Nanomaterials Designed for Antiviral Drug Delivery Transport across Biological Barriers

 , ,
, ,  ,
,  , and
, and
Abstract
:1. Social Impact and Economic Burden of Viral Infectious Diseases
- Dependence of viruses replication on host cell biosynthetic machinery [19], that leads to a limited number of virus-specific metabolic functions can be targeted by antiviral drugs without any damage to the host;
- the viruses’ functions are specific to each virus, preventing the development of a broad-spectrum antivirals fighting against different viruses that cause similar symptoms. Antivirals developed for some viruses (as HSV and HIV) can treat the acute illness, but do not cure the latent infection. This leads to recurrent or chronic diseases that require treatment for longer periods of time [18].
2. Viruses: Types, Current Therapy and Observed Drawbacks
- I—dsDNA viruses (e.g., adenoviruses, herpesviruses, poxviruses): enter to the host nucleus and are dependent by host cell polymerases to replicate viral genome. The virus may induce the cell to forcefully undergo cell division, which may lead to transformation of the cell and, ultimately, to cancer.
- II—ssDNA viruses (+ strand or “sense”) DNA (e.g., parvoviruses), consists of viruses that have a single-stranded DNA genome of the same polarity as the mRNA. Excepting Parvoviruses, most of them have circular genomes and are replicating within nucleus.
- III—dsRNA viruses (e.g., reoviruses): not dependent by host replication polymerases and their replication (monocistronic) is realized into capsid (in cytoplasm).
- IV—(+)ssRNA viruses (+ strand or sense) RNA (e.g., picornaviruses, Togaviruses): the RNA can be directly accessed by ribosomes of the host to form proteins, and use a simple reproduction pathway (viruses with polycistronic mRNA) or a more complex transcription pathway (for which subgenomic mRNAs, ribosomal frameshifting, and proteolytic processing of polyproteins may be used).
- V—(−)ssRNA viruses (− strand or antisense) RNA (e.g., orthomyxoviruses, rhabdoviruses), that first must be transcribed by viral polymerases (positive-sense) before can be directly accessed by host ribosomes to form proteins.
- VI—ssRNA-RT viruses (+ strand or sense) RNA with DNA intermediate in life-cycle (e.g., retroviruses), which use the reverse transcriptase to convert the positive-sense RNA into DNA. They are using DNA to create the templates and those are spliced into host genome by integrase.
- VII—dsDNA-RT viruses DNA with RNA intermediate in life-cycle (e.g., hepadnaviruses), dsDNA viruses that replicate through a single-stranded RNA intermediate, which use a pregenome RNA as a template and conversion to DNA is done by a viral reverse transcriptase.
3. Biological Barriers Security System
3.1. Mucus
3.2. Skin
3.3. Cell Membrane
3.4. Blood-Brain Barrier
- polymeric polybutylcyanoacrylate (PBCA) nanoparticles with two incorporeated antiretroviral drugs (AZT and lamivudine) showed a 8–20 and 10–18 fold increase in BBB permeation, by three possible mechanisms as presented by the authors: prolonged interaction interval between drug-loaded nanoparticles and brain-microvascular endothelial cells elevated the concentration gradient between blood and the brain, Polysorbate 80 covering on the periphery of nanoparticles was able to be absorbed and degraded nanoparticles improved drug absorption [112];
- spherical transferrin coated-PEGylated albumin nanoparticles encapsulating AZT prepared by ultra-emulsification method using chemical cross-linking by glutaraldehyde gained an access across the BBB through the transferrin receptor mediated endocytosis on the membrane [113];
- transferrin-conjugated quantum rod nanoparticles conjugated with saquinavir crossed an in vitro BBB model by exploiting a receptor-mediated transport [114];
- magnetic liposomal nanoformulations of azidothymidine 5′-triphosphate (the active form of azidothymidine) migrate across BBB in vitro, either directly or by a monocyte-mediated transport, under the influence of an external magnetic field [115];
- novel nanodrug consisting of an iron oxide nanoparticle coated with PMA amphiphilic polymer and functionalized with the antiretroviral peptide enfuvirtide crossed the BBB by a passive diffusion, probably mediated by the absorption of the amphiphilic coating on the cell membrane [116].
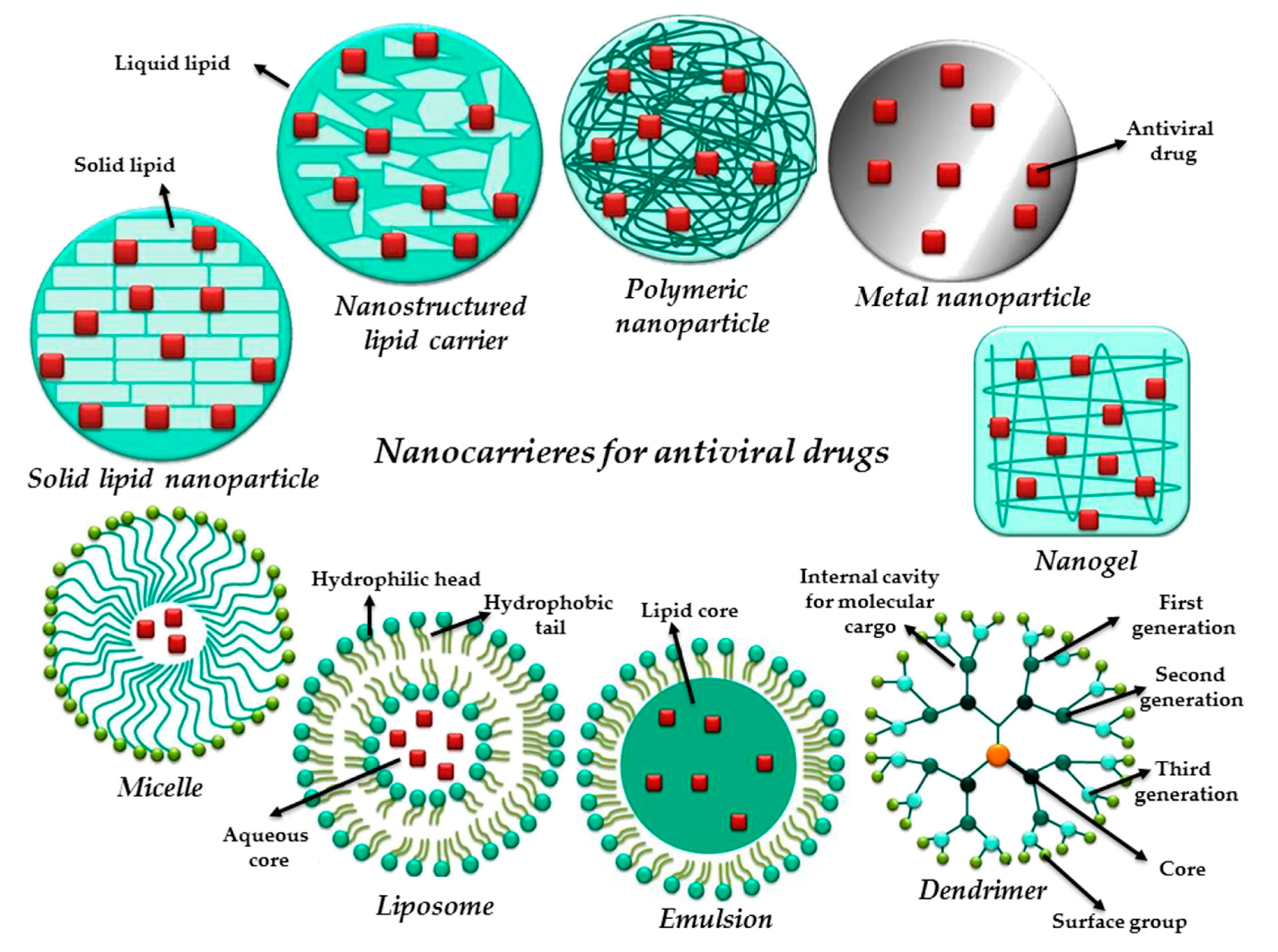
4. Nanotechnology: How Does It Face the Antiviral Therapy?
5. Current Overview of Nanotechnology Use in Antiviral Therapy: Virus Related
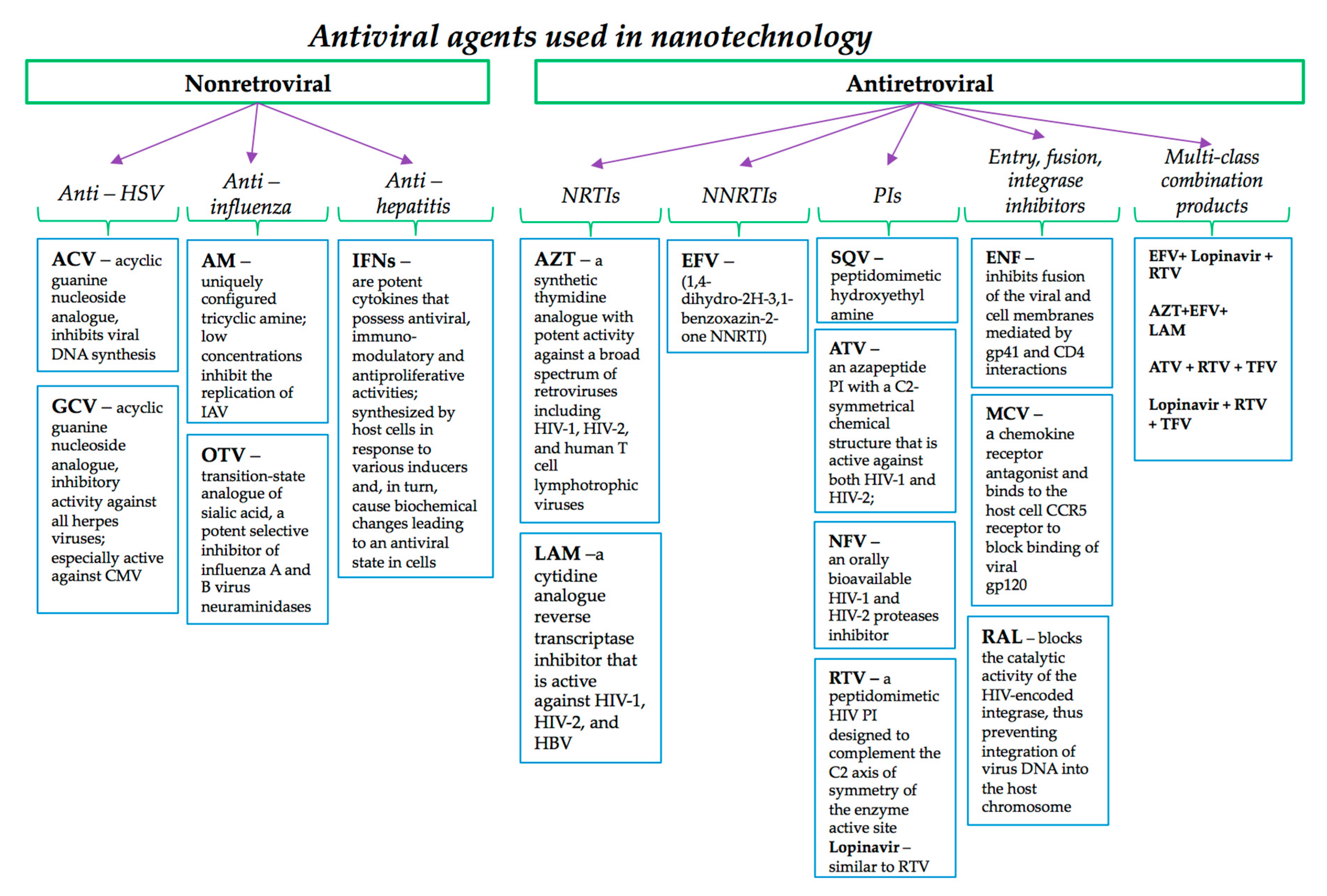
5.1. Nanomaterials Designed for Non-Retroviral Antiviral Agents
5.1.1. Nano-Based Antiviral Agents against Herpes Viruses
5.1.2. Nanomaterials with Antiviral Intrinsic Activity
Gold and Silver NPs Using Seaweed Sargassum wightii with Anti-Herpetic Activity
Lipid Nanoemulsions Encapsulating Coumestrol as Topical Treatment of Herpes Simplex
5.2. Nanomaterials Designed for Antiretroviral Drug Delivery
5.2.1. NRTIs and NNRTIs
5.2.2. PIs
5.2.3. Fusion Inhibitors, Entry Inhibitors and Integrase Inhibitors
6. Progress in Nanomedicine: Antiviral Nanotherapeutics Approved or under Evaluation
- Fluquit (STP 702) from Sirnaomics Inc. currently under preclinical evaluation, a polymer-based nanotherapeutic that incorporates siRNA and targeting the H5N1 (avian flu), H1N1 (swine flu) influenza, and newly emerging H7N9; and cervisil (STP909), a nanobased drug candidate, which incorporates siRNA for the treatment of HPV16 and HPV18;
- DermaVir from Genetic Immunity, a synthetic pathogen-like nanomedicine that incorporates single plasmid DNA expressing 15 HIV antigens that assemble to HIV-like particles; DermaVir vaccine completed Phase I/II randomized, placebo-controlled, dose-finding, double-blinded, multicenter study to assess the safety, tolerability and immune response in HIV-1-infected adults who are currently receiving anti-HIV treatment (number NCT00270205) [194];
- Doravirine (MK-1439), from Merck, a novel, next generation NNRTI described as solid drug nanoparticle formulation tested for HIV; currently doravirine completed the pharmacokinetic trial of the bioavailability of four MK-1439 nano formulations in healthy adults (number NCT02549040) [195];
- Lipid nanoparticles of ARB-001467 TKM-HBV containing three RNAi therapeutics for HBV genome targeting from Arbutus Biopharma; in 2018 the company completed the phase 2a, single blind, randomized, placebo controlled, study evaluating the safety, anti-viral activity, and pharmacokinetics (PK) following multiple doses of intravenous ARB-001467 (number NCT02631096) [196].
7. Authors’ Perspective to Design Next Generation of Nano-Based Antivirals for Clinical Translation
- Clinical outcome, since patients need safe, effective, targeted, available and affordable therapy, as they are our inspiration;
- From the clinical perspective, the future antiviral candidates should improve the efficacy of the fused/encapsulated drug, reduce the intake frequency and time, restrict adverse side effects and reduce therapy costs;
- Design consideration for the nanoplatforms that will allow targeted delivery of the drugs in sustained released manner and improves efficacy, safety and patient convenience; therefore, from a chemist point of view, hybrid nanosystems can gather all the necessary features in terms of composition, shape and size by overcoming limitations of individual systems and offers greater advantages. Starting with the composition, the chosen materials should be biodegradable, biocompatible, and non-toxic, for example polymers are very attractive since they offer the possibility for chemical modifications over the surface or backbone. In addition to these advantages, the second component from the hybrid architecture (in the shape of potential liposomes) should offer besides advanced barrier penetration, higher encapsulation efficiency for the intended drug, which in combination with the polymeric piece will be able to modulate the release kinetics, the stability and prolong drug release. When thinking about the shape, we have in mind targeting capabilities as impact. As we already know, the shape is linked with size and surface charge and density, therefore a complex puzzle that must be solved. The surface charge and density should be carefully chosen during the nanoplatforms design through the surface modification possibility. The ideal candidate here from our perspective is PEG due to its versatility to exhibit various charges, shapes and sizes but also to enhance tolerability, reduce clearance, and lengthen circulation time. The size influences the biodistribution and the uptake rate therefore the “nominee” has to be in the submicron size range, recommended to be under 200 nm.
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- McMichael, A.J. Environmental and social influences on emerging infectious diseases: Past, present and future. Philos. Trans. R. Soc. Lond. B 2004, 359, 1049–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szucs, P.A.; Richman, P.B.; Mandell, M. Triage Nurse Application of the Ottawa Knee Rule. Acad. Emerg. Med. 2001, 8, 112–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Joint United Nations Programme on HIV/AIDS. Available online: https://www.unaids.org/en (accessed on 27 November 2019).
- UNAIDS. Global Statistics Report 2019. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 27 November 2019).
- WHO. Global Hepatits Report. 2017. Available online: https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en (accessed on 25 November 2019).
- WHO. Herpes Simplex Virus Key Facts. 2017. Available online: https://www.who.int/news-room/fact-sheets/detail/herpes-simplex-virus (accessed on 20 November 2019).
- Szucs, T.D.; Berger, K.; Fisman, D.N.; Harbarth, S. The estimated economic burden of genital herpes in the United States. An analysis using two costing approaches. BMC Infect. Dis. 2001, 1, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalo, T.; García Goñi, M.; Muñoz-Fernández, M.A. Socio-economic impact of antiretroviral treatment in HIV patients. An economic review of cost savings after introduction of HAART. AIDS Rev. 2009, 11, 79–90. [Google Scholar]
- Tseng, A.; Seet, J.; Phillips, E.J. The evolution of three decades of antiretroviral therapy: Challenges, triumphs and the promise of the future: Three decades of antiretroviral therapy. Br. J. Clin. Pharmacol. 2015, 79, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Fleßa, S.; Marschall, P. Socio-Economic Impact of Antiviral Intervention. In Antiviral Strategies; Kräusslich, H.-G., Bartenschlager, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 189, pp. 347–374. ISBN 978-3-540-79085-3. [Google Scholar] [CrossRef]
- Woolston, S.L.; Kim, N. Cost and Access to Direct-Acting Antiviral Agents. Hepatoma Res. 2018, 4, 3. [Google Scholar]
- Iyengar, S.; Tay-Teo, K.; Vogler, S.; Beyer, P.; Wiktor, S.; de Joncheere, K.; Hill, S. Prices, Costs, and Affordability of New Medicines for Hepatitis C in 30 Countries: An Economic Analysis. PLoS Med. 2016, 13, e1002032. [Google Scholar] [CrossRef]
- Noyes, K.; Holloway, R.G. Evidence from cost-effectiveness research. Neurotherapeutics 2004, 1, 348–355. [Google Scholar] [CrossRef]
- Byford, S. Economic Note: Cost of illness studies. BMJ 2000, 320, 1335. [Google Scholar] [CrossRef]
- Drummond, M.F.; Sculpher, M.J.; Claxton, K.; Stoddart, G.L.; Torrance, G.W. Methods for the Economic Evaluation of Health Care Programmes; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Jo, C. Cost-of-illness studies: Concepts, scopes, and methods. Clin. Mol. Hepatol. 2014, 20, 327. [Google Scholar] [CrossRef]
- Reports and Data. Available online: www.globenewswire.com (accessed on 10 November 2019).
- Lembo, D.; Cavalli, R. Nanoparticulate Delivery Systems for Antiviral Drugs. Antivir. Chem. Chemother. 2010, 21, 53–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenreich, W.; Rudel, T.; Heesemann, J.; Goebel, W. How Viral and Intracellular Bacterial Pathogens Reprogram the Metabolism of Host Cells to Allow Their Intracellular Replication. Front. Cell. Infect. Microbiol. 2019, 9, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Regenmortel, M.H.V. Virus Species. In Genetics and Evolution of Infectious Dissease; Elsevier: Amsterdam, The Netherlands, 2011; pp. 3–19. [Google Scholar]
- Heikkinen, T.; Järvinen, A. The common cold. Lancet 2003, 361, 51–59. [Google Scholar] [CrossRef]
- Faith, S.C.; Durrani, A.F.; Jhanji, V. Cytomegalovirus keratitis. Curr. Opin. Ophthalmol. 2018, 29, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Kalezic, T.; Mazen, M.; Kuklinski, E.; Asbell, P. Herpetic eye disease study: Lessons learned. Curr. Opin. Ophthalmol. 2018, 29, 340–346. [Google Scholar] [CrossRef]
- Jhanji, V.; Chan, T.C.Y.; Li, E.Y.M.; Agarwal, K.; Vajpayee, R.B. Adenoviral keratoconjunctivitis. Surv. Ophthalmol. 2015, 60, 435–443. [Google Scholar] [CrossRef]
- Venkatesan, A.; Murphy, O.C. Viral Encephalitis. Neurol. Clin. 2018, 36, 705–724. [Google Scholar] [CrossRef]
- Ruuskanen, O.; Lahti, E.; Jennings, L.C.; Murdoch, D.R. Viral pneumonia. Lancet 2011, 377, 1264–1275. [Google Scholar] [CrossRef]
- Hékimian, G.; Combes, A. Myocardites. La Revue de Médecine Interne 2017, 38, 531–538. [Google Scholar] [CrossRef]
- Rose, N.R. Viral myocarditis. Curr. Opin. Rheumatol. 2016, 28, 383–389. [Google Scholar] [CrossRef] [Green Version]
- Op de Beeck, A.; Eizirik, D.L. Viral infections in type 1 diabetes mellitus—Why the β cells? Nat. Rev. Endocrinol. 2016, 12, 263–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thuener, J. Hepatitis A and B Infections. Prim. Care 2017, 44, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, J.; Hepatitis, C. BMJ 358, j2861. Available online: https://bestpractice.bmj.com/topics/en-us/128 (accessed on 31 October 2019).
- Rizzetto, M. Hepatitis D Virus: Introduction and Epidemiology. Cold Spring Harb. Perspect. Med. 2015, 5, a021576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khuroo, M.S.; Khuroo, M.S.; Khuroo, N.S. Hepatitis E: Discovery, global impact, control and cure. WJG 2016, 22, 7030. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.; Gershon, A. Clinical Features of Varicella-Zoster Virus Infection. Viruses 2018, 10, 609. [Google Scholar] [CrossRef] [Green Version]
- Zerboni, L.; Sen, N.; Oliver, S.L.; Arvin, A.M. Molecular mechanisms of varicella zoster virus pathogenesis. Nat. Rev. Microbiol. 2014, 12, 197–210. [Google Scholar] [CrossRef] [Green Version]
- Agut, H.; Bonnafous, P.; Gautheret-Dejean, A. Human Herpesviruses 6A, 6B, and 7. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Ramdass, P.; Mullick, S.; Farber, H.F. Viral Skin Diseases. Prim. Care Clin. Off. Pract. 2015, 42, 517–567. [Google Scholar] [CrossRef]
- Schaffer, J.V.; Berger, E.M. Molluscum Contagiosum. JAMA Dermatol. 2016, 152, 1072. [Google Scholar] [CrossRef] [Green Version]
- Krenzer, M.E. Viral Gastroenteritis in the Adult Population. Crit. Care Nurs. Clin. N. Am. 2012, 24, 541–553. [Google Scholar] [CrossRef]
- Eckardt, A.J.; Baumgart, D.C. Viral Gastroenteritis in Adults. PRI 2011, 6, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Thongprachum, A.; Khamrin, P.; Maneekarn, N.; Hayakawa, S.; Ushijima, H. Epidemiology of gastroenteritis viruses in Japan: Prevalence, seasonality, and outbreak: Epidemiology of Gastroenteritis Viruses in Japan. J. Med. Virol. 2016, 88, 551–570. [Google Scholar] [CrossRef] [PubMed]
- Cone, M.M.; Whitlow, C.B. Sexually Transmitted and Anorectal Infectious Diseases. Gastroenterol. Clin. N. Am. 2013, 42, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Virus Taxonomy: 2018b Release. Available online: https://talk.ictvonline.org/taxonomy (accessed on 30 November 2019).
- Baltimore, D. Expression of animal virus genomes. Bacteriol. Rev. 1971, 35, 235–241. [Google Scholar] [CrossRef]
- Brunton, L.L.; Hilal-Dandan, R.; Knollmann, B. The Pharmacological Basis of Therapeutics, 13th ed.; McGraw-Hill Education-Europe: New York, NY, USA, 2017; ISBN 978-1-259-58473-2. [Google Scholar]
- Sacks, S.L.; Wilson, B. Famciclovir/Penciclovir. In Antiviral Chemotherapy 5; Mills, J., Volberding, P.A., Corey, L., Eds.; Springer: Boston, MA, USA, 1999; Volume 458, pp. 135–147. ISBN 978-1-4613-7150-2. [Google Scholar]
- Hitchcock, M.J.M.; Jaffe, H.S.; Martin, J.C.; Stagg, R.J. Cidofovir, a New Agent with Potent Anti-Herpesvirus Activity. Antivir. Chem. Chemother. 1996, 7, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Perry, C.M.; Barman Balfour, J.A. Fomivirsen. Drugs 1999, 57, 375–380. [Google Scholar] [CrossRef]
- Mathiesen, S.; Dam, E.; Roge, B.; Joergensen, L.B.; Laursen, A.L.; Gerstoft, J.; Clavel, F. Long-term foscarnet therapy remodels thymidine analogue mutations and alters resistance to zidovudine and lamivudine in HIV-1. Antivir. Ther. 2007, 12, 335–343. [Google Scholar]
- Leung, D.T.; Sacks, S.L. Docosanol: A topical antiviral for herpes labialis. Exp. Opin. Pharmacother. 2004, 5, 2567–2571. [Google Scholar] [CrossRef]
- Miller, W.H.; Miller, R.L. Phosphorylation of acyclovir diphosphate by cellular enzymes. Biochem. Pharmacol. 1982, 31, 3879–3884. [Google Scholar] [CrossRef]
- King, D.H. History, pharmacokinetics, and pharmacology of acyclovir. J. Am. Acad. Dermatol. 1988, 18, 176–179. [Google Scholar] [CrossRef]
- O’Brien, K.L.; Baggett, H.C.; Brooks, W.A.; Feikin, D.R.; Hammitt, L.L.; Higdon, M.M.; Howie, S.R.C.; Deloria Knoll, M.; Kotloff, K.L.; Levine, O.S.; et al. Causes of severe pneumonia requiring hospital admission in children without HIV infection from Africa and Asia: The PERCH multi-country case-control study. Lancet 2019, 394, 757–779. [Google Scholar] [CrossRef] [Green Version]
- Strasfeld, L.; Chou, S. Antiviral Drug Resistance: Mechanisms and Clinical Implications. Infect. Dis. Clin. N. Am. 2010, 24, 413–437. [Google Scholar] [CrossRef] [Green Version]
- McGavin, J.K.; Goa, K.L. Ganciclovir: An Update of its Use in the Prevention of Cytomegalovirus Infection and Disease in Transplant Recipients. Drugs 2001, 61, 1153–1183. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.; Härter, G.; Schubert, A.; Bunjes, D.; Mertens, T.; Michel, D. Antiviral treatment of cytomegalovirus infection and resistant strains. Exp. Opin. Pharmacother. 2009, 10, 191–209. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Antiviral agents active against influenza a viruses. Nat. Rev. Drug Discov. 2006, 5, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-W.; Cheng, J.X.; Liu, M.-T.; King, K.; Peng, J.-Y.; Zhang, X.-Q.; Wang, C.-H.; Shresta, S.; Schooley, R.T.; Liu, Y.-T. Inhibitory and combinatorial effect of diphyllin, a v-ATPase blocker, on influenza viruses. Antivir. Res. 2013, 99, 371–382. [Google Scholar] [CrossRef] [Green Version]
- Huss, M.; Wieczorek, H. Inhibitors of V-ATPases: Old and new players. J. Exp. Biol. 2009, 212, 341–346. [Google Scholar] [CrossRef] [Green Version]
- Yeganeh, B.; Ghavami, S.; Kroeker, A.L.; Mahood, T.H.; Stelmack, G.L.; Klonisch, T.; Coombs, K.M.; Halayko, A.J. Suppression of influenza A virus replication in human lung epithelial cells by noncytotoxic concentrations bafilomycin A1. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L270–L286. [Google Scholar] [CrossRef]
- Ison, M.G. Antiviral Treatments. Clin. Chest Med. 2017, 38, 139–153. [Google Scholar] [CrossRef]
- Jefferson, T.; Jones, M.A.; Doshi, P.; Del Mar, C.B.; Hama, R.; Thompson, M.J.; Spencer, E.A.; Onakpoya, I.J.; Mahtani, K.R.; Nunan, D.; et al. Neuraminidase inhibitors for preventing and treating influenza in adults and children. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef]
- De Jong, M.D.; Thanh, T.T.; Khanh, T.H.; Hien, V.M.; Smith, G.J.D.; Chau, N.V.; Cam, B.V.; Qui, P.T.; Ha, D.Q.; Guan, Y.; et al. Oseltamivir Resistance during Treatment of Influenza A (H5N1) Infection. N. Engl. J. Med. 2005, 353, 2667–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Te, H.S.; Randall, G.; Jensen, D.M. Mechanism of action of ribavirin in the treatment of chronic hepatitis C. Gastroenterol. Hepatol. 2007, 3, 218–225. [Google Scholar]
- Krečmerová, M. Amino Acid Ester Prodrugs of Nucleoside and Nucleotide Antivirals. MRMC 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Clinical Potential of the Acyclic Nucleoside Phosphonates Cidofovir, Adefovir, and Tenofovir in Treatment of DNA Virus and Retrovirus Infections. Clin. Microbiol. Rev. 2003, 16, 569–596. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.W.; Park, J.Y.; Ahn, S.H. An evaluation of entecavir for the treatment of chronic hepatitis B infection in adults. Exp. Rev. Gastroenterol. Hepatol. 2016, 10, 177–186. [Google Scholar] [CrossRef]
- Matthews, S. Telbivudine for the management of chronic hepatitis B virus infection. Clin. Ther. 2007, 29, 2635–2653. [Google Scholar] [CrossRef]
- Amarapurkar, D.N. Telbivudine: A new treatment for chronic hepatitis B. WJG 2007, 13, 6150. [Google Scholar] [CrossRef]
- Antiretroviral Drugs Used in the Treatment of HIV Infection. Available online: https://www.fda.gov/patients/hiv-treatment/antiretroviral-drugs-used-treatment-hiv-infection (accessed on 26 October 2019).
- FDA-Approved HIV Medicines. Available online: https://aidsinfo.nih.gov/understanding-hiv-aids/fact-sheets/21/58/fda-approved-hiv-medicines (accessed on 26 October 2019).
- Gallant, J.E.; Gerondelis, P.Z.; Wainberg, M.A.; Shulman, N.S.; Haubrich, R.H.; St Clair, M.; Lanier, E.R.; Hellmann, N.S.; Richman, D.D. Nucleoside and nucleotide analogue reverse transcriptase inhibitors: A clinical review of antiretroviral resistance. Antivir. Ther. 2003, 8, 489–506. [Google Scholar]
- Perry, C.M.; Faulds, D. Lamivudine: A Review of its Antiviral Activity, Pharmacokinetic Properties and Therapeutic Efficacy in the Management of HIV Infection. Drugs 1997, 53, 657–680. [Google Scholar] [CrossRef]
- Margolis, A.M.; Heverling, H.; Pham, P.A.; Stolbach, A. A Review of the Toxicity of HIV Medications. J. Med. Toxicol. 2014, 10, 26–39. [Google Scholar] [CrossRef] [Green Version]
- McConville, C.; Boyd, P.; Major, I. Efficacy of Tenofovir 1% Vaginal Gel in Reducing the Risk of HIV-1 and HSV-2 Infection. Clin. Med. Insights Women’s Health 2014, 7. [Google Scholar] [CrossRef] [Green Version]
- Sluis-Cremer, N.; Temiz, N.; Bahar, I. Conformational Changes in HIV-1 Reverse Transcriptase Induced by Nonnucleoside Reverse Transcriptase Inhibitor Binding. CHR 2004, 2, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Lv, Z.; Chu, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV 2015, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latinovic, O.; Kuruppu, J.; Davis, C.; Le, N.; Heredia, A. Pharmacotherapy of HIV-1 Infection: Focus on CCR5 Antagonist Maraviroc. Clin. Med. Ther. 2009, 1, CMT.S2365. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.; Morris-Natschke, S.L.; Lee, K.-H. HIV entry inhibitors and their potential in HIV therapy. Med. Res. Rev. 2009, 29, 369–393. [Google Scholar] [CrossRef]
- Lai, W.; Huang, L.; Chen, C. HIV entry inhibitors: Progress in development and application. Yao Xue Xue Bao 2010, 45, 131–140. [Google Scholar]
- Hajimahdi, Z.; Zarghi, A. Progress in HIV-1 Integrase Inhibitors: A Review of their Chemical Structure Diversity. Iran. J. Pharm. Res. 2016, 15, 595–628. [Google Scholar]
- Steigbigel, R.T.; Cooper, D.A.; Kumar, P.N.; Eron, J.E.; Schechter, M.; Markowitz, M.; Loutfy, M.R.; Lennox, J.L.; Gatell, J.M.; Rockstroh, J.K.; et al. Raltegravir with Optimized Background Therapy for Resistant HIV-1 Infection. N. Engl. J. Med. 2008, 359, 339–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocohoba, J.; Dong, B.J. Raltegravir: The first HIV integrase inhibitor. Clin. Ther. 2008, 30, 1747–1765. [Google Scholar] [CrossRef]
- Hicks, C.; Gulick, R.M. Raltegravir: The First HIV Type 1 Integrase Inhibitor. Clin. Infect. Dis. 2009, 48, 931–939. [Google Scholar] [CrossRef]
- Avila-Olias, M.; Pegoraro, C.; Battaglia, G.; Canton, I. Inspired by nature: Fundamentals in nanotechnology design to overcome biological barriers. Ther. Deliv. 2013, 4, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Kreyling, W.G.; Hirn, S.; Möller, W.; Schleh, C.; Wenk, A.; Celik, G.; Lipka, J.; Schäffler, M.; Haberl, N.; Johnston, B.D.; et al. Air–Blood Barrier Translocation of Tracheally Instilled Gold Nanoparticles Inversely Depends on Particle Size. ACS Nano 2014, 8, 222–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, H.; Leong, W.; Leong, K.W.; Chen, C.; Zhao, Y. Walking the line: The fate of nanomaterials at biological barriers. Biomaterials 2018, 174, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.; Aalinkeel, R.; Law, W.-C.; Reynolds, J.; Nair, B.B.; Sykes, D.E.; Yong, K.-T.; Roy, I.; Prasad, P.; Schwartz, S. Anti-HIV-1 nanotherapeutics: Promises and challenges for the future. IJN 2012, 5301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyva-Gómez, G.; Piñón-Segundo, E.; Mendoza-Muñoz, N.; Zambrano-Zaragoza, M.; Mendoza-Elvira, S.; Quintanar-Guerrero, D. Approaches in Polymeric Nanoparticles for Vaginal Drug Delivery: A Review of the State of the Art. IJMS 2018, 19, 1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herpetic Eye Disease Study Group. Oral acyclovir for herpes simplex virus eye disease: Effect on prevention of epithelial keratitis and stromal keratitis. Arch. Ophthalmol. 2000, 118, 1030–1036. [Google Scholar]
- Durai, R. Drug delivery approaches of an antiviral drug: A comprehensive review. Asian J. Pharm. 2015, 9. [Google Scholar] [CrossRef]
- Wood, A.J.J.; Whitley, R.J.; Gnann, J.W. Acyclovir: A Decade Later. N. Engl. J. Med. 1992, 327, 782–789. [Google Scholar] [CrossRef]
- Yaldiz, M.; Solak, B.; Kara, R.O.; Cosansu, N.; Erdem, M.T. Comparison of Famciclovir, Valaciclovir, and Brivudine Treatments in Adult Immunocompetent Patients with Herpes Zoster. Am. J. Ther. 2018, 25, e626–e634. [Google Scholar] [CrossRef]
- Cuggino, J.C.; Blanco, E.R.O.; Gugliotta, L.M.; Alvarez Igarzabal, C.I.; Calderón, M. Crossing biological barriers with nanogels to improve drug delivery performance. J. Control. Release 2019, 307, 221–246. [Google Scholar] [CrossRef]
- Raskin, M.M.; Schlachet, I.; Sosnik, A. Mucoadhesive nanogels by ionotropic crosslinking of chitosan-g-oligo(NiPAam) polymeric micelles as novel drug nanocarriers. Nanomedicine 2016, 11, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Heyden, R.J. Layers of the Skin. In Anatomy and Physiology. Available online: https://opentextbc.ca/anatomyandphysiology (accessed on 11 November 2019).
- Schneider, M.; Stracke, F.; Hansen, S.; Schaefer, U.F. Nanoparticles and their interactions with the dermal barrier. Dermat. Endocrinol. 2009, 1, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Xu, Z.; Grice, J.; Zvyagin, A.; Roberts, M.; Liu, X. Penetration of Nanoparticles into Human Skin. CPD 2013, 19, 6353–6366. [Google Scholar] [CrossRef] [PubMed]
- Ross, K.A.; Brenza, T.M.; Binnebose, A.M.; Phanse, Y.; Kanthasamy, A.G.; Gendelman, H.E.; Salem, A.K.; Bartholomay, L.C.; Bellaire, B.H.; Narasimhan, B. Nano-enabled delivery of diverse payloads across complex biological barriers. J. Control. Release 2015, 219, 548–559. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, P.; Patravale, V.; Joshi, M. Nanocarriers for Effective Topical Delivery of Anti-Infectives. CNANO 2012, 8, 491–503. [Google Scholar] [CrossRef]
- Heyden, R.J. The Cell Membrane. In Anatomy and Physiology. Available online: https://opentextbc.ca/anatomyandphysiology/chapter/the-cell-membrane (accessed on 11 November 2019).
- McCaffrey, G.; Davis, T.P. Physiology and Pathophysiology of the Blood-Brain Barrier: P-Glycoprotein and Occludin Trafficking as Therapeutic Targets to Optimize Central Nervous System Drug Delivery. J. Investig. Med. 2012, 60, 1131–1140. [Google Scholar] [CrossRef]
- Ek, C.J.; Wong, A.; Liddelow, S.A.; Johansson, P.A.; Dziegielewska, K.M.; Saunders, N.R. Efflux mechanisms at the developing brain barriers: ABC-transporters in the fetal and postnatal rat. Toxicol. Lett. 2010, 197, 51–59. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Leandro, K.; Bicker, J.; Alves, G.; Falcão, A.; Fortuna, A. ABC transporters in drug-resistant epilepsy: Mechanisms of upregulation and therapeutic approaches. Pharmacol. Res. 2019, 144, 357–376. Available online: https://linkinghub.elsevier.com/retrieve/pii/S1043661819300726 (accessed on 18 February 2020). [CrossRef]
- Mahringer, A.; Fricker, G. ABC transporters at the blood–brain barrier. Exp. Opin. Drug Metab. Toxicol. 2016, 12, 499–508. [Google Scholar] [CrossRef]
- Miller, D. Regulation of ABC transporters at the blood-brain barrier. Clin. Pharmacol. Ther. 2015, 97, 395–403. [Google Scholar] [CrossRef]
- Liu, H.; Qiu, K.; He, Q.; Lei, Q.; Lu, W. Mechanisms of Blood-Brain Barrier Disruption in Herpes Simplex Encephalitis. J. Neuroimmune Pharmacol. 2019, 14, 157–172. [Google Scholar] [CrossRef]
- Al-Obaidi, M.M.J.; Bahadoran, A.; Wang, S.M.; Manikam, R.; Raju, C.S.; Sekaran, S.D. Disruption of the blood brain barrier is vital property of neurotropic viral infection of the central nervous system. Acta Virol. 2018, 62, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Sagar, V.; Pilakka-Kanthikeel, S.; Pottathil, R.; Saxena, S.K.; Nair, M. Towards nanomedicines for neuroAIDS: Nanomedicines for neuroAIDS. Rev. Med. Virol. 2014, 24, 103–124. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tang, X.P.; McArthur, J.C.; Scott, J.; Gartner, S. Analysis of human immunodeficiency virus type 1 gp160 sequences from a patient with HIV dementia: Evidence for monocyte trafficking into brain. J. Neurovirol. 2000, 6 (Suppl. 1), S70–S81. [Google Scholar]
- Kuo, Y.-C.; Chen, H.-H. Effect of nanoparticulate polybutylcyanoacrylate and methylmethacrylate–sulfopropylmethacrylate on the permeability of zidovudine and lamivudine across the in vitro blood–brain barrier. Int. J. Pharm. 2006, 327, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.; Mahor, S.; Rawat, A.; Gupta, P.N.; Dubey, P.; Khatri, K.; Vyas, S.P. Targeted brain delivery of AZT via transferrin anchored pegylated albumin nanoparticles. J. Drug Target. 2006, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.R.; Shusta, E.V. Blood–Brain Barrier Transport of Therapeutics via Receptor-Mediation. Pharm. Res. 2007, 24, 1759–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiyed, Z.; Gandhi, N.; Nair, M. Magnetic nanoformulation of azidothymidine 5′-triphosphate for targeted delivery across the blood & ndash; brain barrier. IJN 2010, 157. [Google Scholar] [CrossRef] [Green Version]
- Fiandra, L.; Colombo, M.; Mazzucchelli, S.; Truffi, M.; Santini, B.; Allevi, R.; Nebuloni, M.; Capetti, A.; Rizzardini, G.; Prosperi, D.; et al. Nanoformulation of antiretroviral drugs enhances their penetration across the blood brain barrier in mice. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1387–1397. [Google Scholar] [CrossRef] [Green Version]
- McNeil, S.E. (Ed.) Unique Benefits of Nanotechnology to Drug Delivery and Diagnostics. In Characterization of Nanoparticles Intended for Drug Delivery; Humana Press: Totowa, NJ, USA, 2011; Volume 697, pp. 3–8. ISBN 978-1-60327-197-4. [Google Scholar]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.; Langer, R.; Jia, X. Nanostructured materials for applications in drug delivery and tissue engineering. J. Biomater. Sci. Polym. Ed. 2007, 18, 241–268. [Google Scholar] [CrossRef] [Green Version]
- Gagliardi, M. Biomimetic and bioinspired nanoparticles for targeted drug delivery. Ther. Deliv. 2017, 8, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Muthu, M.S.; Singh, S. Targeted nanomedicines: Effective treatment modalities for cancer, AIDS and brain disorders. Nanomedicine 2009, 4, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, C. Role of nanotechnology in HIV/AIDS vaccine development. Adv. Drug Deliv. Rev. 2016, 103, 76–89. [Google Scholar] [CrossRef]
- Milovanovic, M.; Arsenijevic, A.; Milovanovic, J.; Kanjevac, T.; Arsenijevic, N. Nanoparticles in Antiviral Therapy. In Antimicrobial Nanoarchitectonics; Elsevier: Amsterdam, The Netherlands, 2017; pp. 383–410. ISBN 978-0-323-52733-0. [Google Scholar]
- Cao, S.; Woodrow, K.A. Nanotechnology approaches to eradicating HIV reservoirs. Eur. J. Pharm. Biopharm. 2019, 138, 48–63. [Google Scholar] [CrossRef]
- Shen, Y.; Tu, J. Preparation and ocular pharmacokinetics of ganciclovir liposomes. AAPS J. 2007, 9, E371. [Google Scholar] [CrossRef] [Green Version]
- Feng, M.; Cai, Q.; Shi, X.; Huang, H.; Zhou, P.; Guo, X. Recombinant high-density lipoprotein complex as a targeting system of nosiheptide to liver cells. J. Drug Target. 2008, 16, 502–508. [Google Scholar] [CrossRef]
- Feng, M. Liver targeting and anti-HBV activity of reconstituted HDL–acyclovir palmitate complex. Eur. J. Pharm. Biopharm. 2008, 68, 688–693. [Google Scholar] [CrossRef]
- Kim, S.I.; Shin, D.; Lee, H.; Ahn, B.-Y.; Yoon, Y.; Kim, M. Targeted delivery of siRNA against hepatitis C virus by apolipoprotein A-I-bound cationic liposomes. J. Hepatol. 2009, 50, 479–488. [Google Scholar] [CrossRef]
- Kim, S.-S.; Peer, D.; Kumar, P.; Subramanya, S.; Wu, H.; Asthana, D.; Habiro, K.; Yang, Y.-G.; Manjunath, N.; Shimaoka, M.; et al. RNAi-mediated CCR5 Silencing by LFA-1-targeted Nanoparticles Prevents HIV Infection in BLT Mice. Mol. Ther. 2010, 18, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Pollock, S.; Dwek, R.A.; Burton, D.R.; Zitzmann, N. N-Butyldeoxynojirimycin is a broadly effective anti-HIV therapy significantly enhanced by targeted liposome delivery. AIDS 2008, 22, 1961–1969. [Google Scholar] [CrossRef] [PubMed]
- Gagné, J.-F.; Désormeaux, A.; Perron, S.; Tremblay, M.J.; Bergeron, M.G. Targeted delivery of indinavir to HIV-1 primary reservoirs with immunoliposomes. BBA Biomembr. 2002, 1558, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Clayton, R.; Ohagen, A.; Nicol, F.; Del Vecchio, A.M.; Jonckers, T.H.M.; Goethals, O.; Van Loock, M.; Michiels, L.; Grigsby, J.; Xu, Z.; et al. Sustained and specific in vitro inhibition of HIV-1 replication by a protease inhibitor encapsulated in gp120-targeted liposomes. Antivir. Res. 2009, 84, 142–149. [Google Scholar] [CrossRef]
- Yadavalli, T.; Ames, J.; Agelidis, A.; Suryawanshi, R.; Jaishankar, D.; Hopkins, J.; Thakkar, N.; Koujah, L.; Shukla, D. Drug-encapsulated carbon (DECON): A novel platform for enhanced drug delivery. Sci. Adv. 2019, 5, eaax0780. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Shah, S.J.; Wang, Z.; Agrahari, V.; Pal, D.; Mitra, A.K. Nanoparticle-based topical ophthalmic formulation for sustained release of stereoisomeric dipeptide prodrugs of ganciclovir. Drug Deliv. 2015, 1–11. [Google Scholar] [CrossRef]
- Li, Y.; Lin, Z.; Guo, M.; Zhao, M.; Xia, Y.; Wang, C.; Xu, T.; Zhu, B. Inhibition of H1N1 influenza virus-induced apoptosis by functionalized selenium nanoparticles with amantadine through ROS-mediated AKT signaling pathways. IJN 2018, 13, 2005–2016. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lin, Z.; Guo, M.; Xia, Y.; Zhao, M.; Wang, C.; Xu, T.; Chen, T.; Zhu, B. Inhibitory activity of selenium nanoparticles functionalized with oseltamivir on H1N1 influenza virus. IJN 2017, 12, 5733–5743. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lin, Z.; Zhao, M.; Guo, M.; Xu, T.; Wang, C.; Xia, H.; Zhu, B. Reversal of H1N1 influenza virus-induced apoptosis by silver nanoparticles functionalized with amantadine. RSC Adv. 2016, 6, 89679–89686. [Google Scholar] [CrossRef]
- Hu, C.-M.J.; Chen, Y.-T.; Fang, Z.-S.; Chang, W.-S.; Chen, H.-W. Antiviral efficacy of nanoparticulate vacuolar ATPase inhibitors against influenza virus infection. IJN 2018, 13, 8579–8593. [Google Scholar] [CrossRef] [Green Version]
- Chiellini, E.E.; Chiellini, F.; Solaro, R. Bioerodible Polymeric Nanoparticles for Targeted Delivery of Proteic Drugs. J. Nanosci. Nanotechnol. 2006, 6, 3040–3047. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Lakshmi, Y.S.; Bhaskar, C.; Golla, K.; Kondapi, A.K. Improved Safety, Bioavailability and Pharmacokinetics of Zidovudine through Lactoferrin Nanoparticles during Oral Administration in Rats. PLoS ONE 2015, 10, e0140399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirimlioğlu, G.Y.; Öztürk, A.A. Preparation and in vitro characterization of lamivudine loaded nanoparticles prepared by acid and/or ester terminated PLGA for effective oral anti-retroviral therapy. JRP 2019, 23, 897–913. [Google Scholar] [CrossRef] [Green Version]
- Sankar, V.; Madhura Keerthi, L.; Nilaykumar, P. Formulation and In-vitro Evaluation of Zidovudine-Lamivudine Nanoparticles. Indian J. Pharm. Educ. Res. 2012, 46, 192–196. [Google Scholar]
- Shah, L.K.; Amiji, M.M. Intracellular Delivery of Saquinavir in Biodegradable Polymeric Nanoparticles for HIV/AIDS. Pharm. Res. 2006, 23, 2638–2645. [Google Scholar] [CrossRef]
- Tang, X.; Liang, Y.; Liu, X.; Zhou, S.; Liu, L.; Zhang, F.; Xie, C.; Cai, S.; Wei, J.; Zhu, Y.; et al. PLGA-PEG Nanoparticles Coated with Anti-CD45RO and Loaded with HDAC Plus Protease Inhibitors Activate Latent HIV and Inhibit Viral Spread. Nanoscale Res. Lett. 2015, 10, 413. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, D.N.; Baskaran, M.; Karri, V.V.S.R.; Mannemala, S.S.; Radhakrishna, K.; Goti, S. Fabrication and in vivo evaluation of Nelfinavir loaded PLGA nanoparticles for enhancing oral bioavailability and therapeutic effect. Saudi Pharm. J. 2015, 23, 667–674. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Lakshmi, Y.; Kondapi, A. An oral formulation of efavirenz-loaded lactoferrin nanoparticles with improved biodistribution and pharmacokinetic profile. HIV Med. 2017, 18, 452–462. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G. The mixed lineage kinase-3 inhibitor URMC-099 improves therapeutic outcomes for long-acting antiretroviral therapy. Nanomedicine 2016, 12, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Savage, A.C.; Tatham, L.M.; Siccardi, M.; Scott, T.; Vourvahis, M.; Clark, A.; Rannard, S.P.; Owen, A. Improving maraviroc oral bioavailability by formation of solid drug nanoparticles. Eur. J. Pharm. Biopharm. 2019, 138, 30–36. [Google Scholar] [CrossRef]
- Shibata, A.; McMullen, E.; Pham, A.; Belshan, M.; Sanford, B.; Zhou, Y.; Goede, M.; Date, A.A.; Destache, C.J. Polymeric Nanoparticles Containing Combination Antiretroviral Drugs for HIV Type 1 Treatment. AIDS Res. Hum. Retrovir. 2013, 29, 746–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prathipati, P.K.; Mandal, S.; Pon, G.; Vivekanandan, R.; Destache, C.J. Pharmacokinetic and Tissue Distribution Profile of Long Acting Tenofovir Alafenamide and Elvitegravir Loaded Nanoparticles in Humanized Mice Model. Pharm. Res. 2017, 34, 2749–2755. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Lakshmi, Y.S.; Kondapi, A.K. Triple Drug Combination of Zidovudine, Efavirenz and Lamivudine Loaded Lactoferrin Nanoparticles: An Effective Nano First-Line Regimen for HIV Therapy. Pharm. Res. 2017, 34, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Lauster, D.; Glanz, M.; Bardua, M.; Ludwig, K.; Hellmund, M.; Hoffmann, U.; Hamann, A.; Böttcher, C.; Haag, R.; Hackenberger, C.P.R.; et al. Multivalent Peptide-Nanoparticle Conjugates for Influenza-Virus Inhibition. Angew. Chem. Int. Ed. 2017, 56, 5931–5936. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Balachandran, Y.L.; Hao, Y.; Hao, X.; Li, R.; Nan, Z.; Zhang, H.; Shao, Y.; Liu, Y. Amantadine Surface-Modified Silver Nanorods Improves Immunotherapy of HIV Vaccine Against HIV-Infected Cells. ACS Appl. Mater. Interfaces 2018, 10, 28494–28501. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.D.; Roy, I.; Xu, G.; Yong, K.-T.; Ding, H.; Aalinkeel, R.; Reynolds, J.L.; Sykes, D.E.; Nair, B.B.; Lin, E.Y.; et al. Enhancing the Delivery of Anti Retroviral Drug “Saquinavir”; Across the Blood Brain Barrier Using Nanoparticles. CHR 2010, 8, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Donalisio, M.; Leone, F.; Civra, A.; Spagnolo, R.; Ozer, O.; Lembo, D.; Cavalli, R. Acyclovir-Loaded Chitosan Nanospheres from Nano-Emulsion Templating for the Topical Treatment of Herpesviruses Infections. Pharmaceutics 2018, 10, 46. [Google Scholar] [CrossRef] [Green Version]
- Patel, G.V.; Patel, V.B.; Pathak, A.; Rajput, S.J. Nanosuspension of efavirenz for improved oral bioavailability: Formulation optimization, in vitro, in situ and in vivo evaluation. Drug Dev. Ind. Pharm. 2014, 40, 80–91. [Google Scholar] [CrossRef]
- Akhter, S.; Talegaonkar, S.; Khan, Z.I.; Jain, G.K.; Khar, R.K.; Ahmad, F.J. Assessment of Ocular Pharmacokinetics and Safety of Ganciclovir Loaded Nanoformulations. J. Biomed. Nanotechnol. 2011, 7, 144–145. [Google Scholar] [CrossRef]
- Ren, J.; Zou, M.; Gao, P.; Wang, Y.; Cheng, G. Tissue distribution of borneol-modified ganciclovir-loaded solid lipid nanoparticles in mice after intravenous administration. Eur. J. Pharm. Biopharm. 2013, 83, 141–148. [Google Scholar] [CrossRef]
- Chattopadhyay, N.; Zastre, J.; Wong, H.-L.; Wu, X.Y.; Bendayan, R. Solid Lipid Nanoparticles Enhance the Delivery of the HIV Protease Inhibitor, Atazanavir, by a Human Brain Endothelial Cell Line. Pharm. Res. 2008, 25, 2262–2271. [Google Scholar] [CrossRef] [PubMed]
- Kovochich, M.; Marsden, M.D.; Zack, J.A. Activation of Latent HIV Using Drug-Loaded Nanoparticles. PLoS ONE 2011, 6, e18270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.; Freeling, J.P.; Koehn, J.; Shu, C.; Ho, R.J.Y. Evaluation of Atazanavir and Darunavir Interactions with Lipids for Developing pH-Responsive Anti-HIV Drug Combination Nanoparticles. J. Pharm. Sci. 2014, 103, 2520–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeling, J.P.; Koehn, J.; Shu, C.; Sun, J.; Ho, R.J.Y. Anti-HIV Drug-Combination Nanoparticles Enhance Plasma Drug Exposure Duration as Well as Triple-Drug Combination Levels in Cells Within Lymph Nodes and Blood in Primates. AIDS Res. Hum. Retrovir. 2015, 31, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Arca-Lafuente, S.; Martínez-Román, P.; Mate-Cano, I.; Madrid, R.; Briz, V. Nanotechnology: A reality for diagnosis of HCV infectious disease. J. Infect. 2019. [Google Scholar] [CrossRef] [Green Version]
- Farzin, L.; Shamsipur, M.; Samandari, L.; Sheibani, S. HIV biosensors for early diagnosis of infection: The intertwine of nanotechnology with sensing strategies. Talanta 2020, 206, 120201. [Google Scholar] [CrossRef]
- Muxika, A.; Etxabide, A.; Uranga, J.; Guerrero, P.; de la Caba, K. Chitosan as a bioactive polymer: Processing, properties and applications. Int. J. Biol. Macromol. 2017, 105, 1358–1368. [Google Scholar] [CrossRef]
- Dhanasezhian, A.; Srivani, S.; Govindaraju, K.; Preetam, P.; Sasikala, S.; Ramesh Kumar, M.R. Anti-Herpes Simplex Virus (HSV-1 and HSV-2) activity of biogenic gold and silver nanoparticles using seaweed Sargassum wightii. Indian J. Geo Mar. Sci. 2019, 48, 1252–1257. [Google Scholar]
- Singaravelu, G.; Arockiamary, J.S.; Kumar, V.G.; Govindaraju, K. A novel extracellular synthesis of monodisperse gold nanoparticles using marine alga, Sargassum wightii Greville. Colloids Surf. B 2007, 57, 97–101. [Google Scholar] [CrossRef]
- Cagno, V.; Andreozzi, P.; D’Alicarnasso, M.; Jacob Silva, P.; Mueller, M.; Galloux, M.; Le Goffic, R.; Jones, S.T.; Vallino, M.; Hodek, J.; et al. Broad-spectrum non-toxic antiviral nanoparticles with a virucidal inhibition mechanism. Nat. Mater. 2018, 17, 195–203. [Google Scholar] [CrossRef]
- Klimyte, E.M.; Smith, S.E.; Oreste, P.; Lembo, D.; Dutch, R.E. Inhibition of Human Metapneumovirus Binding to Heparan Sulfate Blocks Infection in Human Lung Cells and Airway Tissues. J. Virol. 2016, 90, 9237–9250. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Camacho, L.C.; Haag, R. Pathogen Inhibition by Multivalent Ligand Architectures. J. Am. Chem. Soc. 2016, 138, 8654–8666. [Google Scholar] [CrossRef]
- Verma, A.; Uzun, O.; Hu, Y.; Hu, Y.; Han, H.-S.; Watson, N.; Chen, S.; Irvine, D.J.; Stellacci, F. Surface-structure-regulated cell-membrane penetration by monolayer-protected nanoparticles. Nat. Mater. 2008, 7, 588–595. [Google Scholar] [CrossRef]
- Argenta, D.F.; Silva, I.T.; Bassani, V.L.; Koester, L.S.; Teixeira, H.F.; Simões, C.M.O. Antiherpes evaluation of soybean isoflavonoids. Arch. Virol. 2015, 160, 2335–2342. [Google Scholar] [CrossRef]
- Argenta, D.F.; Bidone, J.; Koester, L.S.; Bassani, V.L.; Simões, C.M.O.; Teixeira, H.F. Topical Delivery of Coumestrol from Lipid Nanoemulsions Thickened with Hydroxyethylcellulose for Antiherpes Treatment. AAPS PharmSciTech 2018, 19, 192–200. [Google Scholar] [CrossRef]
- Hoeller, S.; Sperger, A.; Valenta, C. Lecithin based nanoemulsions: A comparative study of the influence of non-ionic surfactants and the cationic phytosphingosine on physicochemical behaviour and skin permeation. Int. J. Pharm. 2009, 370, 181–186. [Google Scholar] [CrossRef]
- Grande, F.; Ioele, G.; Occhiuzzi, M.A.; De Luca, M.; Mazzotta, E.; Ragno, G.; Garofalo, A.; Muzzalupo, R. Reverse Transcriptase Inhibitors Nanosystems Designed for Drug Stability and Controlled Delivery. Pharmaceutics 2019, 11, 197. [Google Scholar] [CrossRef] [Green Version]
- Joshy, K.S.; George, A.; Jose, J.; Kalarikkal, N.; Pothen, L.A.; Thomas, S. Novel dendritic structure of alginate hybrid nanoparticles for effective anti-viral drug delivery. Int. J. Biol. Macromol. 2017, 103, 1265–1275. [Google Scholar] [CrossRef]
- Sneha, R.; Vedha Hari, B.N.; Ramya Devi, D. Design of antiretroviral drug-polymeric nanoparticles laden buccal films for chronic HIV therapy in paediatrics. Colloid Interface Sci. Commun. 2018, 27, 49–59. [Google Scholar] [CrossRef]
- Urbaniak, T.; Musiał, W. Influence of Solvent Evaporation Technique Parameters on Diameter of Submicron Lamivudine-Poly-ε-Caprolactone Conjugate Particles. Nanomaterials 2019, 9, 1240. [Google Scholar] [CrossRef] [Green Version]
- Tshweu, L.; Katata, L.; Kalombo, L.; Swai, H. Nanoencapsulation of water-soluble drug, lamivudine, using a double emulsion spray-drying technique for improving HIV treatment. J. Nanopart. Res. 2013, 15, 2040. [Google Scholar] [CrossRef]
- Reddy, P.P.; Reddy, B.M.; Prakash, J.; Latha, K.; Prashanthi, D. Formulation and characterisation of chitosan based lamivudine nanoparticles. EJPMR 2014, 4, 377–383. [Google Scholar]
- Sanchez, C.G.; Molinski, S.V.; Gongora, R.; Sosulski, M.; Fuselier, T.; MacKinnon, S.S.; Mondal, D.; Lasky, J.A. The Antiretroviral Agent Nelfinavir Mesylate: A Potential Therapy for Systemic Sclerosis. Arthritis Rheumatol. 2018, 70, 115–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, M.-C.; Ballard, T.E.; Ackerson, C.J.; Feldheim, D.L.; Margolis, D.M.; Melander, C. Inhibition of HIV Fusion with Multivalent Gold Nanoparticles. J. Am. Chem. Soc. 2008, 130, 6896–6897. [Google Scholar] [CrossRef] [Green Version]
- Vijayakumar, S.; Ganesan, S. Gold Nanoparticles as an HIV Entry Inhibitor. CHR 2012, 10, 643–646. [Google Scholar] [CrossRef]
- Garrido, C.; Simpson, C.A.; Dahl, N.P.; Bresee, J.; Whitehead, D.C.; Lindsey, E.A.; Harris, T.L.; Smith, C.A.; Carter, C.J.; Feldheim, D.L.; et al. Gold nanoparticles to improve HIV drug delivery. Future Med. Chem. 2015, 7, 1097–1107. [Google Scholar] [CrossRef]
- Singh, L.; Kruger, H.G.; Maguire, G.E.M.; Govender, T.; Parboosing, R. The role of nanotechnology in the treatment of viral infections. Ther. Adv. Infect. 2017, 4, 105–131. [Google Scholar] [CrossRef]
- Bovier, P.A. Epaxal®: A virosomal vaccine to prevent hepatitis A infection. Exp. Rev. Vaccines 2008, 7, 1141–1150. [Google Scholar] [CrossRef]
- He, H.; Yuan, D.; Wu, Y.; Cao, Y. Pharmacokinetics and Pharmacodynamics Modeling and Simulation Systems to Support the Development and Regulation of Liposomal Drugs. Pharmaceutics 2019, 11, 110. [Google Scholar] [CrossRef] [Green Version]
- Herzog, C.; Hartmann, K.; Künzi, V.; Kürsteiner, O.; Mischler, R.; Lazar, H.; Glück, R. Eleven years of Inflexal® V—A virosomal adjuvanted influenza vaccine. Vaccine 2009, 27, 4381–4387. [Google Scholar] [CrossRef]
- Package Insert PEG-IntronTM (Peginterferon alfa-2b) Powder for Injection, Schering Corporation. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2001/pegsche080701LB.htm (accessed on 16 January 2020).
- Ventola, C.L. Progress in Nanomedicine: Approved and Investigational Nanodrugs. Pharm. Ther. 2017, 42, 742–755. [Google Scholar]
- Package Insert PEGASYS® (Peginterferon Alfa-2a) Hoffmann-La Roche Inc. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2002/pegihof101602LB.htm (accessed on 16 January 2020).
- Influvac (Influenza Vaccine). Available online: https://bodyandhealth.canada.com/drug/getdrug/influvac (accessed on 16 January 2020).
- VivaGel® BV launched in Europe. Available online: https://starpharma.com/assets/asxannouncements/190627%20VivaGel%20BV%20launched%20in%20Europe%20-%20final%20.pdf (accessed on 16 January 2020).
- Safety. Tolerability and Immune Response to LC002, an Experimental Therapeutic Vaccine, in Adults Receiving HAART-ClinicalTrials.gov Identifier: NCT00270205. Available online: https://clinicaltrials.gov/ct2/show/NCT00270205?cond=NCT00270205&draw=2&rank=1 (accessed on 18 January 2020).
- Bioavailability of MK-1439 Experimental Nano Formulations in Healthy Adults (MK-1439-046)-ClinicalTrials.gov Identifier: NCT02549040. Available online: https://clinicaltrials.gov/ct2/show/study/NCT02549040?term=NCT02549040&draw=2&rank=1 (accessed on 18 January 2020).
- Study of ARB-001467 in Subjects with Chronic HBV Infection Receiving Nucleos(t)Ide Analogue Therapy-ClinicalTrials.gov Identifier: NCT02631096. Available online: https://clinicaltrials.gov/ct2/show/NCT02631096?term=NCT02631096&draw=2&rank=1 (accessed on 18 January 2020).
- Odiba, A.; Ottah, V.; Ottah, C.; Anunobi, O.; Ukegbu, C.; Edeke, A.; Uroko, R.; Omeje, K. Therapeutic nanomedicine surmounts the limitations of pharmacotherapy. Open Med. 2017, 12. [Google Scholar] [CrossRef]
- Parboosing, R.; Maguire, G.E.M.; Govender, P.; Kruger, H.G. Nanotechnology and the Treatment of HIV Infection. Viruses 2012, 4, 488–520. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Al-Jamal, K.T.; Kostarelos, K.; Reineke, J. Physiologically Based Pharmacokinetic Modeling of Nanoparticles. ACS Nano 2010, 4, 6303–6317. [Google Scholar] [CrossRef]
- Sanvicens, N.; Marco, M.P. Multifunctional nanoparticles—Properties and prospects for their use in human medicine. Trends Biotechnol. 2008, 26, 425–433. [Google Scholar] [CrossRef]
- Müller, R.H.; Gohla, S.; Keck, C.M. State of the art of nanocrystals—Special features, production, nanotoxicology aspects and intracellular delivery. Eur. J. Pharm. Biopharm. 2011, 78, 1–9. [Google Scholar] [CrossRef]
- Ochekpe, N.A.; Olorunfemi, P.O.; Ngwuluka, N.C. Nanotechnology and drug delivery part 2: Nanostructures for drug delivery. Trop. J. Pharm. Res. 2009, 8. [Google Scholar] [CrossRef]
- Dey, P.; Bergmann, T.; Cuellar-Camacho, J.L.; Ehrmann, S.; Chowdhury, M.S.; Zhang, M.; Dahmani, I.; Haag, R.; Azab, W. Multivalent Flexible Nanogels Exhibit Broad-Spectrum Antiviral Activity by Blocking Virus Entry. ACS Nano 2018, 12, 6429–6442. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viral Infection | Viruses | References |
|---|---|---|
| Common cold | rhinoviruses, parainfluenza viruses, respiratory syncytial viruses coronaviruses, Influenza viruses, adenoviruses, enteroviruses, metapneumovirus, unknown | [21] |
| Eye infections | herpes simplex virus, adenovirus, cytomegalovirus | [22,23,24] |
| Encephalitis or meningitis | as JC virus, measles, LCM virus, arbovirus, rabies | [25] |
| Pneumonia | influenza virus (A and B), parainfluenza virus, respiratory syncytial virus, adenovirus, SARS coronavirus | [26] |
| Cardiovascular and pancreas disease | coxsackie virus; | [27,28,29] |
| Hepatitis | hepatitis viruses types A, B, C, D, E | [30,31,32,33] |
| Skin infections | varicella-zoster virus, human herpesvirus 6, smallpox, molluscum contagiosum, human papillomavirus, parvovirus B19, rubella, measles, coxsackie A virus | [34,35,36,37,38] |
| Gastroenteritis | adenoviruses, rotaviruses, noroviruses, astroviruses, coronaviruses | [39,40,41] |
| Sexually transmitted diseases | herpes simplex type 2, human papillomavirus HIV | [42] |
| Nanoplatform Type | Nanoplatform Characteristics (Size, Morphology, Toxicity etc.) | Drug | Virus Type | REF |
|---|---|---|---|---|
| Liposomes | ||||
| - Reverse phase evaporation |
| GCV | HSV | [125] |
| - rHDL |
| Nosiheptide | HBV | [126] |
| ACV | [127] | ||
| - cationic |
| siRNA | HCV | [128] |
| - immunoliposomes |
| HIV gp 120 Folding inhibitor | HIV | [129,130,131] |
| anti-CCR5 siRNA | |||
| Indinavir | |||
| - pegylated |
| PIs | [132] | |
| Nanoparticles | ||||
| - HPAC |
| ACV | HSV | [133] |
| - PLGA |
| GCV | HSV-1 | [134] |
| - Se |
| AM | H1N1 | [135] |
| OTV | [136] | ||
| - Ag |
| AM | [137] | |
| - PEG-PLGA |
| Diphyllin and Bafilomycin | [138] | |
| - Human serum albumin + copolymers of maleic anhydride/alkyl vinyl ethers of oligo (ethylene glycol) |
| INFs-α | HIV | [139] |
| - Tf-Albumin-PEG |
| AZT | [113] | |
| - Lactoferrin |
| [140] | ||
| - PLGA NPs |
| LAM | [141] | |
| - Hybrid NPs (PLGA, MMA-SPM, PLA and PMMA) |
| LAM+AZT | [142] | |
| - PEO-PCL |
| SQV | [143] | |
| - PLGA-PEG |
| SAHA NFV | [144] | |
| - PLGA |
| NFV | [145] | |
| - Lactoferrin |
| EFV | [146] | |
| - Folic acid-conjugated-P407 |
| ATV+RTV | [147] | |
| - PMA coated MNP |
| ENF | [116] | |
| - PVA-AOT |
| MCV | [148] | |
| - pMBA-Au NPs |
| RAL | [130] | |
| - PLGA |
| EFV+Lopinavir+RTV | [149] | |
| - PLGA+Pluronic F127 |
| TAF+EVG | [150] | |
| - Lactoferrin |
| AZT+EFV+LAM | [151] | |
| Dendrimers | ||||
| - PG |
| Peptides | IAV | [152] |
| - Alginate-PEG |
| AZT | HIV | [118] |
| Nanorods | ||||
| - PVP-PEG coated with Ag |
| AM | HIV | [153] |
| - Tf-conjugated QRs |
| SQV | [154] | |
| Nanospheres | ||||
| - Cs |
| ACV | HSV | [155] |
| Micelles | ||||
| - Cs-g-oligo(NiPAam) |
| EFV | HIV | [95] |
| Nanosuspensions | ||||
| - zirconium oxide beads stabilized with PVP, poloxamers and SLS |
| EFV | HIV | [156] |
| Nanoemulsions | ||||
| - Mucoadhesive NEs |
| GCV | HSV | [157] |
| SLNs | ||||
| - Borneol |
| GCV | CMV | [158] |
| - Stearic acid + Pluronic F68) |
| AZT | HIV | [159] |
| Lipid nanoparticles | ||||
| - bryostatin-2 |
| NFV | HIV | [160] |
| - PEG and phospholipids |
| ATV+RTV | [161] | |
| ATV+RTV+TFV | ||||
| - DSPC+MPEG+DSPE |
| Liponavir+RTV+TFV | [162] | |
| Name | Company/Approval Year/Country/Organization | Nanoplatform. Benefits | Virus | Route of Administration | REF |
|---|---|---|---|---|---|
| Epaxal® | Crucell (former Berna Biotech Ltd.); 1994 Switzerland | Virosomes (around 150 nm spherical liposomal vesicles)—intrinsic adjuvant properties; reduced toxicity and superior tolerability; | HAV | Intramuscular vaccine | [186,187] |
| Inflexal® V | Crucell (former Berna Biotech Ltd.); 1997 Switzerland | Virosomes (around 150 nm spherical liposomal vesicles)—biodegradable and biocompatible adjuvant systems; unwanted side effects; superior immune response; | Influenza | Intramuscular vaccine | [187,188] |
| PegIntron® | Schering Corporation, 2001, U.S., FDA | PEG-interferon alfa-2b (polymeric NPs) —31.000 Daltons molecules; superior protein stability; | HCV | Subcutaneous | [189] |
| Pegasys® | Genentech, 2002, U.S. FDA | PEG-interferon alfa-2a (polymeric NPs)—31.000 Daltons molecules; superior protein stability; | HBV, HCV | Subcutaneous | [190,191] |
| Influvac® Plus | BGP Pharma ULC, 2005, Canada | Virosome vaccine | Influenza | Intramuscular vaccine | [192] |
| VivaGel® BV | Starpharma, Australia; Mundipharma, Europe, 2019 | Dendrimer (astodrimer sodium—SPL7013) incorporated in a water-based vaginal gel, acting as a targeting antiviral biofilm. | HIV, HSV | Topically Applied (Vaginal gel) | [193] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cojocaru, F.-D.; Botezat, D.; Gardikiotis, I.; Uritu, C.-M.; Dodi, G.; Trandafir, L.; Rezus, C.; Rezus, E.; Tamba, B.-I.; Mihai, C.-T. Nanomaterials Designed for Antiviral Drug Delivery Transport across Biological Barriers. Pharmaceutics 2020, 12, 171. https://doi.org/10.3390/pharmaceutics12020171
Cojocaru F-D, Botezat D, Gardikiotis I, Uritu C-M, Dodi G, Trandafir L, Rezus C, Rezus E, Tamba B-I, Mihai C-T. Nanomaterials Designed for Antiviral Drug Delivery Transport across Biological Barriers. Pharmaceutics. 2020; 12(2):171. https://doi.org/10.3390/pharmaceutics12020171
Chicago/Turabian StyleCojocaru, Florina-Daniela, Doru Botezat, Ioannis Gardikiotis, Cristina-Mariana Uritu, Gianina Dodi, Laura Trandafir, Ciprian Rezus, Elena Rezus, Bogdan-Ionel Tamba, and Cosmin-Teodor Mihai. 2020. "Nanomaterials Designed for Antiviral Drug Delivery Transport across Biological Barriers" Pharmaceutics 12, no. 2: 171. https://doi.org/10.3390/pharmaceutics12020171
APA StyleCojocaru, F. -D., Botezat, D., Gardikiotis, I., Uritu, C. -M., Dodi, G., Trandafir, L., Rezus, C., Rezus, E., Tamba, B. -I., & Mihai, C. -T. (2020). Nanomaterials Designed for Antiviral Drug Delivery Transport across Biological Barriers. Pharmaceutics, 12(2), 171. https://doi.org/10.3390/pharmaceutics12020171