 | 1:1:8 | 60 |
| F18 | 6 | - | 12 | 42 | 1:2:7 | 60 |
| F19 | 6 | - | 18 | 36 | 1:3:6 | 60 |
| F20 | 12 | - | 18 | 30 | 2:3:5 | 60 |
| NO. | pH | GA | BSD | MG-TSD | LA-TSD |
|---|---|---|---|---|---|
| 1 | 6.5 | 7.17 | 7.02 | 6.42 | 6.72 |
| 2 | 7.0 | 7.18 | 7.01 | 6.44 | 6.73 |
| 3 | 7.5 | 7.17 | 7.07 | 6.44 | 6.71 |
| 4 | 8.0 | 7.16 | 7.09 | 6.43 | 6.73 |
| Average value of pKa | 7.17 | 7.05 | 6.43 | 6.72 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, L.; Mai, Y.; Liu, Q.; Zhang, W.; Yang, J. Mechanism and Improved Dissolution of Glycyrrhetinic Acid Solid Dispersion by Alkalizers. Pharmaceutics 2020, 12, 82. https://doi.org/10.3390/pharmaceutics12010082
Dong L, Mai Y, Liu Q, Zhang W, Yang J. Mechanism and Improved Dissolution of Glycyrrhetinic Acid Solid Dispersion by Alkalizers. Pharmaceutics. 2020; 12(1):82. https://doi.org/10.3390/pharmaceutics12010082
Chicago/Turabian StyleDong, Luning, Yaping Mai, Qiang Liu, Wannian Zhang, and Jianhong Yang. 2020. "Mechanism and Improved Dissolution of Glycyrrhetinic Acid Solid Dispersion by Alkalizers" Pharmaceutics 12, no. 1: 82. https://doi.org/10.3390/pharmaceutics12010082
APA StyleDong, L., Mai, Y., Liu, Q., Zhang, W., & Yang, J. (2020). Mechanism and Improved Dissolution of Glycyrrhetinic Acid Solid Dispersion by Alkalizers. Pharmaceutics, 12(1), 82. https://doi.org/10.3390/pharmaceutics12010082