Optimising PLGA-PEG Nanoparticle Size and Distribution for Enhanced Drug Targeting to the Inflamed Intestinal Barrier

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
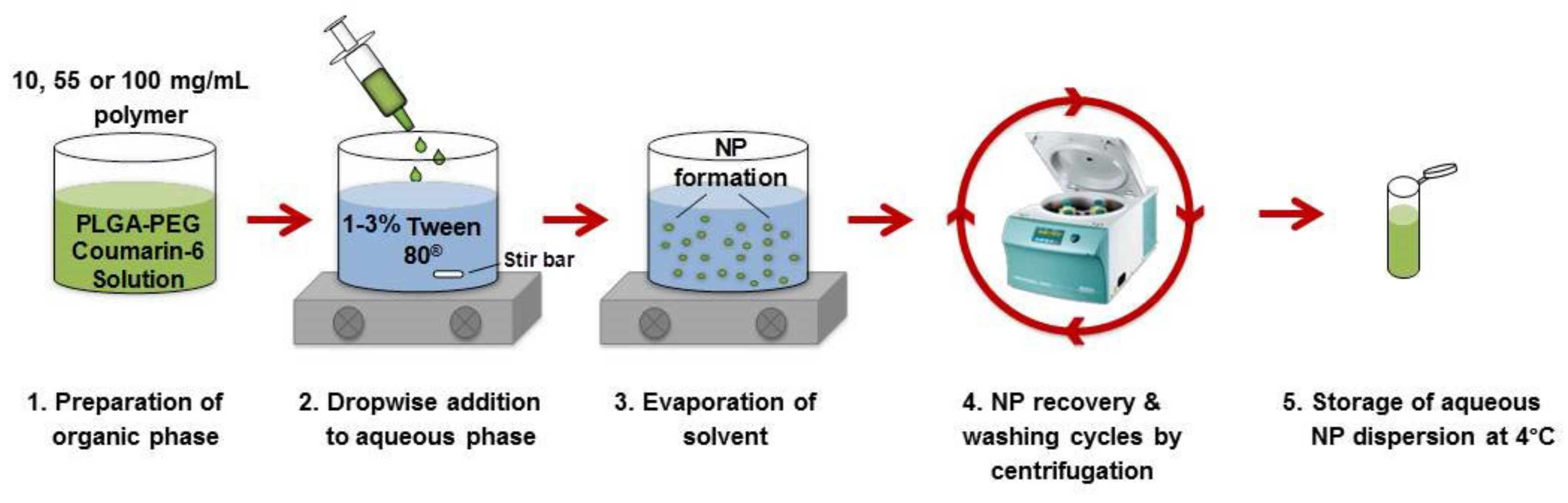
2.2. Formulation of PLGA-PEG NPs
2.3. NP Characterisation
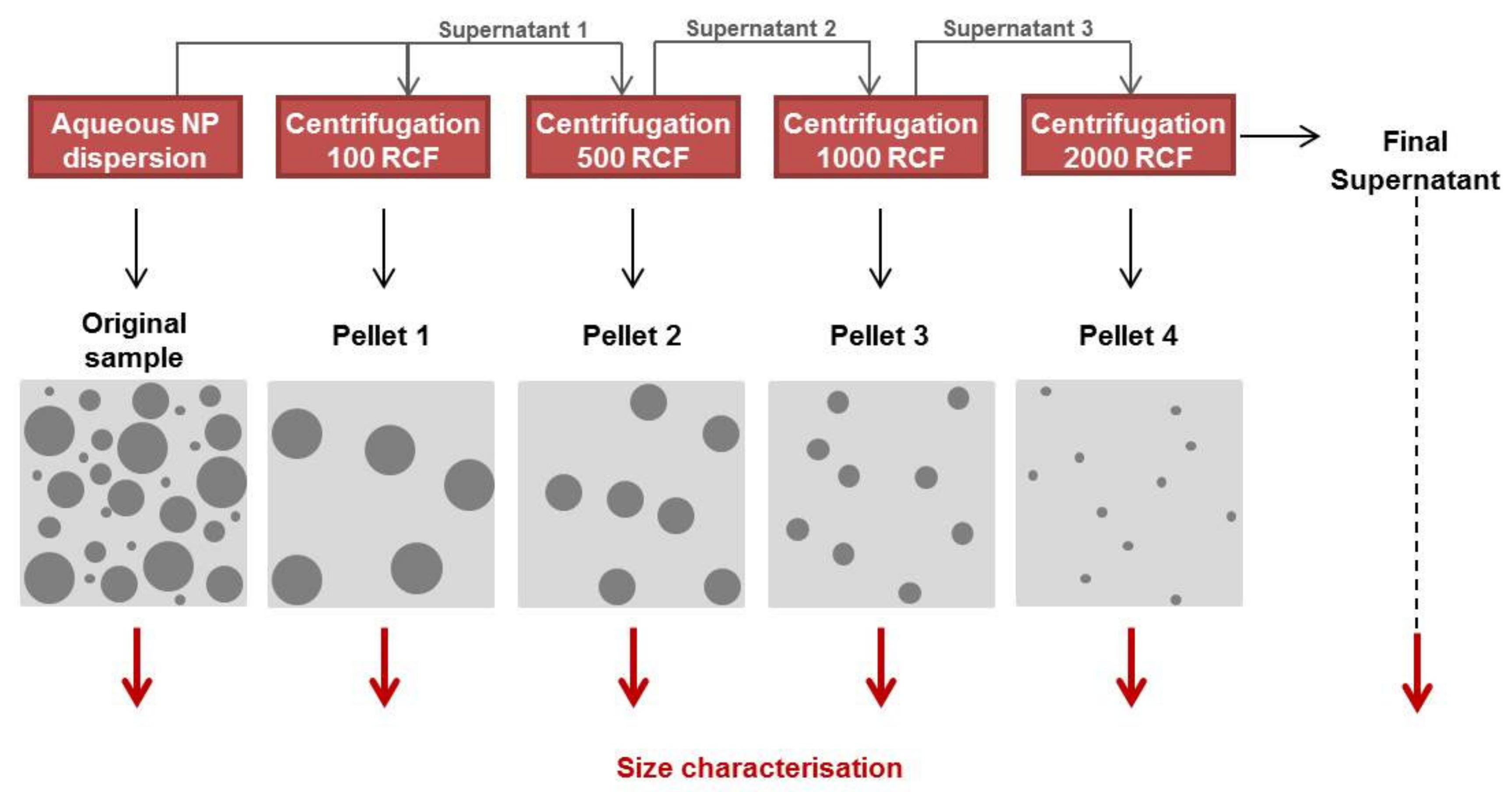
2.4. Size Distribution Analysis of NPs Using Differential Centrifugation
2.5. Cell Culture
2.6. In Vitro Interaction of PLGA-PEG NPs With Caco-2 Cells
2.7. Statistical Analysis
3. Results and Discussion
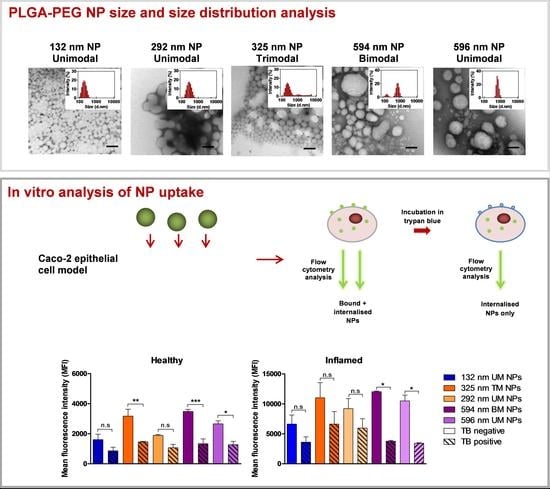
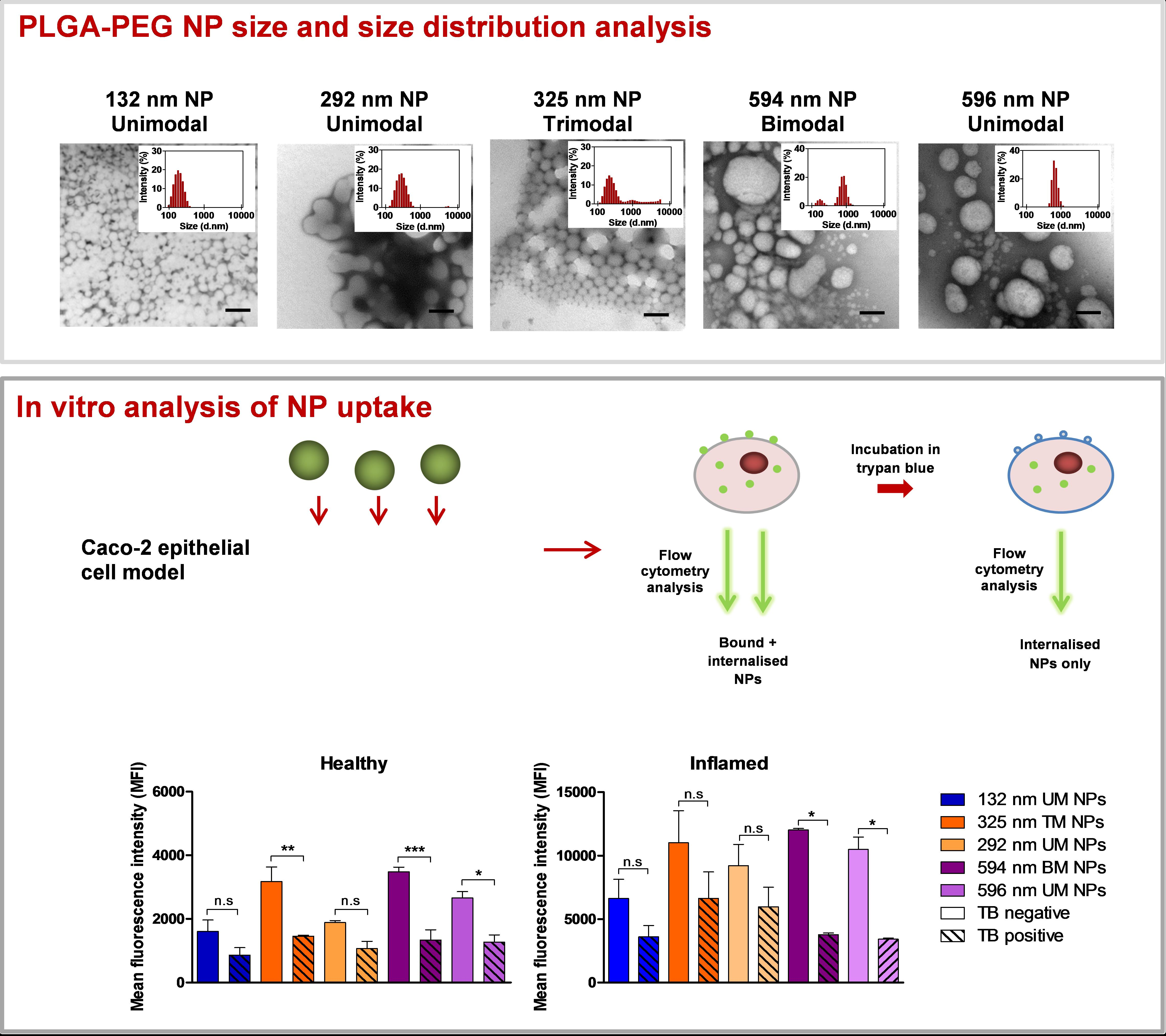
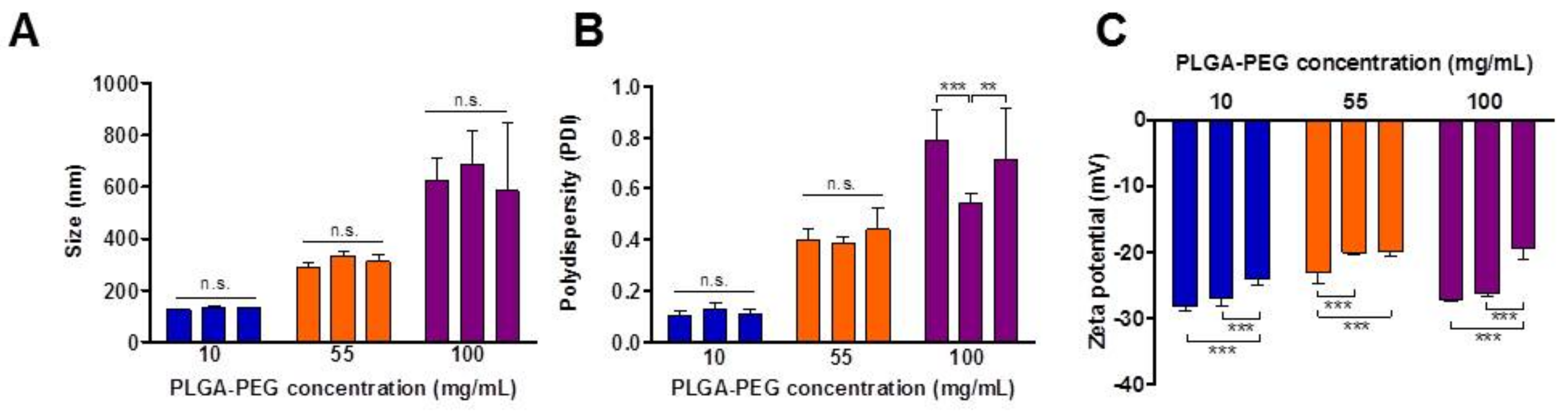
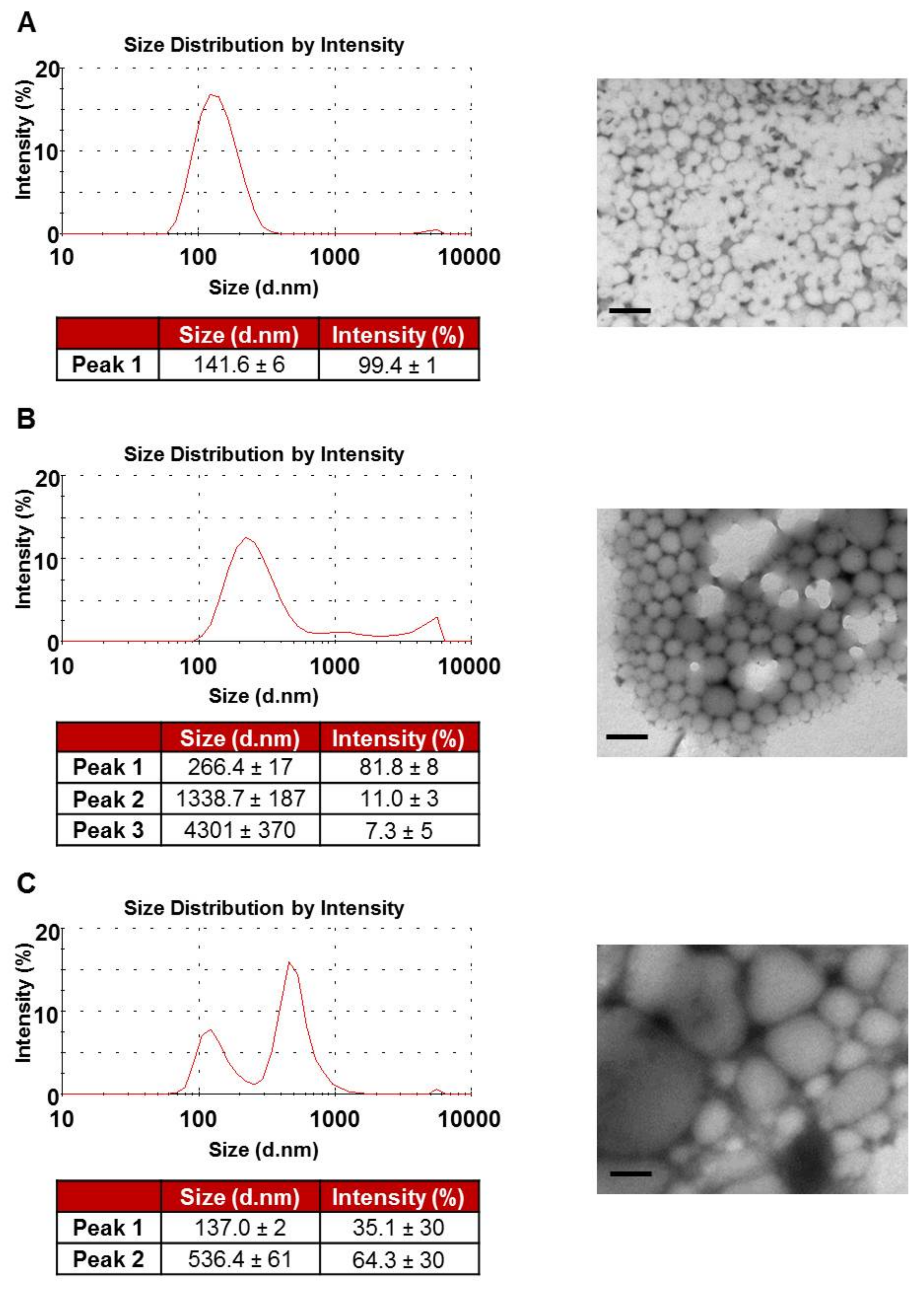
3.1. NP Characterisation
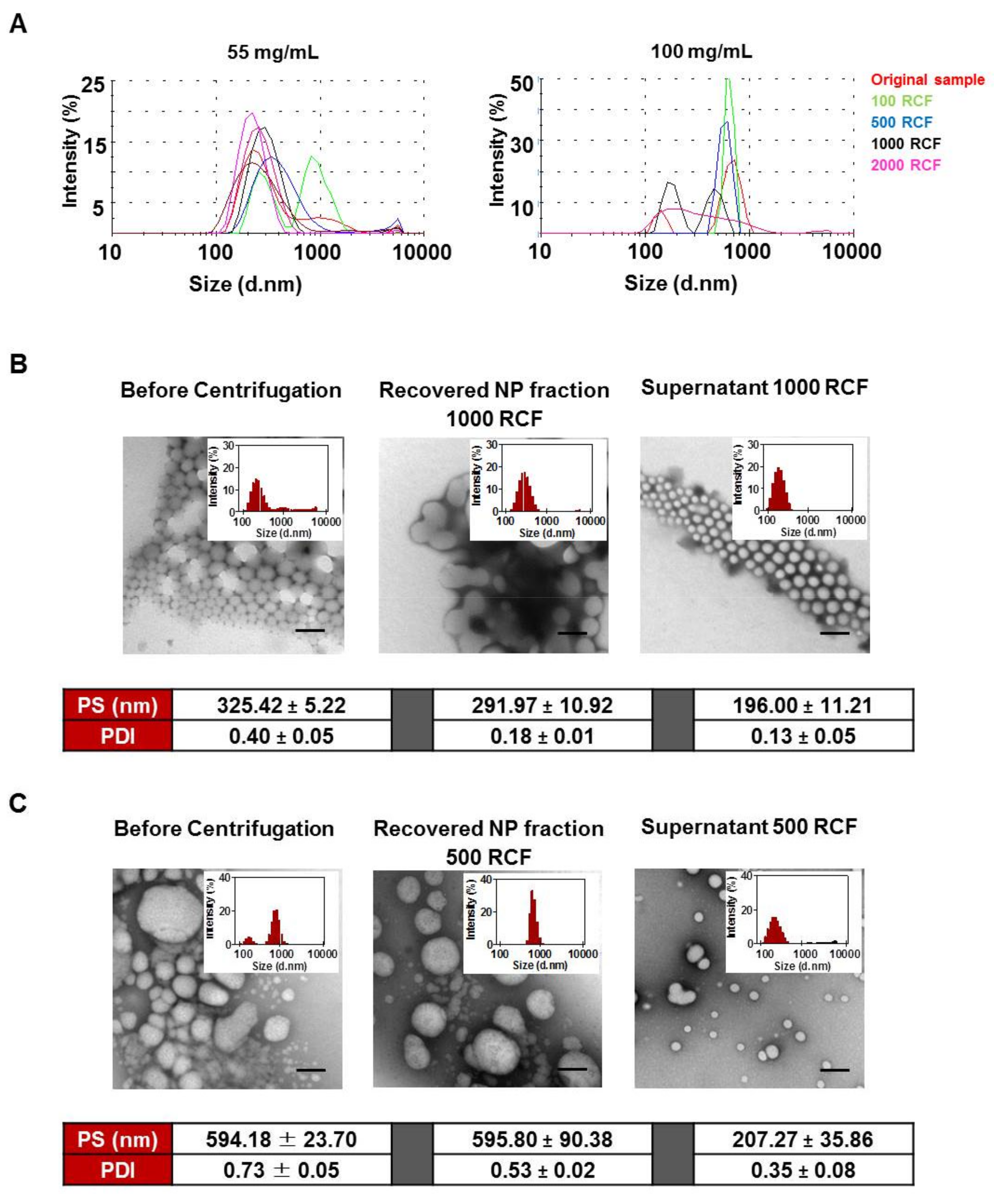
3.2. Size Distribution Analysis of NPs Using Differential Centrifugation
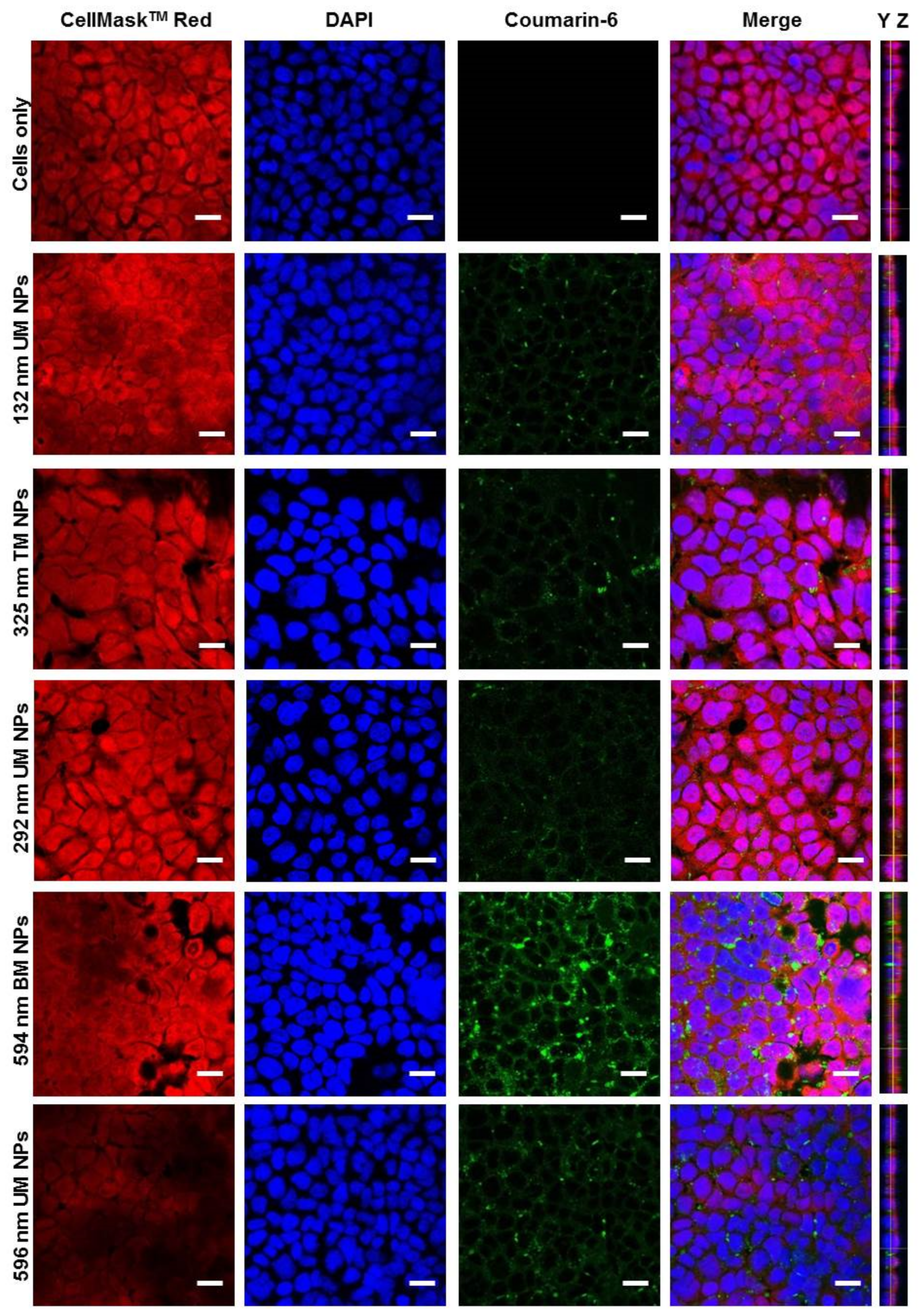
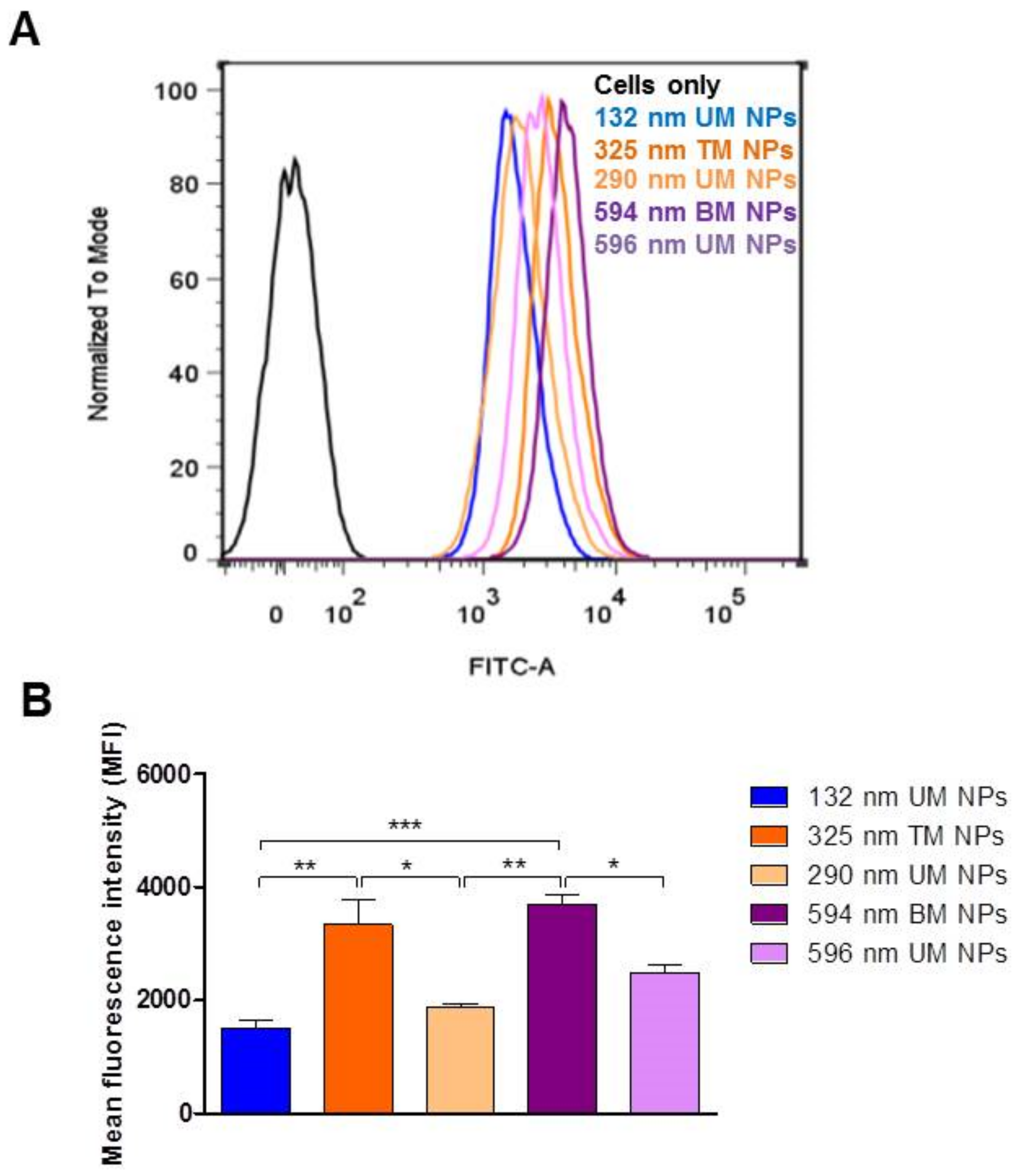
3.3. Effect of PS and PDI on NP Uptake/Interaction in Caco-2 Cells
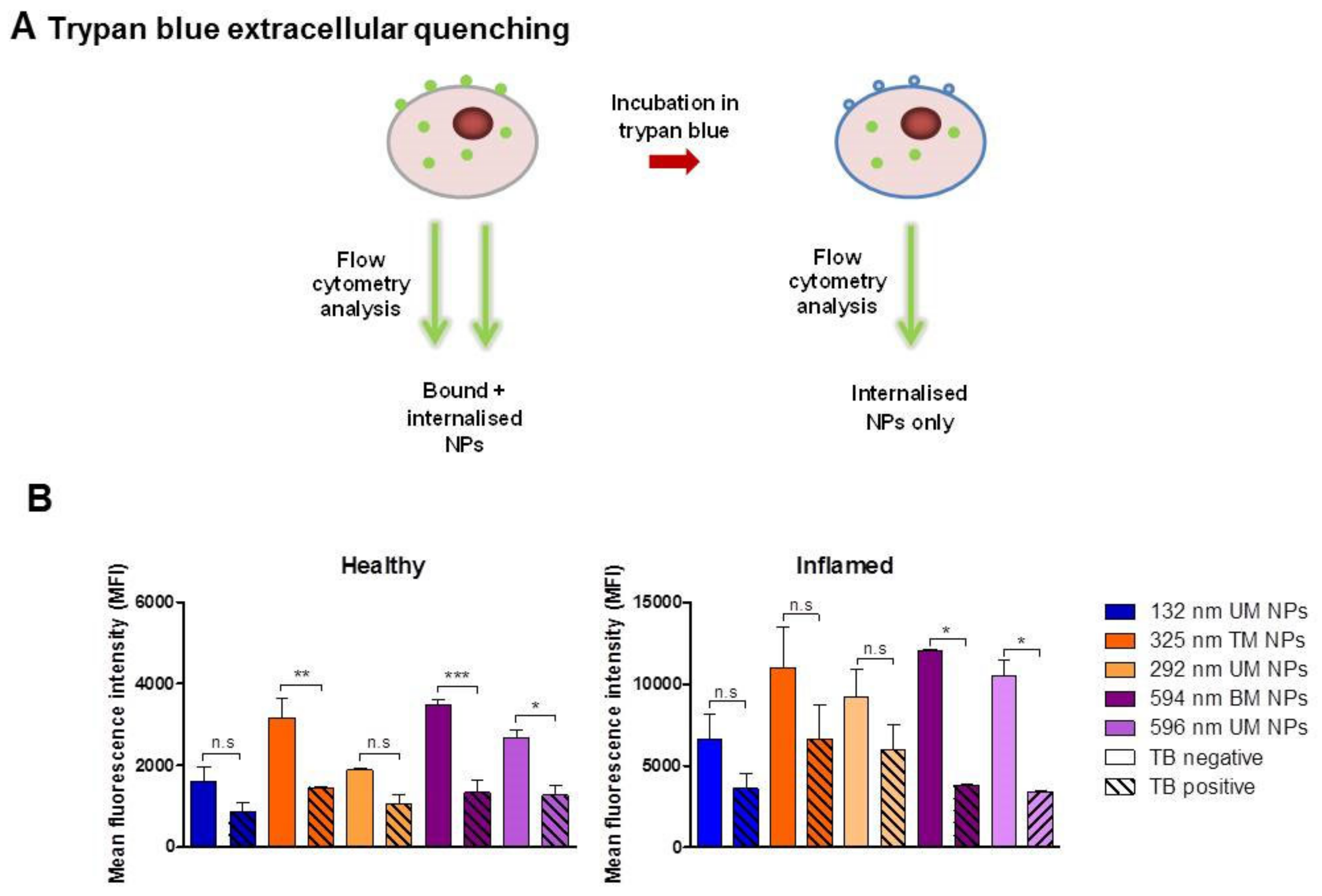
3.4. Evaluation of NP Internalisation in Healthy and Inflamed Epithelial Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burisch, J.; Munkholm, P. The epidemiology of inflammatory bowel disease. Scand. J. Gastroenterol. 2015, 50, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Mohan, L.J.; Daly, J.S.; Ryan, B.M.; Ramtoola, Z. The future of nanomedicine in optimising the treatment of inflammatory bowel disease. Scand. J. Gastroenterol. 2019, 54, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Sastry, S.V.; Nyshadham, J.R.; Fix, J.A. Recent technological advances in oral drug delivery—A review. Pharm. Sci. Technol. Today 2000, 3, 138–145. [Google Scholar] [CrossRef]
- Choonara, B.F.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; Du Toit, L.C.; Pillay, V. A review of advanced oral drug delivery technologies facilitating the protection and absorption of protein and peptide molecules. Biotechnol. Adv. 2014, 32, 1269–1282. [Google Scholar] [CrossRef]
- Singh, R.; Lillard, J.W. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Si, X.Y.; Merlin, D.; Xiao, B. Recent advances in orally administered cell-specific nanotherapeutics for inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 7718–7726. [Google Scholar] [CrossRef]
- Lamprecht, A.; Schafer, U.; Lehr, C.M. Size-dependent bioadhesion of micro- and nanoparticulate carriers to the inflamed colonic mucosa. Pharm. Res. 2001, 18, 788–793. [Google Scholar] [CrossRef]
- Kulkarni, S.A.; Feng, S.-S. Effects of Particle Size and Surface Modification on Cellular Uptake and Biodistribution of Polymeric Nanoparticles for Drug Delivery. Pharm. Res. 2013, 30, 2512–2522. [Google Scholar] [CrossRef]
- Win, K.Y.; Feng, S.S. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials 2005, 26, 2713–2722. [Google Scholar] [CrossRef]
- Dos Santos, T.; Varela, J.; Lynch, I.; Salvati, A.; Dawson, K.A. Quantitative assessment of the comparative nanoparticle-uptake efficiency of a range of cell lines. Small 2011, 7, 3341–3349. [Google Scholar] [CrossRef] [Green Version]
- Ramtoola, Z.; Lyons, P.; Keohane, K.; Kerrigan, S.W.; Kirby, B.P.; Kelly, J.G. Investigation of the interaction of biodegradable micro- and nanoparticulate drug delivery systems with platelets. J. Pharm. Pharmacol. 2011, 63, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- McClean, S.; Prosser, E.; Meehan, E.; O’Malley, D.; Clarke, N.; Ramtoola, Z.; Brayden, D. Binding and uptake of biodegradable poly-dl-lactide micro- and nanoparticles in intestinal epithelia. Eur. J. Pharm. Sci. 1998, 6, 153–163. [Google Scholar] [CrossRef]
- Gaumet, M.; Gurny, R.; Delie, F. Localization and quantification of biodegradable particles in an intestinal cell model: The influence of particle size. Eur. J. Pharm. Sci. 2009, 36, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, C.-G.; Yu, Y. Separation of monodisperse alginate nanoparticles and effect of particle size on transport of vitamin E. Carbohydr. Polym. 2015, 124, 274–279. [Google Scholar] [CrossRef]
- Lautenschläger, C.; Schmidt, C.; Lehr, C.M.; Fischer, D.; Stallmach, A. PEG-functionalized microparticles selectively target inflamed mucosa in inflammatory bowel disease. Eur. J. Pharm. Biopharm. 2013, 85, 578–586. [Google Scholar] [CrossRef]
- Kirby, B.P.; Pabari, R.; Chen, C.N.; Al Baharna, M.; Walsh, J.; Ramtoola, Z. Comparative evaluation of the degree of pegylation of poly(lactic-co-glycolic acid) nanoparticles in enhancing central nervous system delivery of loperamide. J. Pharm. Pharmacol. 2013, 65, 1473–1481. [Google Scholar] [CrossRef]
- Beloqui, A.; Solinís, M.Á; Gascón, A.R.; Del Pozo-Rodríguez, A.; Des Rieux, A.; Préat, V. Mechanism of transport of saquinavir-loaded nanostructured lipid carriers across the intestinal barrier. J. Control. Release 2013, 166, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Pabari, R.M.; Mattu, C.; Partheeban, S.; Almarhoon, A.; Boffito, M.; Ciardelli, G.; Ramtoola, Z. Novel Polyurethane-based nanoparticles of infliximab to reduce inflammation in an in-vitro intestinal epithelial barrier model. Int. J. Pharm. 2019. [Google Scholar] [CrossRef]
- Van De Walle, J.; Hendrickx, A.; Romier, B.; Larondelle, Y.; Schneider, Y.J. Inflammatory parameters in Caco-2 cells: Effect of stimuli nature, concentration, combination and cell differentiation. Toxicol. In Vitro 2010, 24, 1441–1449. [Google Scholar] [CrossRef]
- Vranic, S.; Boggetto, N.; Contremoulins, V.; Mornet, S.; Reinhardt, N.; Marano, F.; Baeza-Squiban, A.; Boland, S. Deciphering the mechanisms of cellular uptake of engineered nanoparticles by accurate evaluation of internalization using imaging flow cytometry. Part. Fibre Toxicol. 2013, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Teply, B.A.; Sherifi, I.; Sung, J.; Luther, G.; Gu, F.X.; Levy-Nissenbaum, E.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. Formulation of functionalized PLGA–PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 2007, 28, 869–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draheim, C.; De Crécy, F.; Hansen, S.; Collnot, E.M.; Lehr, C.M. A Design of Experiment Study of Nanoprecipitation and Nano Spray Drying as Processes to Prepare PLGA Nano- and Microparticles with Defined Sizes and Size Distributions. Pharm. Res. 2015, 32, 2609–2624. [Google Scholar] [CrossRef] [PubMed]
- Nasef, A.; Gardouh, A.R.; Ghorab, M. Polymeric nanoparticles: Influence of polymer, surfactant and composition of manufacturing vehicle on particle size. World J. Pharm. Sci. 2015, 3, 2308–2322. [Google Scholar]
- Guhagarkar, S.A.; Malshe, V.C.; Devarajan, P.V. Nanoparticles of Polyethylene Sebacate: A New Biodegradable Polymer. AAPS Pharmscitech 2009, 10, 935. [Google Scholar] [CrossRef] [Green Version]
- Rafiei, P.; Haddadi, A. Docetaxel-loaded PLGA and PLGA-PEG nanoparticles for intravenous application: Pharmacokinetics and biodistribution profile. Int. J. Nanomed. 2017, 12, 935–947. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Lung, P.S.; Zhao, S.; Chu, Z.; Chrzanowski, W.; Li, Q. Shape dependent cytotoxicity of PLGA-PEG nanoparticles on human cells. Sci. Rep. 2017, 7, 7315. [Google Scholar] [CrossRef]
- Clogston, J.D.; Patri, A.K. Zeta Potential Measurement, in Characterization of Nanoparticles Intended for Drug Delivery; McNeil, S.E., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 63–70. [Google Scholar]
- Nel, A.E.; Parak, W.J.; Chan, W.C.; Xia, T.; Hersam, M.C.; Brinker, C.J.; Zink, J.I.; Pinkerton, K.E.; Baer, D.R.; Weiss, P.S. Where Are We Heading in Nanotechnology Environmental Health and Safety and Materials Characterization? ACS Nano 2015, 9, 5627–5630. [Google Scholar] [CrossRef]
- Robertson, J.D.; Rizzello, L.; Avila-Olias, M.; Gaitzsch, J.; Contini, C.; Magoń, M.S.; Renshaw, S.A.; Battaglia, G. Purification of Nanoparticles by Size and Shape. Sci. Rep. 2016, 6, 27494. [Google Scholar] [CrossRef]
- Zhou, G.; Zhang, J.; Pan, C.; Liu, N.; Wang, Z.; Zhang, J. Enhanced Uptake of Fe3O4; Nanoparticles by Intestinal Epithelial Cells in a State of Inflammation. Molecules 2017, 22, 1240. [Google Scholar] [CrossRef]
- Clark, R.T.; Hope, A.; Lopez-Fraga, M.; Schiller, N.; Lo, D.D. Bacterial Particle Endocytosis by Epithelial Cells Is Selective and Enhanced by Tumor Necrosis Factor Receptor Ligands. Clin. Vaccine Immunol. 2009, 16, 397–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruewer, M.; Luegering, A.; Kucharzik, T.; Parkos, C.A.; Madara, J.L.; Hopkins, A.M.; Nusrat, A. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J. Immunol. 2003, 171, 6164–6172. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment No. | PLGA-PEG (mg/mL) | Tween 80® (% w/v) | PS ± SD (nm) | PDI ± SD | ZP ± SD (mV) |
|---|---|---|---|---|---|
| 1 | 100 | 1 | 600.86 ± 192.94 a | 0.76 ± 0.11 a | −22.62 ± 0.49 b |
| 2 | 10 | 3 | 145.76 ± 4.60 e | 0.20 ± 0.02 d | −23.40 ± 1.39 e |
| 3 | 10 | 2 | 145.50 ± 3.53 d,e | 0.28 ± 0.03 d | −22.82 ± 2.72 e |
| 4 | 100 | 3 | 417.42 ± 237.27 b | 0.63 ± 0.19 a | −20.00 ± 0.90 c |
| 5 | 10 | 1 | 167.72 ± 8.13 d | 0.32 ± 0.04 d | −22.32 ± 2.67 e |
| 6 | 55 | 1 | 293.48 ± 27.38 c | 0.51 ± 0.03 b | −23.18 ± 0.51 d |
| 7 | 55 | 3 | 177.66 ± 5.22 c | 0.29 ± 0.05 c | −22.08 ± 0.74 d |
| 8 | 100 | 2 | 577.48 ± 134.93 a,b | 0.69 ± 0.11 a | −26.52 ± 1.79 a |
| 9 | 55 | 2 | 319.62 ± 13.65 c | 0.35 ± 0.03 c | −24.24 ± 0.73 d |
| PLGA-PEG (mg/mL) | PS ± SD | PDI ± SD | ZP ± SD |
|---|---|---|---|
| 10 | 131.74 ± 3.67 c | 0.15 ± 0.04 c | −25.85 ± 4.38 |
| 55 | 312.01 ± 21.87 b | 0.41 ± 0.03 b | −20.97 ± 1.81 |
| 100 | 630.85 ± 52.50 a | 0.68 ± 0.13 a | −24.25 ± 4.21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohan, L.J.; McDonald, L.; Daly, J.S.; Ramtoola, Z. Optimising PLGA-PEG Nanoparticle Size and Distribution for Enhanced Drug Targeting to the Inflamed Intestinal Barrier. Pharmaceutics 2020, 12, 1114. https://doi.org/10.3390/pharmaceutics12111114
Mohan LJ, McDonald L, Daly JS, Ramtoola Z. Optimising PLGA-PEG Nanoparticle Size and Distribution for Enhanced Drug Targeting to the Inflamed Intestinal Barrier. Pharmaceutics. 2020; 12(11):1114. https://doi.org/10.3390/pharmaceutics12111114
Chicago/Turabian StyleMohan, Lauren J., Lauren McDonald, Jacqueline S. Daly, and Zebunnissa Ramtoola. 2020. "Optimising PLGA-PEG Nanoparticle Size and Distribution for Enhanced Drug Targeting to the Inflamed Intestinal Barrier" Pharmaceutics 12, no. 11: 1114. https://doi.org/10.3390/pharmaceutics12111114
APA StyleMohan, L. J., McDonald, L., Daly, J. S., & Ramtoola, Z. (2020). Optimising PLGA-PEG Nanoparticle Size and Distribution for Enhanced Drug Targeting to the Inflamed Intestinal Barrier. Pharmaceutics, 12(11), 1114. https://doi.org/10.3390/pharmaceutics12111114