Targeted Therapy of Hepatocellular Carcinoma Using Gemcitabine-Incorporated GPC3 Aptamer

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Aptamers
2.3. Serum Stability
2.4. Flow Cytometry
2.5. Confocal Fluorescence Microscopy
2.6. Lysosome Labeling
2.7. Cell Viability
2.8. γ-H2AX Immunostaining
2.9. Cell-Cycle Analysis
2.10. Animal Model
2.11. In Vivo Tumor Growth Inhibition
2.12. Statistical Analyses
3. Results
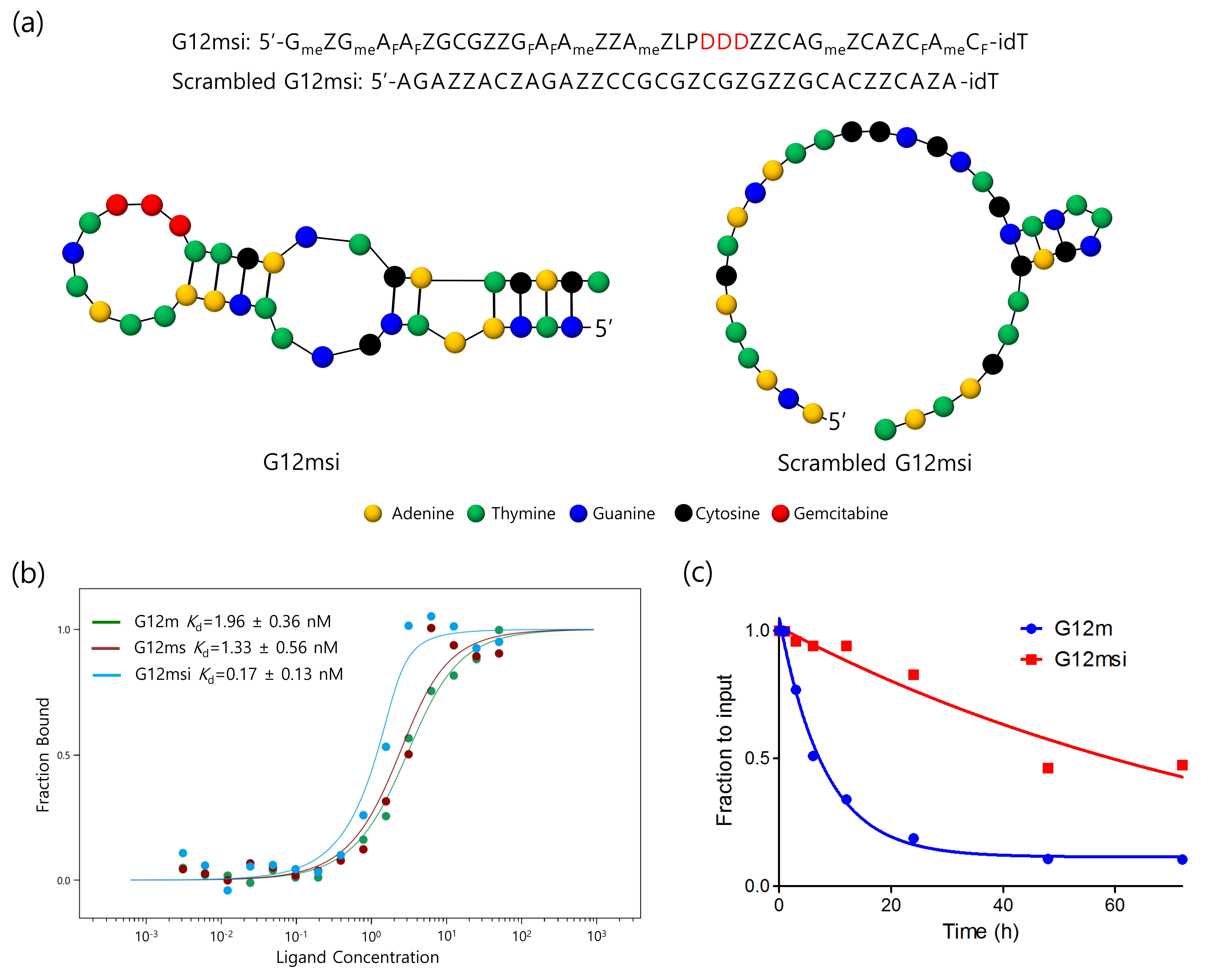
3.1. Synthesis and Characterization of G12msi Aptamer
3.2. Binding Specificity of G12msi Aptamer
3.3. Intracellular Localization of G12msi Aptamer
3.4. Tumor Cell Cytotoxicity and DNA Damage Effects of G12msi Aptamer
3.5. Effects of Nucleoside Transporter Inhibition on G12msi Aptamer
3.6. Regulation of Cell Cycle by G12msi Aptamer
3.7. In Vivo Anti-Tumor Activity of G12msi Aptamer
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Balogh, J.; Victor, D., III; Asham, E.H.; Burroughs, S.G.; Boktour, M.; Saharia, A.; Li, X.; Ghobrial, R.M.; Monsour, H.P., Jr. Hepatocellular carcinoma: A review. J. Hepatocell. Carcinoma 2016, 3, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Kumari, R.; Sahu, M.K.; Tripathy, A.; Uthansingh, K.; Behera, M. Hepatocellular carcinoma treatment: Hurdles, advances and prospects. Hepat. Oncol. 2018, 5, HEP08. [Google Scholar] [CrossRef] [PubMed]
- Daher, S.; Massarwa, M.; Benson, A.A.; Khoury, T. Current and future treatment of hepatocellular carcinoma: An updated comprehensive review. J. Clin. Transl. Hepatol. 2018, 6, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.W.; Talati, C.; Kim, R. Hepatocellular carcinoma (HCC): Beyond sorafenib-chemotherapy. J. Gastrointest. Oncol. 2017, 8, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Deng, G.L.; Zeng, S.; Shen, H. Chemotherapy and target therapy for hepatocellular carcinoma: New advances and challenges. World J. Hepatol. 2015, 7, 787–798. [Google Scholar] [CrossRef]
- Toschi, L.; Finocchiaro, G.; Bartolini, S.; Gioia, V.; Cappuzzo, F. Role of gemcitabine in cancer therapy. Future Oncol. 2005, 1, 7–17. [Google Scholar] [CrossRef]
- Yang, T.S.; Lin, Y.C.; Chen, J.S.; Wang, H.M.; Wang, C.H. Phase II study of gemcitabine in patients with advanced hepatocellular carcinoma. Cancer 2000, 89, 750–756. [Google Scholar] [CrossRef]
- Hammond, J.S.; Franko, J.; Holloway, S.E.; Heckman, J.T.; Orons, P.D.; Gamblin, T.C. Gemcitabine transcatheter arterial chemoembolization for unresectable hepatocellular carcinoma. Hepatogastroenterology 2014, 61, 1339–1343. [Google Scholar] [PubMed]
- Cuneo, K.C.; Davis, M.A.; Feng, M.U.; Novelli, P.M.; Ensminger, W.D.; Lawrence, T.S. Low dose rate radiosensitization of hepatocellular carcinoma in vitro and in patients. Transl. Oncol. 2014, 7, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Dyawanapelly, S.; Kumar, A.; Chourasia, M.K. Lessons learned from gemcitabine: Impact of therapeutic carrier systems and gemcitabine’s drug conjugates on cancer therapy. Crit Rev. Ther. Drug Carr. Syst. 2017, 34, 63–96. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Ren, T.; Zhao, S.; Shi, M.; Gu, J. Comparative pharmacokinetic study of PEGylated gemcitabine and gemcitabine in rats by LC-MS/MS coupled with pre-column derivatization and MSALL technique. Talanta 2020, 206, 120184. [Google Scholar] [CrossRef]
- Sloat, B.R.; Sandoval, M.A.; Li, D.; Chung, W.G.; Lansakara, D.S.; Proteau, P.J.; Kiguchi, K.; DiGiovanni, J.; Cui, Z. In vitro and in vivo anti-tumor activities of a gemcitabine derivative carried by nanoparticles. Int. J. Pharm. 2011, 409, 278–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federico, C.; Morittu, V.M.; Britti, D.; Trapasso, E.; Cosco, D. Gemcitabine-loaded liposomes: Rationale, potentialities and future perspectives. Int. J. Nanomed. 2012, 7, 5423–5436. [Google Scholar]
- Koshkina, N.V.; Kleinerman, E.S. Aerosol gemcitabine inhibits the growth of primary osteosarcoma and osteosarcoma lung metastases. Int. J. Cancer 2005, 116, 458–463. [Google Scholar] [CrossRef]
- Yoon, S.; Huang, K.W.; Reebye, V.; Spalding, D.; Przytycka, T.M.; Wang, Y.; Swiderski, P.; Li, L.; Armstrong, B.; Reccia, I.; et al. Aptamer-drug conjugates of active metabolites of nucleoside analogs and cytotoxic agents inhibit pancreatic tumor cell growth. Mol. Ther. Nucleic Acids 2017, 6, 80–88. [Google Scholar] [CrossRef]
- Karampelas, T.; Skavatsou, E.; Argyros, O.; Fokas, D.; Tamvakopoulos, C. Gemcitabine based peptide conjugate with improved metabolic properties and dual mode of efficacy. Mol. Pharm. 2017, 14, 674–685. [Google Scholar] [CrossRef]
- Xuan, W.; Peng, Y.; Deng, Z.; Peng, T.; Kuai, H.; Li, Y.; He, J.; Jin, C.; Liu, Y.; Wang, R.; et al. A basic insight into aptamer-drug conjugates (ApDCs). Biomaterials 2018, 182, 216–226. [Google Scholar] [CrossRef]
- Zhang, Y.; Lai, B.S.; Juhas, M. Recent advances in aptamer discovery and applications. Molecules 2019, 24, 941. [Google Scholar] [CrossRef] [Green Version]
- Richardson, F.C.; Richardson, K.K.; Kroin, J.S.; Hertel, L.W. Synthesis and restriction enzyme analysis of oligodeoxyribonucleotides containing the anti-cancer drug 2’,2’-difluoro-2’-deoxycytidine. Nucleic Acids Res. 1992, 20, 1763–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Niu, G.; Chen, X. Aptamer-Drug Conjugates. Bioconjug. Chem. 2015, 26, 2186–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.Y.; Cho, Y.L.; Chae, J.R.; Moon, S.H.; Cho, W.G.; Choi, Y.J.; Lee, S.J.; Kang, W.J. Gemcitabine-incorporated G-quadruplex aptamer for targeted drug delivery into pancreas cancer. Mol. Ther. Nucleic Acids 2018, 12, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Nakatsura, T.; Yoshitake, Y.; Senju, S.; Monji, M.; Komori, H.; Motomura, Y.; Hosaka, S.; Beppu, T.; Ishiko, T.; Kamohara, H.; et al. Glypican-3, overexpressed specifically in human hepatocellular carcinoma, is a novel tumor marker. Biochem. Biophys. Res. Commun. 2003, 306, 16–25. [Google Scholar] [CrossRef]
- Yamauchi, N.; Watanabe, A.; Hishinuma, M.; Ohashi, K.; Midorikawa, Y.; Morishita, Y.; Niki, T.; Shibahara, J.; Mori, M.; Makuuchi, M.; et al. The glypican 3 oncofetal protein is a promising diagnostic marker for hepatocellular carcinoma. Mod. Pathol. 2005, 18, 1591–1598. [Google Scholar] [CrossRef]
- Dong, L.; Zhou, H.; Zhao, M.; Gao, X.; Liu, Y.; Liu, D.; Guo, W.; Hu, H.; Xie, Q.; Fan, J.; et al. Phosphorothioate-modified AP613-1 specifically targets GPC3 when used for hepatocellular carcinoma cell imaging. Mol. Ther. Nucleic Acids 2018, 13, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Dong, L.; Liu, Z.; Yang, S.; Wu, W.; Lin, J. In vivo fluorescence imaging of hepatocellular carcinoma using a novel GPC3-specific aptamer probe. Quant. Imaging Med. Surg. 2018, 8, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Davies, D.R.; Gelinas, A.D.; Zhang, C.; Rohloff, J.C.; Carter, J.D.; O’Connell, D.; Waugh, S.M.; Wolk, S.K.; Mayfield, W.S.; Burgin, A.B.; et al. Unique motifs and hydrophobic interactions shape the binding of modified DNA ligands to protein targets. Proc. Natl. Acad. Sci. USA 2012, 109, 19971–19976. [Google Scholar] [CrossRef] [Green Version]
- Yunn, N.O.; Koh, A.; Han, S.; Lim, J.H.; Park, S.; Lee, J.; Kim, E.; Jang, S.K.; Berggren, P.O.; Ryu, S.H. Agonistic aptamer to the insulin receptor leads to biased signaling and functional selectivity through allosteric modulation. Nucleic Acids Res. 2015, 43, 7688–7701. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 440. [Google Scholar] [CrossRef] [Green Version]
- Ni, S.; Yao, H.; Wang, L.; Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Chemical modifications of nucleic acid aptamers for therapeutic purposes. Int. J. Mol. Sci. 2017, 18, 1683. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Ozer, A.; Pagano, J.M.; Lis, J.T. New technologies provide quantum changes in the scale, speed, and success of SELEX methods and aptamer characterization. Mol. Ther. Nucleic Acids 2014, 3, e183. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Kang, H.; Ryu, S.H.; Lee, D.S.; Lee, J.H.; Kim, S. Bioimaging of nucleolin aptamer-containing 5-(N-benzylcarboxyamide)-2’-deoxyuridine more capable of specific binding to targets in cancer cells. J. Biomed. Biotechnol. 2010, 2010, 168306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odeh, F.; Nsairat, H.; Alshaer, W.; Ismail, M.A.; Esawi, E.; Qaqish, B.; Bawab, A.A.; Ismail, S.I. Aptamers chemistry: Chemical modifications and conjugation strategies. Molecules 2019, 25, 3. [Google Scholar] [CrossRef] [Green Version]
- Cedergren, R.; Lang, B.F.; Gravel, D. A mechanism for the RNA-catalyzed formation of 5’-phosphates. The origin of nucleases. FEBS Lett. 1987, 226, 63–66. [Google Scholar] [CrossRef] [Green Version]
- Trujillo, C.A.; Nery, A.A.; Alves, J.M.; Martins, A.H.; Ulrich, H. Development of the anti-VEGF aptamer to a therapeutic agent for clinical ophthalmology. Clin. Ophthalmol. 2007, 1, 393–402. [Google Scholar]
- Dougan, H.; Lyster, D.M.; Vo, C.V.; Stafford, A.; Weitz, J.I.; Hobbs, J.B. Extending the lifetime of anticoagulant oligodeoxynucleotide aptamers in blood. Nucl. Med. Biol. 2000, 27, 289–297. [Google Scholar] [CrossRef]
- Shum, K.T.; Tanner, J.A. Differential inhibitory activities and stabilisation of DNA aptamers against the SARS coronavirus helicase. Chembiochem. 2008, 9, 3037–3045. [Google Scholar] [CrossRef]
- Serdjebi, C.; Milano, G.; Ciccolini, J. Role of cytidine deaminase in toxicity and efficacy of nucleosidic analogs. Expert Opin. Drug Metab. Toxicol. 2015, 11, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Lohitesh, K.; Chowdhury, R.; Mukherjee, S. Resistance a major hindrance to chemotherapy in hepatocellular carcinoma: An insight. Cancer Cell Int. 2018, 18, 44. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.Y.; Chae, J.R.; Cho, Y.L.; Kim, Y.; Lee, D.; Lee, J.K.; Kang, W.J. Targeted Therapy of Hepatocellular Carcinoma Using Gemcitabine-Incorporated GPC3 Aptamer. Pharmaceutics 2020, 12, 985. https://doi.org/10.3390/pharmaceutics12100985
Park JY, Chae JR, Cho YL, Kim Y, Lee D, Lee JK, Kang WJ. Targeted Therapy of Hepatocellular Carcinoma Using Gemcitabine-Incorporated GPC3 Aptamer. Pharmaceutics. 2020; 12(10):985. https://doi.org/10.3390/pharmaceutics12100985
Chicago/Turabian StylePark, Jun Young, Ju Ri Chae, Ye Lim Cho, Youndong Kim, Dasom Lee, Jeong Kyun Lee, and Won Jun Kang. 2020. "Targeted Therapy of Hepatocellular Carcinoma Using Gemcitabine-Incorporated GPC3 Aptamer" Pharmaceutics 12, no. 10: 985. https://doi.org/10.3390/pharmaceutics12100985
APA StylePark, J. Y., Chae, J. R., Cho, Y. L., Kim, Y., Lee, D., Lee, J. K., & Kang, W. J. (2020). Targeted Therapy of Hepatocellular Carcinoma Using Gemcitabine-Incorporated GPC3 Aptamer. Pharmaceutics, 12(10), 985. https://doi.org/10.3390/pharmaceutics12100985