Dendritic Cells and Immunogenic Cancer Cell Death: A Combination for Improving Antitumor Immunity

 ,
,  and
and
Abstract
:
1. Introduction
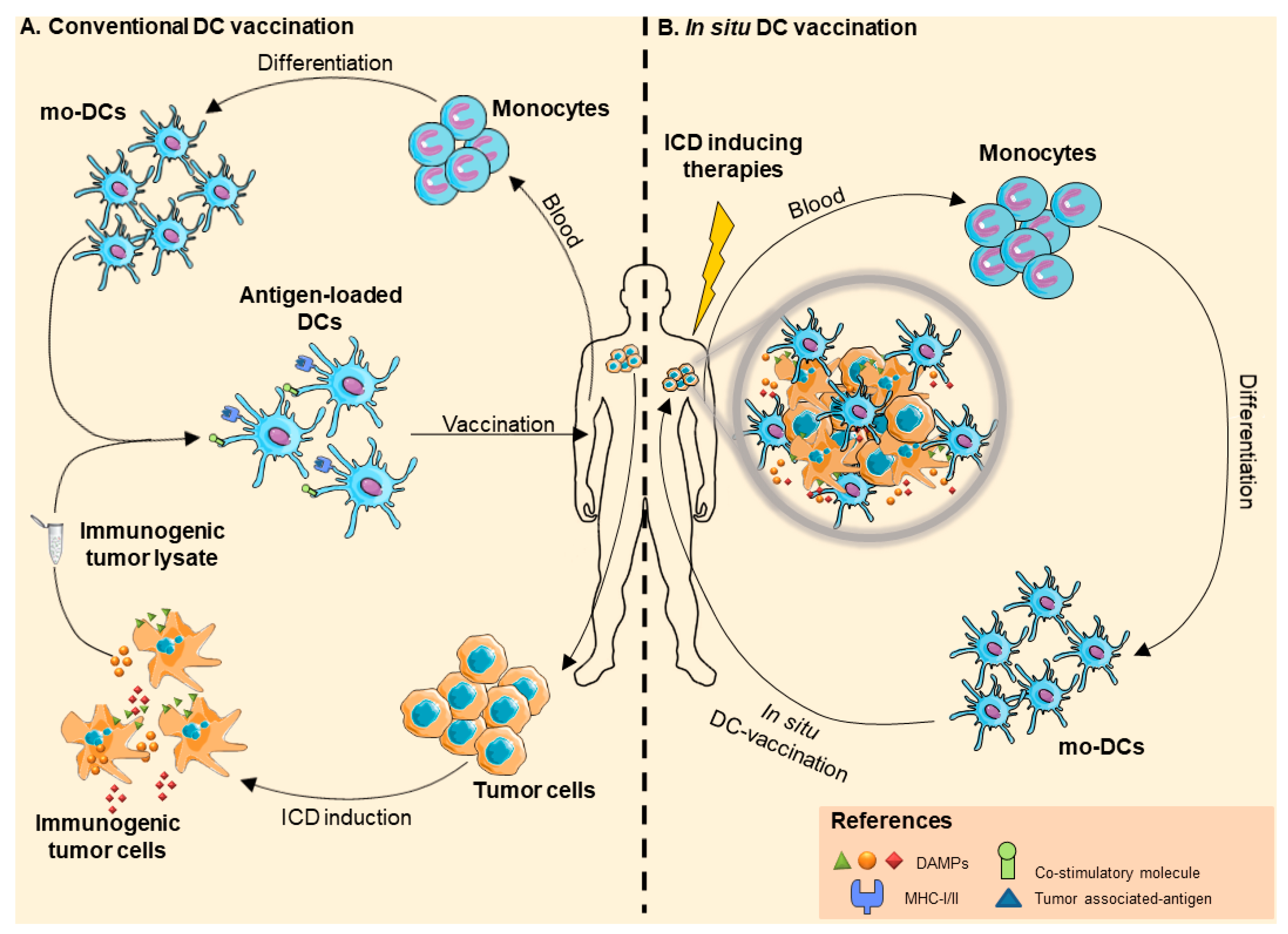
2. Dendritic Cell-Based Anticancer Immunotherapies
2.1. DC-Based Vaccines
2.2. In Situ Vaccination
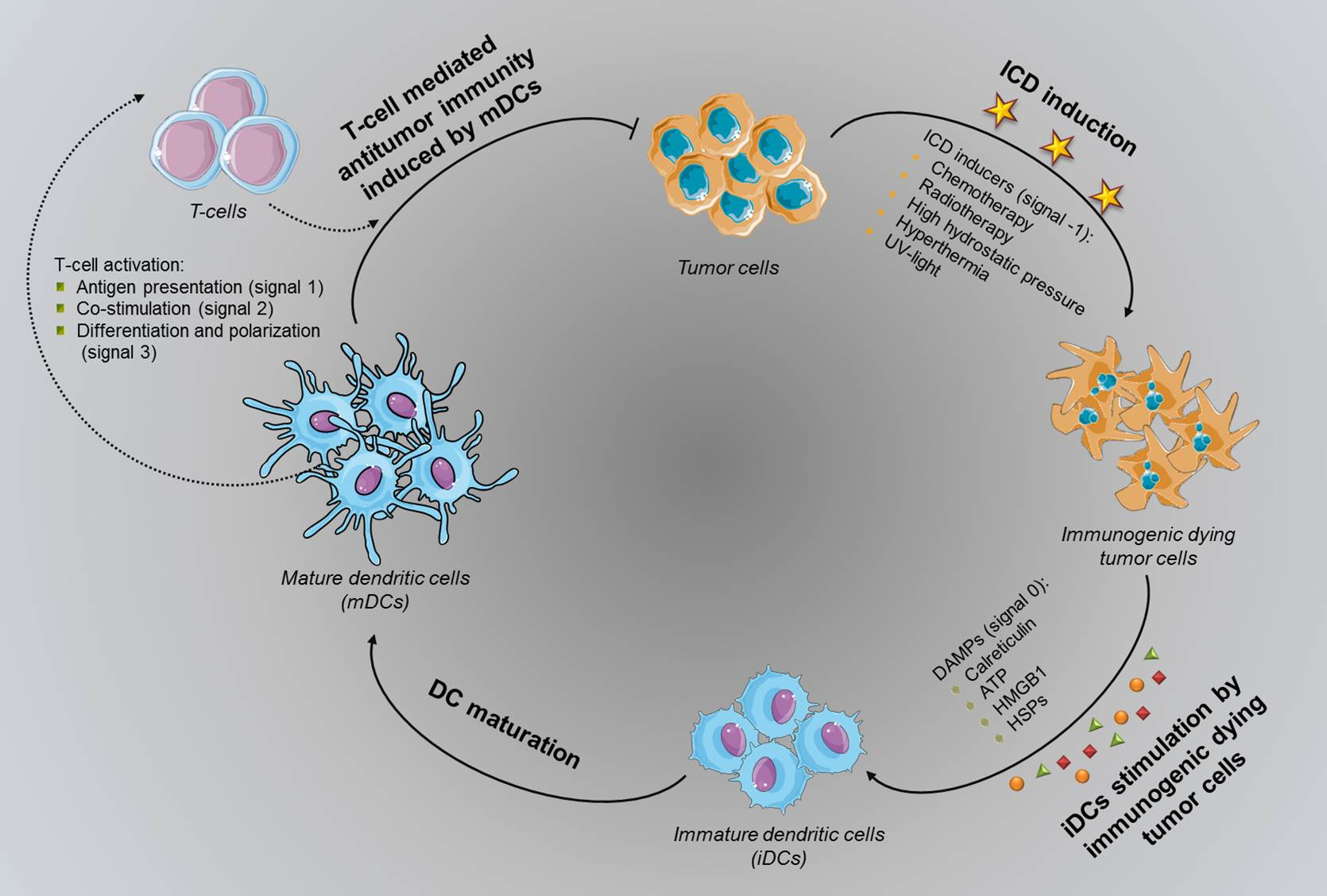
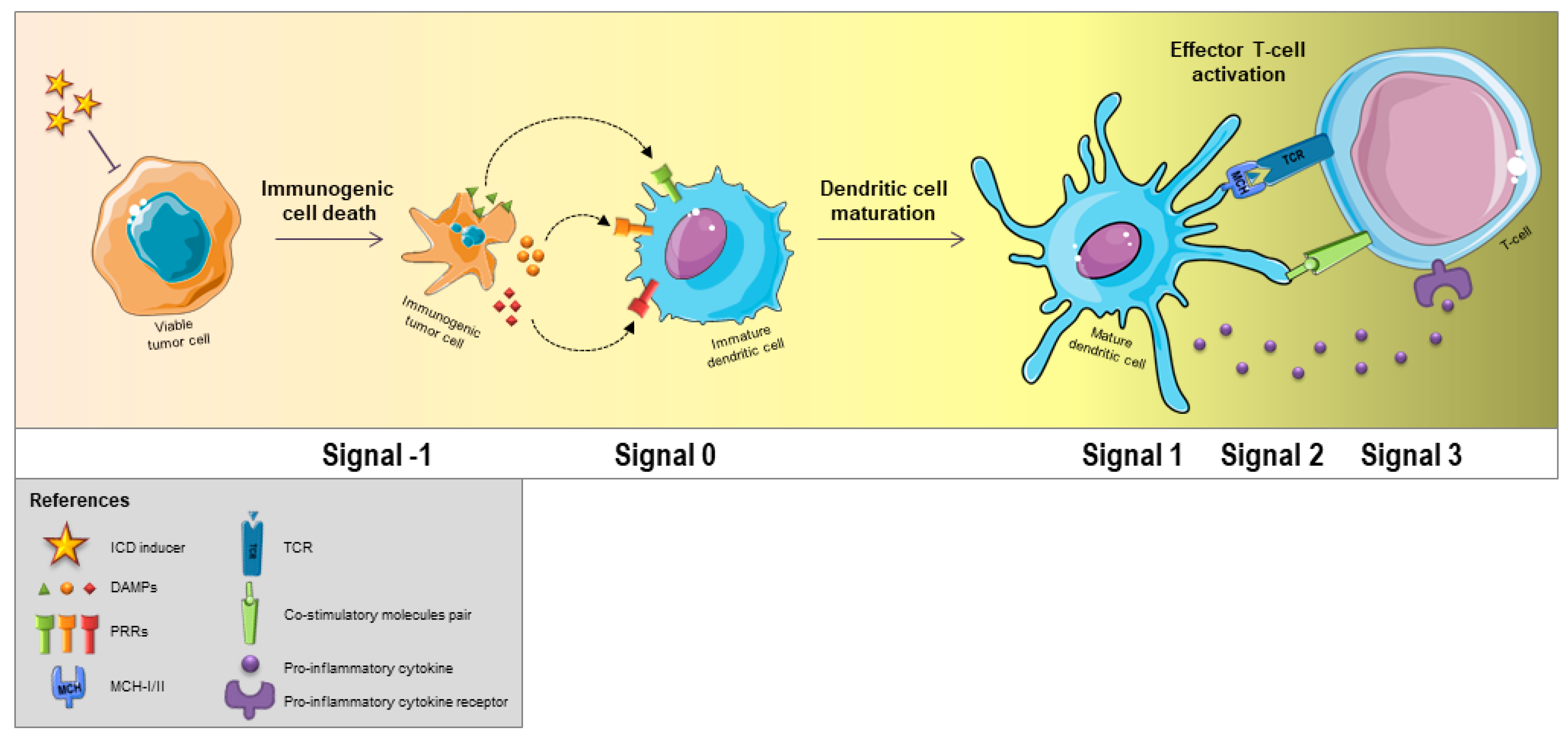
3. Effects of ICD Hallmarks on Immune Cells in Tumor Microenvironment
3.1. CRT
3.2. HSP70 and HSP90
3.3. HMGB1
3.4. ATP
4. Tumor ICD Handling in DC-Based Vaccine Development
4.1. Chemotherapy
4.1.1. Doxorubicin
4.1.2. IFN-α
4.1.3. Shikonin
4.1.4. Colchicine
4.2. Physical Therapeutic Modalities
4.2.1. High Hydrostatic Pressure
4.2.2. Photodynamic Therapy
4.2.3. Radiotherapy
4.2.4. UV-Light
4.2.5. Hyperthermia
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| ICD Inductor | Tumor Type | Type of Study | DCs Source | Vaccine Preparation | Regimen | Administration of Vaccine | Effectiveness |
|---|---|---|---|---|---|---|---|
| Chemotherapy | |||||||
| Doxorubicin (Komorowski et al. 2018) | - NXS2: murine neuroblastoma cell line | Pre-clinical | Origin: BM precursors Differentiation media: Complete RPMI-1640 + 10 ng/mL GM-CSF Incubation in differentiation media: 7 days (replenished every 2–3 days). Cells harvested: Non-adherent and loosely adherent. | Ratio DC/TC: 1:1 Co-culture time: 12 h Extra maturation stimuli: LPS (1 µg/mL, 1 h) | Prophylactic Tumor challenge*: +8 Doses: 1 | Intradermal (2 × 106 DCs) | Success |
| Pre-clinical | Therapeutic Tumor challenge*: −7 Doses: 2 (Day 0 and 7) | Intradermal(2 × 106 DCs) | Failure | ||||
| RA/IFN-α (Montico et al. 2017) | - Mino: mantle cell lymphoma (MCL) cell line | Pre-clinical | Origin: CD14+ peripheral blood monocytes Differentiation media: CellGenix™ GMP DC + IFN-α 10.000 UI/mL + 50 ng/mL rhGM-CSF Incubation in differentiation media: 3 days | Ratio DC/TC: 1 × 106 DCs/100 µg TCL | Therapeutic Tumor challenge*: −4 Dose: 3 (Day 0, 7 and 14) | Intraperitoneal (2 × 106 DCs) | Success |
| Shikonin (Chen et al. 2012) | - B16F10 (B16): murine melanoma cell line | Pre-clinical | Origin: BM precursors Differentiation media: Complete RPMI-1640 + 20 ng/mL GM-CSF + 50 μM 2-mercaptoethanol or 20 ng/mL IL-4 Incubation in differentiation media: 7 days (replenished every 2–3 days). Cells harvested: Non-adherent and loosely adherent. | Ratio DC/TC: 1 × 106 DCs + 200 μg protein of TCL/0.5 ml Co-culture time: 24 h Extra maturation stimuli: LPS | Therapeutic Tumor challenge*: −7 Dose: 3 (Day 0, 10 and 13) | Subcutaneous (1 × 106 DCs) | Success |
| Shikonin (Lin et al. 2015) | - 4T1: mammary tumor cells - 4 T1-luc2: 4 T1 cells transfected with a firefly luciferase cDNA expression vector | Pre-clinical | Origin: BM precursors Differentiation media: Complete RPMI-1640 + 20 ng/mL GM-CSF + 50 μM 2-mercaptoethanol or 20 ng/mL IL-4 Incubation in differentiation media: 7 days (replenished every 2–3 days). Cells harvested: Non-adherent and loosely adherent. | Ratio DC/TC: 3 × 106 DCs + 200 μg protein of TCL/ml Co-culture time: 24 h Extra maturation stimuli: LPS | Adjuvant Tumor challenge*: −16 (and then resected) Dose: 3 (Day 0, 7 and 14 after tumor resection) | Intravenous (1 × 106 DCs) | Success |
| Colchicin (Wen et al. 2011) | - B16F10 (B16): murine melanoma cell line | Pre-clinical | Origin: BM precursors Differentiation media: Complete RPMI-1640 + 20 ng/mL GM-CSF + 20 ng/mL IL-4 Incubation in differentiation media: 7 days (replenished every 2–3 days). Cells harvested: Non-adherent | Ratio DC/TC: 2 × 106 DC/3 mL + 400 μg TCL Co-culture time: 24 h Extra maturation stimuli: LPS | Therapeutic Tumor challenge*: −8 Dose: 3 (Day 0, 10 and 13) | Intratumoral (5 × 105 DCs) | Success |
| Physical therapeutic modalities | |||||||
| HHP (200 MPa) (Mikyšková et al. 2016) | - TC-1: murine lung tumor cell line (weakly-immunogenic) - TRAMP-C2: murine prostate tumor cell line (immunogenic) | Pre-clinical | Origin: BM precursors Differentiation media: Complete RPMI-1640 + 10 ng/mL GM-CSF + 10 ng/mL IL-4 + 2 × 10−5 M mercaptoethanol Incubation in differentiation media: 7 days Cells harvested: Non-adherent | Ratio DC/TC: 2:1 Co-culture time: 72 h (on day 5 of differentiation) Extra maturation stimuli: CpG ODN 182 | Prophylactic Tumor challenge*: +10 Doses: 2 (Day −15 and 0) | Subcutaneous (2 × 106 DCs) | Success (in immunogenic tumor model) |
| Pre-clinical | Therapeutic Tumor challenge*: −14 Doses: 3 (Day 0, 14 and 28) * In combination of doxetaclel | Peritumoral (2 × 106 DCs) | Success (in both tumor model) | ||||
| HHP (200 MPa) (Mikyskova et al. 2017) | - TRAMP-C2: murine prostate tumor cell line (immunogenic) | Pre-clinical | Origin: BM precursor Differentiation media: Complete RPMI-1640 + 10 ng/mL GM-CSF + 10 ng/mL IL-4 + 2 × 10−5 M mercaptoethanol Incubation in differentiation media: 7 days Cells harvested: Non-adherent | Ratio DC/TC: 2:1 Co-culture time: 72 h (on day 5 of differentiation) Extra maturation stimuli: CpG ODN 182 | Therapeutic Tumor challenge*: −9 Doses: 4 (Day 0, 2, 4 and 6) *In combination of cyclophosphamide | Subcutaneous (2 × 106 DCs) | Success |
| Pre-clinical | Adjuvant Tumor challenge*: −28 (and then resected) Dose: 2 (Day 7 and 21after tumor resection) | In surgical site (2 × 106 DCs) | Success | ||||
| HT (43 °C, 1 h)/Apoptotic inductor AP20187 (40 nM) (Feng et al. 2001) | - 12B1-D1: murine leukemia cell line, stably expressing FK506 binding protein (FKBP) domains and the Fas death domain (apoptotic response to AP20187) | Pre-clinical | Origin: BM precursor Differentiation media: Complete AIM V medium + 10 ng/mL GM-CSF + 10 ng/mL IL-4 Incubation in differentiation media: 4 days Cells harvested: Non-adherent | Ratio DC/TC: 1:1 Co-culture time: 6 h, in the presence of 40 nM AP20187 (no toxic for DCs). | Adjuvant/Prophylactic Tumor challenge*: DCs were co-administered with tumor cells during tumor challenge Dose: 1 | Subcutaneous (5 × 105 DCs) | Success |
| HT (43 °C, 1 h) + γ-irradiation (200 Gy) (Prasad et al. 2005) | - B16-F10: murine melanoma cell line | Pre-clinical | Origin: BM precursor Differentiation media: Complete IMDM medium + 10 ng/mL GM-CSF + 20 ng/mL IL-4 Incubation in differentiation media: 6–8 days (replenished every 2–3 days). Cells harvested: Non-adherent and loosely adherent | Ratio DC/TC: 1:1 Co-culture time: 48 h Extra maturation stimuli: LPS | Prophylactic: Tumor challenge*: +7 Doses: 1 (Day 0) | Subcutaneous (5 × 105 DCs) | Success |
| HT (46 °C, 1 h) + γ-irradiation (100 Gy) + UV-C (0.5 J/m2 per second, for 25 seconds) (Di Nicola et al. 2009) | - Follicular B-cell non-Hodgkin lymphomas (12 patients)* - Lymphoplasmocytoid lymphoma (6 patients)* *All these patients have relapsed after their previous treatment. | Clinical | Origin: CD14+ peripheral blood monocytes Differentiation media: Complete RPMI medium + 50 ng/mL GM-CSF + 1000 U/mL IL-4 Incubation in differentiation media: 5 days Cells harvested: Non-adherent and loosely adherent | Ratio DC/TC: 1:2 Co-culture time: 48 h Extra maturation stimuli: TNF-α | Therapeutic Doses: 4 (Day 0, 15, 30 and 45) | Subcutaneous injected close to axillary and inguinal lymphoid nodes (45 ± 3 × 106 DCs) | Success (Objective responses were identified in 33.3% of patients) |
| UVB (10 minutes at 1.5 mW/cm2) (Son et al. 2001) | - KLN 205: murine lung squamous cell carcinoma | Pre-clinical | Origin: BM precursor Differentiation media: Complete RPMI+1640 medium + 1000 U/mL GM-CSF + 1000 U/mL IL-4 Incubation in differentiation media: 8 days (replenished at day 4–5). Cell harvested: Non-adherent or loosely adherent * DCs were enriched by means of 14.5% w/v metrizamide gradient separation. | Ratio DC/TC: 2:1 Co-culture time: 48 h (on day 6). | Therapeutic Tumor challenge*: −7 Doses: 5 (Day 0, 10–11, 15–16, 20–21 and 25–26) * In combination with PEG-IL-2 | Subcutaneous (2 × 106 DCs) | Success |
| UVB (Jeong et al. 2007) | - SCCVII (H-2k): murine oral squamous cell carcinoma | Pre-clinical | Origin: BM precursor Differentiation media: Complete RPMI+1640 medium + 1000 U/mL GM-CSF + 1000 U/mL IL-4 Incubation in differentiation media: 8 days (replenished at day 4–5). Cell harvested: Non-adherent or loosely adherent * DCs were enriched by means of 14.5% w/v metrizamide gradient separation. | Ratio DC/TC: 2:1 Co-culture time: 48 h (on day 6). | Prophylactic Tumor challenge*: +15 Doses: 2 (Day −7 and 0) | Subcutaneous (1 × 106 DCs) | Success |
| UVB (Whiteside et al. 2016) | - Squamous cell carcinomas of the head and neck (HNSCC) (12 patients) | Clinical | Origin: Peripheral blood monocytes Differentiation media: AIMV medium + 1000 U/mL GM-CSF + 1000 U/mL IL-4 Incubation in differentiation media: 6 days (replenished on day 3) | Ratio DC/TC: 5:1 Co-culture time: 48 h Extra maturation stimuli: IL-1β, IL-6, TNF-α, PGE2 | Therapeutic Doses: 2 (Day 0 and 42) | Intranodal(2–5 × 106 DC) | Failure |
| UVC (Wculek et al. 2019) | - B16-OVA: OVA-expressing melanoma cells line - B16/F10: melanoma cells line - MC38: colon adenocarcinoma cell line | Pre-clinical | Origin: cDC1s isolated from mouse primary spleen | Ratio DC/TC: 1:2 Co-culture time: 4 h | Therapeutic Tumor challenge*: −5 Doses: 1 (Day 0) | Intradermal (1 × 106 DCs) | Success |
| Pre-clinical | Prophylactic Tumor challenge: +30 Doses: 2 (Day −5 and 0) | Intradermal (0.5 × 106 DCs) | Success | ||||
| Radiotherapy (Strome et al. 2002) | - E.G7 thymoma: murine thymoma cell lines - SCCVII: poorly immunogenic murine squamous cell carcinoma - B16-OVA: melanoma cell line | Pre-clinical | Origin: BM precursors Differentiation media: RPMI-1640 medium + 10 ng/mL GM-CSF + 1 ng/mL IL-4 Incubation in differentiation media: 5 days | Ratio DC/TC: 1:3 Co-culture time: 24 h | Prophylactic Tumor challenge: +7 Doses: 2 (Day −21 and 0) | Intradermal (1 × 106 DCs) | Success |
| Radiotherapy (Goldszmid et al. 2003) | - B16: melanoma cell line | Pre-clinical | Origin: BM precursors Differentiation media: RPMI-1640 medium + 20 g/mL gentamicin + 50 M 2-ME + 10% supernatant from CHO cells transfected with the plasmid pCDNA3 containing the recombinant mouse GM-CSF gene. Incubation in differentiation media: 5 days (replenished on day 3) | Ratio DC/TC: 1:1 Co-culture time: 24 h | Prophilactic Tumor challenge: +7 Doses: 4 (Day −21, −14, −7, and 0) | Subcutaneous (2 × 106 DCs) | Success |
| Radiotherapy (Vandenberk et al. 2015) | - GL261: Methylcholanthrene-induced murine C57BL/6J syngeneic glioma cells | Pre-clinical | Origin: BM precursors Differentiation media: RPMI-1640 medium + 20 ng/mL GM-CSF. Incubation in differentiation media: 7 days (replenished every 2-3 days) | Ratio DC/TC: 2 mg protein of TCL per 10 × 106 DCs per ml culture medium Co-culture time: 24 h Extra maturation stimuli: LPS | Prophylactic Tumor challenge: +7 Doses: 2 (Day −7 and 0) | Intraperitoneal (1 × 106 DCs) | Success |
| Pre-clinical | Therapeutic Tumor challenge*: −2 Doses: 3 (Day 0, 7 and 15) | Intraperitoneal (1 × 106 DCs) | Success | ||||
| Photodynamic Therapy (Ji et al, 2015) | - PECA: Squamous cell carcinoma of the mouse skin | Pre-clinical | Origin: BM precursors Differentiation media: Complete RPMI-1640 medium + 20 ng/mL GM-CSF + 10 ng/mL IL-4. Incubation in differentiation media: 7 days (replenished every 2-3 days) Cell harvested: Loosely adherent | Ratio DC/TC: 1:20 Co-culture time: 24 h | Prophylactic Tumor challenge: +7 Doses: 3 (Day −20, −10 and 0) | Subcutaneous (5 × 106 DCs) | Success |
| Photodynamic Therapy (Zheng et al, 2016) | - LLC: Lewis lung carcinoma cells | Pre-clinical | Origin: BM precursors Differentiation media: Complete RPMI-1640 medium + 10 ng/mL GM-CSF + 10 ng/mL IL-4. Incubation in differentiation media: 7 days (replenished every 2 days) Cell harvested: Non-adherent and loosely adherent | Ratio DC/TC: 1:5 Co-culture time: 24 h | Prophylactic Tumor challenge: +7 Doses: 2 (Day −10 and 0) | Subcutaneous (1 × 106 DCs) | Success |
| Photodynamic Therapy (Garg et al, 2016) | - GL261: Mouse glioblastoma | Pre-clinical | Origin: BM precursors Differentiation media: Complete RPMI-1640 medium + 20 ng/mL GM-CSF Incubation in differentiation media: 7 days (replenished every 2-3 days) | Ratio DC/TC: 2 mg protein of TCL per 10 × 106 DCs per ml culture medium Co-culture time: 24 h Extra maturation stimuli: LPS | Prophylactic Tumor challenge: +7 Doses: 2 (Day −7 and 0) | Intraperitoneal (1 × 106 DCs) | Success |
| Therapeutic Tumor challenge*: −2 Doses: 3 (Day 0, 7 and 14) * In combination with Temozolomide | Intraperitoneal (1 × 106 DCs) | Success (in combination with Temozolomide) | |||||
References
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Cohn, Z.A. Pillars article: Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J. Immunol. 2007, 178, 5–25. [Google Scholar] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258. [Google Scholar] [CrossRef] [Green Version]
- De Vries, I.J.; Krooshoop, D.J.; Scharenborg, N.M.; Lesterhuis, W.J.; Diepstra, J.H.; Van Muijen, G.N.; Strijk, S.P.; Ruers, T.J.; Boerman, O.C.; Oyen, W.J.; et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003, 63, 12–17. [Google Scholar]
- Yatim, N.; Cullen, S.; Albert, M.L. Dying cells actively regulate adaptive immune responses. Nat. Rev. Immunol. 2017, 17, 262–275. [Google Scholar] [CrossRef]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for t cell immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [Green Version]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef]
- Palucka, K.; Ueno, H.; Fay, J.; Banchereau, J. Dendritic cells and immunity against cancer. J. Intern. Med. 2011, 269, 64–73. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Corak, J.; Ciernik, I.F.; Kavanaugh, D.; Carbone, D.P. Decreased antigen presentation by dendritic cells in patients with breast cancer. Clin. Cancer Res. 1997, 3, 483–490. [Google Scholar]
- Kusmartsev, S.; Gabrilovich, D.I. Effect of tumor-derived cytokines and growth factors on differentiation and immune suppressive features of myeloid cells in cancer. Cancer Metastasis Rev. 2006, 25, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Melief, C.J. Cancer immunotherapy by dendritic cells. Immunity 2008, 29, 372–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adkins, I.; Fucikova, J.; Garg, A.D.; Agostinis, P.; Spisek, R. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology 2014, 3, e968434. [Google Scholar] [CrossRef] [PubMed]
- Koks, C.A.; Garg, A.D.; Ehrhardt, M.; Riva, M.; Vandenberk, L.; Boon, L.; De Vleeschouwer, S.; Agostinis, P.; Graf, N.; Van Gool, S.W. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int. J. Cancer 2015, 136, E313–E325. [Google Scholar] [CrossRef] [PubMed]
- Truxova, I.; Hensler, M.; Skapa, P.; Halaska, M.J.; Laco, J.; Ryska, A.; Spisek, R.; Fucikova, J. Rationale for the combination of dendritic cell-based vaccination approaches with chemotherapy agents. Int. Rev. Cell Mol. Biol. 2017, 330, 115–156. [Google Scholar] [PubMed]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rufo, N.; Garg, A.D.; Agostinis, P. The unfolded protein response in immunogenic cell death and cancer immunotherapy. Trends Cancer 2017, 3, 643–658. [Google Scholar] [CrossRef]
- Tesniere, A.; Panaretakis, T.; Kepp, O.; Apetoh, L.; Ghiringhelli, F.; Zitvogel, L.; Kroemer, G. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2008, 15, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and damps in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Cirone, M.; Di Renzo, L.; Lotti, L.V.; Conte, V.; Trivedi, P.; Santarelli, R.; Gonnella, R.; Frati, L.; Faggioni, A. Activation of dendritic cells by tumor cell death. Oncoimmunology 2012, 1, 1218–1219. [Google Scholar] [CrossRef] [Green Version]
- Di Blasio, S.; Wortel, I.M.; van Bladel, D.A.; de Vries, L.E.; Duiveman-de Boer, T.; Worah, K.; de Haas, N.; Buschow, S.I.; de Vries, I.J.; Figdor, C.G.; et al. Human cd1c(+) dcs are critical cellular mediators of immune responses induced by immunogenic cell death. Oncoimmunology 2016, 5, e1192739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fucikova, J.; Becht, E.; Iribarren, K.; Goc, J.; Remark, R.; Damotte, D.; Alifano, M.; Devi, P.; Biton, J.; Germain, C.; et al. Calreticulin expression in human non-small cell lung cancers correlates with increased accumulation of antitumor immune cells and favorable prognosis. Cancer Res. 2016, 76, 1746–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the nlrp3 inflammasome in dendritic cells induces il-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Portela Catani, J.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Adjemian, S.; Yang, H.; Catani, J.P.; Hannani, D.; Martins, I.; Michaud, M.; Kepp, O.; Sukkurwala, A.Q.; Vacchelli, E.; et al. Atp-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology 2013, 2, e24568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, S.K.; Skold, A.E.; Mohanram, V.; Persson, C.; Johansson, U.; Spetz, A.L. Activated apoptotic cells induce dendritic cell maturation via engagement of toll-like receptor 4 (tlr4), dendritic cell-specific intercellular adhesion molecule 3 (icam-3)-grabbing nonintegrin (dc-sign), and beta2 integrins. J. Biol. Chem. 2012, 287, 13731–13742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, T.; Hannani, D.; Poirier-Colame, V.; Ladoire, S.; Locher, C.; Sistigu, A.; Prada, N.; Adjemian, S.; Catani, J.P.; Freudenberg, M.; et al. Defective immunogenic cell death of hmgb1-deficient tumors: Compensatory therapy with tlr4 agonists. Cell Death Differ. 2014, 21, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and activation of cd103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic pd-l1 and braf inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef]
- Sanchez-Paulete, A.R.; Cueto, F.J.; Martinez-Lopez, M.; Labiano, S.; Morales-Kastresana, A.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, M.A.; Quetglas, J.I.; et al. Cancer immunotherapy with immunomodulatory anti-cd137 and anti-pd-1 monoclonal antibodies requires batf3-dependent dendritic cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful anti-pd-1 cancer immunotherapy requires t cell-dendritic cell crosstalk involving the cytokines ifn-gamma and il-12. Immunity 2018, 49, 1148–1161.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Luke, J.J.; Bao, R.; Zha, Y.; Hernandez, K.M.; Li, Y.; Gajewski, A.P.; Andrade, J.; Gajewski, T.F. Density of immunogenic antigens does not explain the presence or absence of the t-cell-inflamed tumor microenvironment in melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, E7759–E7768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, A.; Dammeijer, F.; Aerts, J.; Vroman, H. Current state of dendritic cell-based immunotherapy: Opportunities for in vitro antigen loading of different dc subsets? Front. Immunol. 2018, 9, 2804. [Google Scholar] [CrossRef] [PubMed]
- Macri, C.; Pang, E.S.; Patton, T.; O’Keeffe, M. Dendritic cell subsets. Semin. Cell Dev. Biol. 2018, 84, 11–21. [Google Scholar] [CrossRef]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef] [Green Version]
- Fecek, R.J.; Storkus, W.J. Combination strategies to enhance the potency of monocyte-derived dendritic cell-based cancer vaccines. Immunotherapy 2016, 8, 1205–1218. [Google Scholar] [CrossRef] [Green Version]
- Parlato, S.; Santini, S.M.; Lapenta, C.; Di Pucchio, T.; Logozzi, M.; Spada, M.; Giammarioli, A.M.; Malorni, W.; Fais, S.; Belardelli, F. Expression of ccr-7, mip-3beta, and th-1 chemokines in type i ifn-induced monocyte-derived dendritic cells: Importance for the rapid acquisition of potent migratory and functional activities. Blood 2001, 98, 3022–3029. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L.; Odoux, C. Dendritic cell biology and cancer therapy. Cancer Immunol. Immunother. 2004, 53, 240–248. [Google Scholar] [CrossRef]
- Lee, A.W.; Truong, T.; Bickham, K.; Fonteneau, J.F.; Larsson, M.; Da Silva, I.; Somersan, S.; Thomas, E.K.; Bhardwaj, N. A clinical grade cocktail of cytokines and pge2 results in uniform maturation of human monocyte-derived dendritic cells: Implications for immunotherapy. Vaccine 2002, 20 (Suppl. 4), A8–A22. [Google Scholar] [CrossRef]
- Hanks, B.A. Immune evasion pathways and the design of dendritic cell-based cancer vaccines. Discov. Med. 2016, 21, 135–142. [Google Scholar] [PubMed]
- Mohme, M.; Riethdorf, S.; Pantel, K. Circulating and disseminated tumour cells—Mechanisms of immune surveillance and escape. Nat. Rev. Clin. Oncol. 2017, 14, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Bassani-Sternberg, M.; Braunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific t cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alaniz, L.; Rizzo, M.; Mazzolini, G. Pulsing dendritic cells with whole tumor cell lysates. Methods Mol. Biol. 2014, 1139, 27–31. [Google Scholar]
- Chiang, C.L.; Coukos, G.; Kandalaft, L.E. Whole tumor antigen vaccines: Where are we? Vaccines 2015, 3, 344–372. [Google Scholar] [CrossRef] [Green Version]
- Vandenberk, L.; Belmans, J.; Van Woensel, M.; Riva, M.; Van Gool, S.W. Exploiting the immunogenic potential of cancer cells for improved dendritic cell vaccines. Front. Immunol. 2015, 6, 663. [Google Scholar] [CrossRef] [Green Version]
- Begovic, M.; Herberman, R.B.; Gorelik, E. Ultraviolet light-induced increase in tumor cell susceptibility to tnf-dependent and tnf-independent natural cell-mediated cytotoxicity. Cell Immunol. 1991, 138, 349–359. [Google Scholar] [CrossRef]
- Obeid, M.; Panaretakis, T.; Joza, N.; Tufi, R.; Tesniere, A.; van Endert, P.; Zitvogel, L.; Kroemer, G. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and uvc light-induced apoptosis. Cell Death Differ. 2007, 14, 1848–1850. [Google Scholar] [CrossRef]
- Chiang, C.L.; Kandalaft, L.E.; Tanyi, J.; Hagemann, A.R.; Motz, G.T.; Svoronos, N.; Montone, K.; Mantia-Smaldone, G.M.; Smith, L.; Nisenbaum, H.L.; et al. A dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: From bench to bedside. Clin. Cancer Res. 2013, 19, 4801–4815. [Google Scholar] [CrossRef] [Green Version]
- Kandalaft, L.E.; Chiang, C.L.; Tanyi, J.; Motz, G.; Balint, K.; Mick, R.; Coukos, G. A phase i vaccine trial using dendritic cells pulsed with autologous oxidized lysate for recurrent ovarian cancer. J. Transl. Med. 2013, 11, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandalaft, L.E.; Powell, D.J., Jr.; Chiang, C.L.; Tanyi, J.; Kim, S.; Bosch, M.; Montone, K.; Mick, R.; Levine, B.L.; Torigian, D.A.; et al. Autologous lysate-pulsed dendritic cell vaccination followed by adoptive transfer of vaccine-primed ex vivo co-stimulated t cells in recurrent ovarian cancer. Oncoimmunology 2013, 2, e22664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas-Sepulveda, D.; Tittarelli, A.; Gleisner, M.A.; Avalos, I.; Pereda, C.; Gallegos, I.; Gonzalez, F.E.; Lopez, M.N.; Butte, J.M.; Roa, J.C.; et al. Tumor lysate-based vaccines: On the road to immunotherapy for gallbladder cancer. Cancer Immunol. Immunother. 2018, 67, 1897–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanyi, J.L.; Bobisse, S.; Ophir, E.; Tuyaerts, S.; Roberti, A.; Genolet, R.; Baumgartner, P.; Stevenson, B.J.; Iseli, C.; Dangaj, D.; et al. Personalized cancer vaccine effectively mobilizes antitumor t cell immunity in ovarian cancer. Sci. Transl. Med. 2018, 10, eaao5931. [Google Scholar] [CrossRef] [Green Version]
- Frank, M.J.; Reagan, P.M.; Bartlett, N.L.; Gordon, L.I.; Friedberg, J.W.; Czerwinski, D.K.; Long, S.R.; Hoppe, R.T.; Janssen, R.; Candia, A.F.; et al. In situ vaccination with a tlr9 agonist and local low-dose radiation induces systemic responses in untreated indolent lymphoma. Cancer Discov. 2018, 8, 1258–1269. [Google Scholar] [CrossRef] [Green Version]
- Hammerich, L.; Marron, T.U.; Upadhyay, R.; Svensson-Arvelund, J.; Dhainaut, M.; Hussein, S.; Zhan, Y.; Ostrowski, D.; Yellin, M.; Marsh, H.; et al. Systemic clinical tumor regressions and potentiation of pd1 blockade with in situ vaccination. Nat. Med. 2019, 25, 814–824. [Google Scholar] [CrossRef]
- Suek, N.; Campesato, L.F.; Merghoub, T.; Khalil, D.N. Targeted apc activation in cancer immunotherapy to enhance the abscopal effect. Front. Immunol. 2019, 10, 604. [Google Scholar] [CrossRef]
- Castiello, L.; Arico, E.; D’Agostino, G.; Santodonato, L.; Belardelli, F. In situ vaccination by direct dendritic cell inoculation: The coming of age of an old idea? Front. Immunol. 2019, 10, 2303. [Google Scholar] [CrossRef]
- Aznar, M.A.; Tinari, N.; Rullan, A.J.; Sanchez-Paulete, A.R.; Rodriguez-Ruiz, M.E.; Melero, I. Intratumoral delivery of immunotherapy-act locally, think globally. J. Immunol. 2017, 198, 31–39. [Google Scholar] [CrossRef]
- Sanchez-Paulete, A.R.; Teijeira, A.; Quetglas, J.I.; Rodriguez-Ruiz, M.E.; Sanchez-Arraez, A.; Labiano, S.; Etxeberria, I.; Azpilikueta, A.; Bolanos, E.; Ballesteros-Briones, M.C.; et al. Intratumoral immunotherapy with xcl1 and sflt3l encoded in recombinant semliki forest virus-derived vectors fosters dendritic cell-mediated t-cell cross-priming. Cancer Res. 2018, 78, 6643–6654. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Feng, X.; Wang, J.; Xu, W.; Islam, M.A.; Sun, T.; Xie, Z.; Wang, C.; Ding, J.; Chen, X. Multiantigenic nanoformulations activate anticancer immunity depending on size. Adv. Funct. Mater. 2019, 29, 1903391. [Google Scholar] [CrossRef]
- Li, S.; Liu, J.; Sun, M.; Wang, J.; Wang, C.; Sun, Y. Cell membrane-camouflaged nanocarriers for cancer diagnostic and therapeutic. Front. Pharmacol. 2020, 11, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, B.; Middelberg, A.P.; Gemiarto, A.; MacDonald, K.; Baxter, A.G.; Talekar, M.; Moi, D.; Tullett, K.M.; Caminschi, I.; Lahoud, M.H.; et al. Self-adjuvanting nanoemulsion targeting dendritic cell receptor clec9a enables antigen-specific immunotherapy. J. Clin. Investig. 2018, 128, 1971–1984. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Kusmartsev, S.; Cheng, F.; Paolini, M.; Nefedova, Y.; Sotomayor, E.; Gabrilovich, D. Effective combination of chemotherapy and dendritic cell administration for the treatment of advanced-stage experimental breast cancer. Clin. Cancer Res. 2003, 9, 285–294. [Google Scholar]
- Lee, J.M.; Lee, M.H.; Garon, E.; Goldman, J.W.; Salehi-Rad, R.; Baratelli, F.E.; Schaue, D.; Wang, G.; Rosen, F.; Yanagawa, J.; et al. Phase i trial of intratumoral injection of ccl21 gene-modified dendritic cells in lung cancer elicits tumor-specific immune responses and cd8(+) t-cell infiltration. Clin. Cancer Res. 2017, 23, 4556–4568. [Google Scholar] [CrossRef] [Green Version]
- Subbiah, V.; Murthy, R.; Hong, D.S.; Prins, R.M.; Hosing, C.; Hendricks, K.; Kolli, D.; Noffsinger, L.; Brown, R.; McGuire, M.; et al. Cytokines produced by dendritic cells administered intratumorally correlate with clinical outcome in patients with diverse cancers. Clin. Cancer Res. 2018, 24, 3845–3856. [Google Scholar] [CrossRef] [Green Version]
- Cox, M.C.; Castiello, L.; Mattei, M.; Santodonato, L.; D’Agostino, G.; Muraro, E.; Martorelli, D.; Lapenta, C.; Di Napoli, A.; Di Landro, F.; et al. Clinical and antitumor immune responses in relapsed/refractory follicular lymphoma patients after intranodal injections of ifnalpha-dendritic cells and rituximab: A phase i clinical trial. Clin. Cancer Res. 2019, 25, 5231–5241. [Google Scholar] [CrossRef] [Green Version]
- Hirooka, Y.; Itoh, A.; Kawashima, H.; Hara, K.; Nonogaki, K.; Kasugai, T.; Ohno, E.; Ishikawa, T.; Matsubara, H.; Ishigami, M.; et al. A combination therapy of gemcitabine with immunotherapy for patients with inoperable locally advanced pancreatic cancer. Pancreas 2009, 38, e69–e74. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Guo, J.; Zhu, J.; Sheng, X.; Wang, X.; Qu, L.; Han, Y.; Liu, Y.; Zhang, H.; Huo, L.; Zhang, S.; et al. Intratumoral injection of dendritic cells in combination with local hyperthermia induces systemic antitumor effect in patients with advanced melanoma. Int. J. Cancer 2007, 120, 2418–2425. [Google Scholar] [CrossRef]
- Yang, W.; Zhu, G.; Wang, S.; Yu, G.; Yang, Z.; Lin, L.; Zhou, Z.; Liu, Y.; Dai, Y.; Zhang, F.; et al. In situ dendritic cell vaccine for effective cancer immunotherapy. ACS Nano 2019, 13, 3083–3094. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Groenendyk, J.; Lynch, J.; Michalak, M. Calreticulin, ca2+, and calcineurin—Signaling from the endoplasmic reticulum. Mol. Cells 2004, 17, 383–389. [Google Scholar] [PubMed]
- Oliver, J.D.; Roderick, H.L.; Llewellyn, D.H.; High, S. Erp57 functions as a subunit of specific complexes formed with the er lectins calreticulin and calnexin. Mol. Biol. Cell 1999, 10, 2573–2582. [Google Scholar] [CrossRef] [Green Version]
- Obeid, M. Erp57 membrane translocation dictates the immunogenicity of tumor cell death by controlling the membrane translocation of calreticulin. J. Immunol. 2008, 181, 2533–2543. [Google Scholar] [CrossRef] [Green Version]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; van Endert, P.; et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef]
- Pawaria, S.; Binder, R.J. Cd91-dependent programming of t-helper cell responses following heat shock protein immunization. Nat. Commun. 2011, 2, 521. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.; et al. A novel pathway combining calreticulin exposure and atp secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [Green Version]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Stoll, G.; Iribarren, K.; Michels, J.; Leary, A.; Zitvogel, L.; Cremer, I.; Kroemer, G. Calreticulin expression: Interaction with the immune infiltrate and impact on survival in patients with ovarian and non-small cell lung cancer. Oncoimmunology 2016, 5, e1177692. [Google Scholar] [CrossRef] [Green Version]
- Chao, M.P.; Jaiswal, S.; Weissman-Tsukamoto, R.; Alizadeh, A.A.; Gentles, A.J.; Volkmer, J.; Weiskopf, K.; Willingham, S.B.; Raveh, T.; Park, C.Y.; et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by cd47. Sci. Transl. Med. 2010, 2, 63ra94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigro, A.; Ricciardi, L.; Salvato, I.; Sabbatino, F.; Vitale, M.; Crescenzi, M.A.; Montico, B.; Triggiani, M.; Pepe, S.; Stellato, C.; et al. Enhanced expression of cd47 is associated with off-target resistance to tyrosine kinase inhibitor gefitinib in nsclc. Front. Immunol. 2020, 10, 3135. [Google Scholar] [CrossRef] [Green Version]
- Genest, O.; Wickner, S.; Doyle, S.M. Hsp90 and hsp70 chaperones: Collaborators in protein remodeling. J. Biol. Chem. 2019, 294, 2109–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Bravo-San Pedro, J.M.; Breckpot, K.; Brough, D.; Chaurio, R.; Cirone, M.; et al. Molecular and translational classifications of damps in immunogenic cell death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flechtner, J.B.; Cohane, K.P.; Mehta, S.; Slusarewicz, P.; Leonard, A.K.; Barber, B.H.; Levey, D.L.; Andjelic, S. High-affinity interactions between peptides and heat shock protein 70 augment cd8+ t lymphocyte immune responses. J. Immunol. 2006, 177, 1017–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh-Jasuja, H.; Scherer, H.U.; Hilf, N.; Arnold-Schild, D.; Rammensee, H.G.; Toes, R.E.; Schild, H. The heat shock protein gp96 induces maturation of dendritic cells and down-regulation of its receptor. Eur. J. Immunol. 2000, 30, 2211–2215. [Google Scholar] [CrossRef]
- Lehner, T.; Wang, Y.; Whittall, T.; McGowan, E.; Kelly, C.G.; Singh, M. Functional domains of hsp70 stimulate generation of cytokines and chemokines, maturation of dendritic cells and adjuvanticity. Biochem. Soc. Trans. 2004, 32, 629–632. [Google Scholar] [CrossRef]
- Lin, T.J.; Lin, H.T.; Chang, W.T.; Mitapalli, S.P.; Hsiao, P.W.; Yin, S.Y.; Yang, N.S. Shikonin-enhanced cell immunogenicity of tumor vaccine is mediated by the differential effects of damp components. Mol. Cancer 2015, 14, 174. [Google Scholar] [CrossRef] [Green Version]
- Salimu, J.; Spary, L.K.; Al-Taei, S.; Clayton, A.; Mason, M.D.; Staffurth, J.; Tabi, Z. Cross-presentation of the oncofetal tumor antigen 5t4 from irradiated prostate cancer cells--a key role for heat-shock protein 70 and receptor cd91. Cancer Immunol. Res. 2015, 3, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Spisek, R.; Dhodapkar, M.V. Towards a better way to die with chemotherapy: Role of heat shock protein exposure on dying tumor cells. Cell Cycle 2007, 6, 1962–1965. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic cell death, damps and anticancer therapeutics: An emerging amalgamation. Biochim. Biophys. Acta 2010, 1805, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.G.; Yan, Z.; et al. Hmgb1 in health and disease. Mol. Asp. Med. 2014, 40, 1–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J., 3rd; et al. Endogenous hmgb1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Messer, J.S.; Wang, Y.; Lin, F.; Cham, C.M.; Chang, J.; Billiar, T.R.; Lotze, M.T.; Boone, D.L.; Chang, E.B. Cytosolic hmgb1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J. Clin. Investig. 2015, 125, 1098–1110. [Google Scholar] [CrossRef] [Green Version]
- Bell, C.W.; Jiang, W.; Reich, C.F., 3rd; Pisetsky, D.S. The extracellular release of hmgb1 during apoptotic cell death. Am. J. Physiol. Cell Physiol. 2006, 291, C1318–C1325. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein hmgb1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. Hmgb1 and rage in inflammation and cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- Yu, M.; Wang, H.; Ding, A.; Golenbock, D.T.; Latz, E.; Czura, C.J.; Fenton, M.J.; Tracey, K.J.; Yang, H. Hmgb1 signals through toll-like receptor (tlr) 4 and tlr2. Shock 2006, 26, 174–179. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Shiratsuchi, A.; Watanabe, I.; Takeuchi, O.; Akira, S.; Nakanishi, Y. Inhibitory effect of toll-like receptor 4 on fusion between phagosomes and endosomes/lysosomes in macrophages. J. Immunol. 2004, 172, 2039–2047. [Google Scholar] [CrossRef] [Green Version]
- Fae, D.A.; Martorelli, D.; Mastorci, K.; Muraro, E.; Dal Col, J.; Franchin, G.; Barzan, L.; Comaro, E.; Vaccher, E.; Rosato, A.; et al. Broadening specificity and enhancing cytotoxicity of adoptive t cells for nasopharyngeal carcinoma immunotherapy. Cancer Immunol. Res. 2016, 4, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; De Marchis, F.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually exclusive redox forms of hmgb1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 2012, 209, 1519–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Lundback, P.; Ottosson, L.; Erlandsson-Harris, H.; Venereau, E.; Bianchi, M.E.; Al-Abed, Y.; Andersson, U.; Tracey, K.J.; Antoine, D.J. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (hmgb1). Mol. Med. 2012, 18, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cauwels, A.; Rogge, E.; Vandendriessche, B.; Shiva, S.; Brouckaert, P. Extracellular atp drives systemic inflammation, tissue damage and mortality. Cell Death Dis. 2014, 5, e1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chekeni, F.B.; Elliott, M.R.; Sandilos, J.K.; Walk, S.F.; Kinchen, J.M.; Lazarowski, E.R.; Armstrong, A.J.; Penuela, S.; Laird, D.W.; Salvesen, G.S.; et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature 2010, 467, 863–867. [Google Scholar] [CrossRef] [Green Version]
- Martins, I.; Michaud, M.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Shen, S.; Kepp, O.; Menger, L.; Vacchelli, E.; Galluzzi, L.; et al. Premortem autophagy determines the immunogenicity of chemotherapy-induced cancer cell death. Autophagy 2012, 8, 413–415. [Google Scholar] [CrossRef]
- Martins, I.; Wang, Y.; Michaud, M.; Ma, Y.; Sukkurwala, A.Q.; Shen, S.; Kepp, O.; Metivier, D.; Galluzzi, L.; Perfettini, J.L.; et al. Molecular mechanisms of atp secretion during immunogenic cell death. Cell Death Differ. 2014, 21, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Martins, I.; Ma, Y.; Kepp, O.; Galluzzi, L.; Kroemer, G. Autophagy-dependent atp release from dying cells via lysosomal exocytosis. Autophagy 2013, 9, 1624–1625. [Google Scholar] [CrossRef] [Green Version]
- Chalmin, F.; Mignot, G.; Bruchard, M.; Chevriaux, A.; Vegran, F.; Hichami, A.; Ladoire, S.; Derangere, V.; Vincent, J.; Masson, D.; et al. Stat3 and gfi-1 transcription factors control th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity 2012, 36, 362–373. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Wu, Y.; Gao, W.; Enjyoji, K.; Csizmadia, E.; Muller, C.E.; Murakami, T.; Robson, S.C. Cd39/entpd1 expression by cd4+foxp3+ regulatory t cells promotes hepatic metastatic tumor growth in mice. Gastroenterology 2010, 139, 1030–1040. [Google Scholar] [CrossRef] [Green Version]
- Bloy, N.; Garcia, P.; Laumont, C.M.; Pitt, J.M.; Sistigu, A.; Stoll, G.; Yamazaki, T.; Bonneil, E.; Buque, A.; Humeau, J.; et al. Immunogenic stress and death of cancer cells: Contribution of antigenicity vs adjuvanticity to immunosurveillance. Immunol. Rev. 2017, 280, 165–174. [Google Scholar] [CrossRef]
- Montico, B.; Nigro, A.; Casolaro, V.; Dal Col, J. Immunogenic apoptosis as a novel tool for anticancer vaccine development. Int. J. Mol. Sci. 2018, 19, E594. [Google Scholar] [CrossRef] [Green Version]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remedios, C.; et al. Cancer cell-autonomous contribution of type i interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Komorowski, M.; Tisonczyk, J.; Kolakowska, A.; Drozdz, R.; Kozbor, D. Modulation of the tumor microenvironment by cxcr4 antagonist-armed viral oncotherapy enhances the antitumor efficacy of dendritic cell vaccines against neuroblastoma in syngeneic mice. Viruses 2018, 10, 455. [Google Scholar] [CrossRef] [Green Version]
- Kirkwood, J.M.; Strawderman, M.H.; Ernstoff, M.S.; Smith, T.J.; Borden, E.C.; Blum, R.H. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: The eastern cooperative oncology group trial est 1684. J. Clin. Oncol. 1996, 14, 7–17. [Google Scholar] [CrossRef]
- Moschos, S.J.; Edington, H.D.; Land, S.R.; Rao, U.N.; Jukic, D.; Shipe-Spotloe, J.; Kirkwood, J.M. Neoadjuvant treatment of regional stage iiib melanoma with high-dose interferon alfa-2b induces objective tumor regression in association with modulation of tumor infiltrating host cellular immune responses. J. Clin. Oncol. 2006, 24, 3164–3171. [Google Scholar] [CrossRef]
- Kirkwood, J.M.; Ibrahim, J.G.; Sosman, J.A.; Sondak, V.K.; Agarwala, S.S.; Ernstoff, M.S.; Rao, U. High-dose interferon alfa-2b significantly prolongs relapse-free and overall survival compared with the gm2-klh/qs-21 vaccine in patients with resected stage iib-iii melanoma: Results of intergroup trial e1694/s9512/c509801. J. Clin. Oncol. 2001, 19, 2370–2380. [Google Scholar] [CrossRef]
- Montico, B.; Lapenta, C.; Ravo, M.; Martorelli, D.; Muraro, E.; Zeng, B.; Comaro, E.; Spada, M.; Donati, S.; Santini, S.M.; et al. Exploiting a new strategy to induce immunogenic cell death to improve dendritic cell-based vaccines for lymphoma immunotherapy. Oncoimmunology 2017, 6, e1356964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigro, A.; Montico, B.; Casolaro, V.; Dal Col, J. A novel dendritic cell-based vaccination protocol to stimulate immunosurveillance of aggressive cancers. Methods Mol. Biol. 2019, 1884, 317–333. [Google Scholar]
- Mastorci, K.; Montico, B.; Fae, D.A.; Sigalotti, L.; Ponzoni, M.; Inghirami, G.; Dolcetti, R.; Dal Col, J. Phospholipid scramblase 1 as a critical node at the crossroad between autophagy and apoptosis in mantle cell lymphoma. Oncotarget 2016, 7, 41913–41928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Col, J.; Mastorci, K.; Fae, D.A.; Muraro, E.; Martorelli, D.; Inghirami, G.; Dolcetti, R. Retinoic acid/alpha-interferon combination inhibits growth and promotes apoptosis in mantle cell lymphoma through akt-dependent modulation of critical targets. Cancer Res. 2012, 72, 1825–1835. [Google Scholar] [CrossRef] [Green Version]
- Lamberti, M.J.; Mentucci, F.M.; Roselli, E.; Araya, P.; Rivarola, V.A.; Rumie Vittar, N.B.; Maccioni, M. Photodynamic modulation of type 1 interferon pathway on melanoma cells promotes dendritic cell activation. Front. Immunol. 2019, 10, 2614. [Google Scholar] [CrossRef] [Green Version]
- Medrano, R.F.V.; Hunger, A.; Mendonca, S.A.; Barbuto, J.A.M.; Strauss, B.E. Immunomodulatory and antitumor effects of type i interferons and their application in cancer therapy. Oncotarget 2017, 8, 71249–71284. [Google Scholar] [CrossRef] [Green Version]
- Vermaelen, K. Vaccine strategies to improve anti-cancer cellular immune responses. Front. Immunol. 2019, 10, 8. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, H.J.; Magesh, V.; Nam, D.; Lee, E.O.; Ahn, K.S.; Jung, M.H.; Ahn, K.S.; Kim, D.K.; Kim, J.Y.; et al. Shikonin, acetylshikonin, and isobutyroylshikonin inhibit vegf-induced angiogenesis and suppress tumor growth in lewis lung carcinoma-bearing mice. Yakugaku Zasshi 2008, 128, 1681–1688. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.M.; Wang, P.H.; Chen, S.S.; Wen, C.C.; Chen, Y.H.; Yang, W.C.; Yang, N.S. Shikonin induces immunogenic cell death in tumor cells and enhances dendritic cell-based cancer vaccine. Cancer Immunol. Immunother. 2012, 61, 1989–2002. [Google Scholar] [CrossRef]
- Lin, S.Y.; Hsieh, S.Y.; Fan, Y.T.; Wei, W.C.; Hsiao, P.W.; Tsai, D.H.; Wu, T.S.; Yang, N.S. Necroptosis promotes autophagy-dependent upregulation of damp and results in immunosurveillance. Autophagy 2018, 14, 778–795. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.J.; Liang, W.M.; Hsiao, P.W.; Mitapalli, S.P.; Wei, W.C.; Lin, H.T.; Yin, S.Y.; Yang, N.S. Rapamycin promotes mouse 4t1 tumor metastasis that can be reversed by a dendritic cell-based vaccine. PLoS ONE 2015, 10, e0138335. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.C.; Chen, H.M.; Chen, S.S.; Huang, L.T.; Chang, W.T.; Wei, W.C.; Chou, L.C.; Arulselvan, P.; Wu, J.B.; Kuo, S.C.; et al. Specific microtubule-depolymerizing agents augment efficacy of dendritic cell-based cancer vaccines. J. Biomed. Sci. 2011, 18, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, B.; Franz, S.; Sheriff, A.; Korn, A.; Bluemelhuber, G.; Gaipl, U.S.; Voll, R.E.; Meyer-Pittroff, R.; Herrmann, M. Hydrostatic pressure induced death of mammalian cells engages pathways related to apoptosis or necrosis. Cell Mol. Biol. 2004, 50, 459–467. [Google Scholar] [PubMed]
- Frey, B.; Janko, C.; Ebel, N.; Meister, S.; Schlucker, E.; Meyer-Pittroff, R.; Fietkau, R.; Herrmann, M.; Gaipl, U.S. Cells under pressure—Treatment of eukaryotic cells with high hydrostatic pressure, from physiologic aspects to pressure induced cell death. Curr. Med. Chem. 2008, 15, 2329–2336. [Google Scholar] [CrossRef]
- Korn, A.; Frey, B.; Sheriff, A.; Gaipl, U.S.; Franz, S.; Meyer-Pittroff, R.; Bluemelhuberh, G.; Herrmann, M. High hydrostatic pressure inactivated human tumour cells preserve their immunogenicity. Cell Mol. Biol. 2004, 50, 469–477. [Google Scholar]
- Fucikova, J.; Moserova, I.; Truxova, I.; Hermanova, I.; Vancurova, I.; Partlova, S.; Fialova, A.; Sojka, L.; Cartron, P.F.; Houska, M.; et al. High hydrostatic pressure induces immunogenic cell death in human tumor cells. Int. J. Cancer 2014, 135, 1165–1177. [Google Scholar] [CrossRef]
- Mikyskova, R.; Stepanek, I.; Indrova, M.; Bieblova, J.; Simova, J.; Truxova, I.; Moserova, I.; Fucikova, J.; Bartunkova, J.; Spisek, R.; et al. Dendritic cells pulsed with tumor cells killed by high hydrostatic pressure induce strong immune responses and display therapeutic effects both in murine tc-1 and tramp-c2 tumors when combined with docetaxel chemotherapy. Int. J. Oncol. 2016, 48, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Hradilova, N.; Sadilkova, L.; Palata, O.; Mysikova, D.; Mrazkova, H.; Lischke, R.; Spisek, R.; Adkins, I. Generation of dendritic cell-based vaccine using high hydrostatic pressure for non-small cell lung cancer immunotherapy. PLoS ONE 2017, 12, e0171539. [Google Scholar] [CrossRef]
- Mikyskova, R.; Indrova, M.; Stepanek, I.; Kanchev, I.; Bieblova, J.; Vosahlikova, S.; Moserova, I.; Truxova, I.; Fucikova, J.; Bartunkova, J.; et al. Dendritic cells pulsed with tumor cells killed by high hydrostatic pressure inhibit prostate tumor growth in tramp mice. Oncoimmunology 2017, 6, e1362528. [Google Scholar] [CrossRef]
- Nath, S.; Obaid, G.; Hasan, T. The course of immune stimulation by photodynamic therapy: Bridging fundamentals of photochemically induced immunogenic cell death to the enrichment of t-cell repertoire. Photochem. Photobiol. 2019, 95, 1288–1305. [Google Scholar] [CrossRef] [Green Version]
- Cogno, I.S.; Vittar, N.B.; Lamberti, M.J.; Rivarola, V.A. Optimization of photodynamic therapy response by survivin gene knockdown in human metastatic breast cancer t47d cells. J. Photochem. Photobiol. B 2011, 104, 434–443. [Google Scholar] [CrossRef]
- Lamberti, M.J.; Vittar, N.B.; da Silva Fde, C.; Ferreira, V.F.; Rivarola, V.A. Synergistic enhancement of antitumor effect of beta-lapachone by photodynamic induction of quinone oxidoreductase (nqo1). Phytomedicine 2013, 20, 1007–1012. [Google Scholar] [CrossRef]
- McCormick, B.P.P.; Pansa, M.F.; Sanabria, L.N.M.; Carvalho, C.M.B.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S.; Vittar, N.B.R.; Rivarola, V.A. Cationic porphyrin derivatives for application in photodynamic therapy of cancer. Laser Phys. 2014, 24, 045603. [Google Scholar] [CrossRef]
- Lamberti, M.J.; Vittar, N.B.; Rivarola, V.A. Breast cancer as photodynamic therapy target: Enhanced therapeutic efficiency by overview of tumor complexity. World J. Clin. Oncol. 2014, 5, 901–907. [Google Scholar] [CrossRef] [Green Version]
- Rumie Vittar, N.B.; Comini, L.; Fernadez, I.M.; Agostini, E.; Nunez-Montoya, S.; Cabrera, J.L.; Rivarola, V.A. Photochemotherapy using natural anthraquinones: Rubiadin and soranjidiol sensitize human cancer cell to die by apoptosis. Photodiagn. Photodyn. Ther. 2014, 11, 182–192. [Google Scholar] [CrossRef] [Green Version]
- Vera, R.E.; Lamberti, M.J.; Rivarola, V.A.; Rumie Vittar, N.B. Developing strategies to predict photodynamic therapy outcome: The role of melanoma microenvironment. Tumour Biol. 2015, 36, 9127–9136. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. Damps and pdt-mediated photo-oxidative stress: Exploring the unknown. Photochem. Photobiol. Sci. 2011, 10, 670–680. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like hsp70 and calreticulin. Cancer Immunol. Immunother. 2012, 61, 215–221. [Google Scholar] [CrossRef]
- Garg, A.D.; Nowis, D.; Golab, J.; Agostinis, P. Photodynamic therapy: Illuminating the road from cell death towards anti-tumour immunity. Apoptosis 2010, 15, 1050–1071. [Google Scholar] [CrossRef]
- Kessel, D. Subcellular targeting as a determinant of the efficacy of photodynamic therapy. Photochem. Photobiol. 2017, 93, 609–612. [Google Scholar] [CrossRef] [Green Version]
- Kessel, D.; Oleinick, N.L. Cell death pathways associated with photodynamic therapy: An update. Photochem. Photobiol. 2018, 94, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Milla Sanabria, L.; Rodriguez, M.E.; Cogno, I.S.; Rumie Vittar, N.B.; Pansa, M.F.; Lamberti, M.J.; Rivarola, V.A. Direct and indirect photodynamic therapy effects on the cellular and molecular components of the tumor microenvironment. Biochim. Biophys. Acta 2013, 1835, 36–45. [Google Scholar] [CrossRef]
- Kessel, D. Apoptosis, paraptosis and autophagy: Death and survival pathways associated with photodynamic therapy. Photochem. Photobiol. 2019, 95, 119–125. [Google Scholar] [CrossRef]
- Korbelik, M.; Zhang, W.; Merchant, S. Involvement of damage-associated molecular patterns in tumor response to photodynamic therapy: Surface expression of calreticulin and high-mobility group box-1 release. Cancer Immunol. Immunother. 2011, 60, 1431–1437. [Google Scholar] [CrossRef]
- Mitra, S.; Giesselman, B.R.; De Jesus-Andino, F.J.; Foster, T.H. Tumor response to mthpc-mediated photodynamic therapy exhibits strong correlation with extracellular release of hsp70. Lasers Surg. Med. 2011, 43, 632–643. [Google Scholar] [CrossRef] [Green Version]
- Panzarini, E.; Inguscio, V.; Fimia, G.M.; Dini, L. Rose bengal acetate photodynamic therapy (rbac-pdt) induces exposure and release of damage-associated molecular patterns (damps) in human hela cells. PLoS ONE 2014, 9, e105778. [Google Scholar] [CrossRef]
- Ji, J.; Fan, Z.; Zhou, F.; Wang, X.; Shi, L.; Zhang, H.; Wang, P.; Yang, D.; Zhang, L.; Chen, W.R.; et al. Improvement of dc vaccine with ala-pdt induced immunogenic apoptotic cells for skin squamous cell carcinoma. Oncotarget 2015, 6, 17135–17146. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Vandenberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Van Gool, S.W.; Agostinis, P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and t cell-driven rejection of high-grade glioma. Sci. Transl. Med. 2016, 8, 328ra327. [Google Scholar] [CrossRef]
- Zheng, Y.; Yin, G.; Le, V.; Zhang, A.; Chen, S.; Liang, X.; Liu, J. Photodynamic-therapy activates immune response by disrupting immunity homeostasis of tumor cells, which generates vaccine for cancer therapy. Int. J. Biol. Sci. 2016, 12, 120–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldszmid, R.S.; Idoyaga, J.; Bravo, A.I.; Steinman, R.; Mordoh, J.; Wainstok, R. Dendritic cells charged with apoptotic tumor cells induce long-lived protective cd4+ and cd8+ t cell immunity against b16 melanoma. J. Immunol. 2003, 171, 5940–5947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strome, S.E.; Voss, S.; Wilcox, R.; Wakefield, T.L.; Tamada, K.; Flies, D.; Chapoval, A.; Lu, J.; Kasperbauer, J.L.; Padley, D.; et al. Strategies for antigen loading of dendritic cells to enhance the antitumor immune response. Cancer Res 2002, 62, 1884–1889. [Google Scholar] [PubMed]
- Hu, J.; Adebali, O.; Adar, S.; Sancar, A. Dynamic maps of uv damage formation and repair for the human genome. Proc. Natl. Acad. Sci. USA 2017, 114, 6758–6763. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.S.; Lee, H.; Ko, Y.; Son, Y.I. Vaccinations with dendritic cells primed with apoptotic tumor cells can elicit preventive antitumor immunity in a poorly immunogenic animal model of squamous cell carcinoma. Laryngoscope 2007, 117, 1588–1593. [Google Scholar] [CrossRef]
- Son, Y.I.; Mailliard, R.B.; Watkins, S.C.; Lotze, M.T. Dendritic cells pulsed with apoptotic squamous cell carcinoma have anti-tumor effects when combined with interleukin-2. Laryngoscope 2001, 111, 1472–1478. [Google Scholar] [CrossRef]
- Whiteside, T.L.; Ferris, R.L.; Szczepanski, M.; Tublin, M.; Kiss, J.; Johnson, R.; Johnson, J.T. Dendritic cell-based autologous tumor vaccines for head and neck squamous cell carcinoma. Head Neck 2016, 38 (Suppl. 1), E494–E501. [Google Scholar] [CrossRef] [Green Version]
- Bachem, A.; Guttler, S.; Hartung, E.; Ebstein, F.; Schaefer, M.; Tannert, A.; Salama, A.; Movassaghi, K.; Opitz, C.; Mages, H.W.; et al. Superior antigen cross-presentation and xcr1 expression define human cd11c+cd141+ cells as homologues of mouse cd8+ dendritic cells. J. Exp. Med. 2010, 207, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human cd141+ (bdca-3)+ dendritic cells (dcs) represent a unique myeloid dc subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Lopez, M.; Iborra, S.; Conde-Garrosa, R.; Sancho, D. Batf3-dependent cd103+ dendritic cells are major producers of il-12 that drive local th1 immunity against leishmania major infection in mice. Eur. J. Immunol. 2015, 45, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Amores-Iniesta, J.; Conde-Garrosa, R.; Khouili, S.C.; Melero, I.; Sancho, D. Effective cancer immunotherapy by natural mouse conventional type-1 dendritic cells bearing dead tumor antigen. J. Immunother. Cancer 2019, 7, 100. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, B.; Wust, P.; Ahlers, O.; Dieing, A.; Sreenivasa, G.; Kerner, T.; Felix, R.; Riess, H. The cellular and molecular basis of hyperthermia. Crit. Rev. Oncol. Hematol. 2002, 43, 33–56. [Google Scholar] [CrossRef]
- Harmon, B.V.; Corder, A.M.; Collins, R.J.; Gobe, G.C.; Allen, J.; Allan, D.J.; Kerr, J.F. Cell death induced in a murine mastocytoma by 42-47 degrees c heating in vitro: Evidence that the form of death changes from apoptosis to necrosis above a critical heat load. Int. J. Radiat. Biol. 1990, 58, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Roti Roti, J.L. Cellular responses to hyperthermia (40-46 degrees c): Cell killing and molecular events. Int. J. Hyperth. 2008, 24, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Di Nicola, M.; Zappasodi, R.; Carlo-Stella, C.; Mortarini, R.; Pupa, S.M.; Magni, M.; Devizzi, L.; Matteucci, P.; Baldassari, P.; Ravagnani, F.; et al. Vaccination with autologous tumor-loaded dendritic cells induces clinical and immunologic responses in indolent b-cell lymphoma patients with relapsed and measurable disease: A pilot study. Blood 2009, 113, 18–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Zeng, Y.; Graner, M.W.; Katsanis, E. Stressed apoptotic tumor cells stimulate dendritic cells and induce specific cytotoxic t cells. Blood 2002, 100, 4108–4115. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Zeng, Y.; Whitesell, L.; Katsanis, E. Stressed apoptotic tumor cells express heat shock proteins and elicit tumor-specific immunity. Blood 2001, 97, 3505–3512. [Google Scholar] [CrossRef] [Green Version]
- Prasad, S.J.; Farrand, K.J.; Matthews, S.A.; Chang, J.H.; McHugh, R.S.; Ronchese, F. Dendritic cells loaded with stressed tumor cells elicit long-lasting protective tumor immunity in mice depleted of cd4+cd25+ regulatory t cells. J. Immunol. 2005, 174, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Zappasodi, R.; Pupa, S.M.; Ghedini, G.C.; Bongarzone, I.; Magni, M.; Cabras, A.D.; Colombo, M.P.; Carlo-Stella, C.; Gianni, A.M.; Di Nicola, M. Improved clinical outcome in indolent b-cell lymphoma patients vaccinated with autologous tumor cells experiencing immunogenic death. Cancer Res. 2010, 70, 9062–9072. [Google Scholar] [CrossRef] [Green Version]
- Boudewijns, S.; Bloemendal, M.; Gerritsen, W.R.; de Vries, I.J.; Schreibelt, G. Dendritic cell vaccination in melanoma patients: From promising results to future perspectives. Hum. Vaccines Immunother. 2016, 12, 2523–2528. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Vara Perez, M.; Schaaf, M.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Dendritic cell-based anticancer immunotherapy. Oncoimmunology 2017, 6, e1328341. [Google Scholar] [CrossRef]
- Barbuto, J.A. Are dysfunctional monocyte-derived dendritic cells in cancer an explanation for cancer vaccine failures? Immunotherapy 2013, 5, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Orsini, G.; Legitimo, A.; Failli, A.; Ferrari, P.; Nicolini, A.; Spisni, R.; Miccoli, P.; Consolini, R. Defective generation and maturation of dendritic cells from monocytes in colorectal cancer patients during the course of disease. Int. J. Mol. Sci. 2013, 14, 22022–22041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, R.N.; Chin, L.S.; Dos Santos, A.P.; Bergami-Santos, P.C.; Laginha, F.; Barbuto, J.A. Monocyte-derived dendritic cells from breast cancer patients are biased to induce cd4+cd25+foxp3+ regulatory t cells. J. Leukoc. Biol. 2012, 92, 673–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinde, P.; Fernandes, S.; Melinkeri, S.; Kale, V.; Limaye, L. Compromised functionality of monocyte-derived dendritic cells in multiple myeloma patients may limit their use in cancer immunotherapy. Sci. Rep. 2018, 8, 5705. [Google Scholar] [CrossRef] [PubMed]
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.M.; Bergami-Santos, P.C.; Barbuto, J.A.M. Human dendritic cells: Their heterogeneity and clinical application potential in cancer immunotherapy. Front. Immunol. 2018, 9, 3176. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamberti, M.J.; Nigro, A.; Mentucci, F.M.; Rumie Vittar, N.B.; Casolaro, V.; Dal Col, J. Dendritic Cells and Immunogenic Cancer Cell Death: A Combination for Improving Antitumor Immunity. Pharmaceutics 2020, 12, 256. https://doi.org/10.3390/pharmaceutics12030256
Lamberti MJ, Nigro A, Mentucci FM, Rumie Vittar NB, Casolaro V, Dal Col J. Dendritic Cells and Immunogenic Cancer Cell Death: A Combination for Improving Antitumor Immunity. Pharmaceutics. 2020; 12(3):256. https://doi.org/10.3390/pharmaceutics12030256
Chicago/Turabian StyleLamberti, María Julia, Annunziata Nigro, Fátima María Mentucci, Natalia Belén Rumie Vittar, Vincenzo Casolaro, and Jessica Dal Col. 2020. "Dendritic Cells and Immunogenic Cancer Cell Death: A Combination for Improving Antitumor Immunity" Pharmaceutics 12, no. 3: 256. https://doi.org/10.3390/pharmaceutics12030256
APA StyleLamberti, M. J., Nigro, A., Mentucci, F. M., Rumie Vittar, N. B., Casolaro, V., & Dal Col, J. (2020). Dendritic Cells and Immunogenic Cancer Cell Death: A Combination for Improving Antitumor Immunity. Pharmaceutics, 12(3), 256. https://doi.org/10.3390/pharmaceutics12030256