Strong and Selective Inhibitory Effects of the Biflavonoid Selamariscina A against CYP2C8 and CYP2C9 Enzyme Activities in Human Liver Microsomes

 ,
, 
Abstract
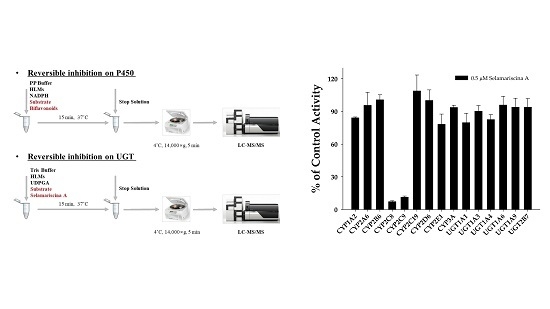
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Inhibitory Effect of Five Biflavonoids against Human Cytochrome P450 Activity
2.3. Kinetic Characterization of Five Biflavonoids on CYP2C8 in Human Liver Microsomes
2.4. Kinetic Characterization of Selamariscina A on Five P450 Enzymes in Human Liver Microsomes
2.5. Time-Dependent Inhibition Assay
2.6. Inhibitory Effect of Selamariscina A against Human UGT Activity
2.7. LC–MS/MS Analysis
2.8. Data Analysis
3. Results and Discussion
3.1. Inhibition of Cytochrome P450 Enzymes Activities by Five Biflaovnoids
3.2. Inhibition of UGT Enzymes Activities by Selamariscina A
3.3. Comparison of the Selectivity of Selamariscina A and Montelukast for CYP2C8 Inhibition
3.4. Evaluation of Drug Interaction Potential of Selamariscina A
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bravo, L. Polyphenols: Chemistry, dietary sources, metabolism, and nutritional significance. Nutr. Rev. 1998, 56, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Cermak, R.; Wolffram, S. The potential of flavonoids to influence drug metabolism and pharmacokinetics by local gastrointestinal mechanisms. Curr. Drug Metab. 2006, 7, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Pandey, A.K. Chemistry and biological activities of flavonoids: An overview. Sci. World J. 2013, 2013, 162750. [Google Scholar] [CrossRef] [Green Version]
- Miron, A.; Aprotosoaie, A.C.; Trifan, A.; Xiao, J. Flavonoids as modulators of metabolic enzymes and drug transporters. Ann. N. Y. Acad. Sci. 2017, 1398, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Kopecna-Zapletalova, M.; Krasulova, K.; Anzenbacher, P.; Hodek, P.; Anzenbacherova, E. Interaction of isoflavonoids with human liver microsomal cytochromes P450: Inhibition of CYP enzyme activities. Xenobiotica 2017, 47, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Scalbert, A.; Morand, C.; Remesy, C.; Jimenez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikkanen, M.J.; Adlercreutz, H. Dietary soy-derived isoflavone phytoestrogens. Could they have a role in coronary heart disease prevention? Biochem. Pharm. 2000, 60, 1–5. [Google Scholar] [CrossRef]
- Cermak, R. Effect of dietary flavonoids on pathways involved in drug metabolism. Expert Opin. Drug Metab. Toxicol. 2008, 4, 17–35. [Google Scholar] [CrossRef]
- Obermeier, M.T.; White, R.E.; Yang, C.S. Effects of bioflavonoids on hepatic P450 activities. Xenobiotica 1995, 25, 575–584. [Google Scholar] [CrossRef]
- Williams, J.A.; Ring, B.J.; Cantrell, V.E.; Campanale, K.; Jones, D.R.; Hall, S.D.; Wrighton, S.A. Differential modulation of UDP-glucuronosyltransferase 1A1 (UGT1A1)-catalyzed estradiol-3-glucuronidation by the addition of UGT1A1 substrates and other compounds to human liver microsomes. Drug Metab. Dispos. 2002, 30, 1266–1273. [Google Scholar] [CrossRef] [Green Version]
- Miniscalco, A.; Lundahl, J.; Regardh, C.G.; Edgar, B.; Eriksson, U.G. Inhibition of dihydropyridine metabolism in rat and human liver microsomes by flavonoids found in grapefruit juice. J. Pharmacol. Exp. Ther. 1992, 261, 1195–1199. [Google Scholar] [PubMed]
- Choi, J.S.; Piao, Y.J.; Kang, K.W. Effects of quercetin on the bioavailability of doxorubicin in rats: Role of CYP3A4 and P-gp inhibition by quercetin. Arch. Pharm. Res. 2011, 34, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Surya Sandeep, M.; Sridhar, V.; Puneeth, Y.; Ravindra Babu, P.; Naveen Babu, K. Enhanced oral bioavailability of felodipine by naringenin in Wistar rats and inhibition of P-glycoprotein in everted rat gut sacs in vitro. Drug Dev. Ind. Pharm. 2014, 40, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Alnaqeeb, M.; Mansor, K.A.; Mallah, E.M.; Ghanim, B.Y.; Idkaidek, N.; Qinna, N.A. Critical pharmacokinetic and pharmacodynamic drug-herb interactions in rats between warfarin and pomegranate peel or guava leaves extracts. BMC Complement. Altern. Med. 2019, 19, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabares-Guevara, J.H.; Lara-Guzman, O.J.; Londono-Londono, J.A.; Sierra, J.A.; Leon-Varela, Y.M.; Alvarez-Quintero, R.M.; Osorio, E.J.; Ramirez-Pineda, J.R. Natural Biflavonoids Modulate Macrophage-Oxidized LDL Interaction In Vitro and Promote Atheroprotection In Vivo. Front. Immunol. 2017, 8, 923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.P.; Park, H.; Son, K.H.; Chang, H.W.; Kang, S.S. Biochemical pharmacology of biflavonoids: Implications for anti-inflammatory action. Arch. Pharm. Res. 2008, 31, 265–273. [Google Scholar] [CrossRef]
- Liu, H.; Ye, M.; Guo, H. An updated review of randomized clinical trials testing the improvement of cognitive function of ginkgo biloba extract in healthy people and Alzheimer’s patients. Front. Pharmacol. 2019, 10, 1688. [Google Scholar] [CrossRef] [Green Version]
- Lv, X.; Zhang, J.B.; Wang, X.X.; Hu, W.Z.; Shi, Y.S.; Liu, S.W.; Hao, D.C.; Zhang, W.D.; Ge, G.B.; Hou, J.; et al. Amentoflavone is a potent broad-spectrum inhibitor of human UDP-glucuronosyltransferases. Chem. Biol. Interact. 2018, 284, 48–55. [Google Scholar] [CrossRef]
- Von Moltke, L.L.; Weemhoff, J.L.; Bedir, E.; Khan, I.A.; Harmatz, J.S.; Goldman, P.; Greenblatt, D.J. Inhibition of human cytochromes P450 by components of Ginkgo biloba. J. Pharm. Pharmacol. 2004, 56, 1039–1044. [Google Scholar] [CrossRef]
- Walsky, R.L.; Obach, R.S.; Gaman, E.A.; Gleeson, J.P.; Proctor, W.R. Selective inhibition of human cytochrome P4502C8 by montelukast. Drug Metab. Dispos. 2005, 33, 413–418. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Ji, D.J.; Han, Y.R.; Choi, J.S.; Rhyu, D.Y.; Min, B.S.; Woo, M.H. Selaginellin and biflavonoids as protein tyrosine phosphatase 1B inhibitors from Selaginella tamariscina and their glucose uptake stimulatory effects. Bioorg. Med. Chem. 2015, 23, 3730–3737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Li, Q.Y.; Yan, L.L.; Shi, Y. Structural characterization and identification of biflavones in Selaginella tamariscina by liquid chromatography-diode-array detection/electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 2173–2186. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.K.; Nguyen, P.H.; Kim, W.C.; Phuc, N.M.; Liu, K.H. Inhibitory effect of selaginellins from Selaginella tamariscina (Beauv.) spring against cytochrome p450 and uridine 5′-diphosphoglucuronosyltransferase isoforms on human liver microsomes. Molecules 2017, 22, 1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Lee, H.; Ji, H.K.; Lee, T.; Liu, K.H. Screening of ten cytochrome P450 enzyme activities with 12 probe substrates in human liver microsomes using cocktail incubation and liquid chromatography-tandem mass spectrometry. Biopharm. Drug Dispos. 2019, 40, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Perloff, E.S.; Mason, A.K.; Dehal, S.S.; Blanchard, A.P.; Morgan, L.; Ho, T.; Dandeneau, A.; Crocker, R.M.; Chandler, C.M.; Boily, N.; et al. Validation of cytochrome P450 time-dependent inhibition assays: A two-time point IC50 shift approach facilitates kinact assay design. Xenobiotica 2009, 39, 99–112. [Google Scholar] [CrossRef]
- Kim, S.J.; You, J.; Choi, H.G.; Kim, J.A.; Jee, J.G.; Lee, S. Selective inhibitory effects of machilin A isolated from Machilus thunbergii on human cytochrome P450 1A and 2B6. Phytomedicine 2015, 22, 615–620. [Google Scholar] [CrossRef]
- Krishnan, S.; Moncrief, S. An evaluation of the cytochrome p450 inhibition potential of lisdexamfetamine in human liver microsomes. Drug Metab. Dispos. 2007, 35, 180–184. [Google Scholar] [CrossRef]
- Joo, J.; Lee, B.; Lee, T.; Liu, K.H. Screening of six UGT enzyme activities in human liver microsomes using liquid chromatography/triple quadrupole mass spectrometry. Rapid Commun. Mass Spectrom. 2014, 28, 2405–2414. [Google Scholar] [CrossRef]
- Shin, J.G.; Soukhova, N.; Flockhart, D.A. Effect of antipsychotic drugs on human liver cytochrome P-450 (CYP) isoforms in vitro: Preferential inhibition of CYP2D6. Drug Metab. Dispos. 1999, 27, 1078–1084. [Google Scholar] [PubMed]
- Lee, E.; Wu, Z.; Shon, J.C.; Liu, K.H. Danazol Inhibits Cytochrome P450 2J2 Activity in a Substrate-independent Manner. Drug Metab. Dispos. 2015, 43, 1250–1253. [Google Scholar] [CrossRef] [Green Version]
- Walsky, R.L.; Gaman, E.A.; Obach, R.S. Examination of 209 drugs for inhibition of cytochrome P450 2C8. J. Clin. Pharmacol. 2005, 45, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Kwara, A.; Greenblatt, D.J. Metabolic interactions between acetaminophen (paracetamol) and two flavonoids, luteolin and quercetin, through in-vitro inhibition studies. J. Pharm. Pharmacol. 2017, 69, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, M.; Qi, H.; Pan, P.; Hou, T.; Li, J.; He, G.; Zhang, H. Pathway-dependent inhibition of paclitaxel hydroxylation by kinase inhibitors and assessment of drug-drug interaction potentials. Drug Metab. Dispos. 2014, 42, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Bjorkman, A.; Andersson, T.B.; Ridderstrom, M.; Masimirembwa, C.M. Amodiaquine clearance and its metabolism to N-desethylamodiaquine is mediated by CYP2C8: A new high affinity and turnover enzyme-specific probe substrate. J. Pharmacol. Exp. Ther. 2002, 300, 399–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khojasteh, S.C.; Prabhu, S.; Kenny, J.R.; Halladay, J.S.; Lu, A.Y. Chemical inhibitors of cytochrome P450 isoforms in human liver microsomes: A re-evaluation of P450 isoform selectivity. Eur. J. Drug Metab. Pharmacol. 2011, 36, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.S.; Galetin, A.; Hallifax, D.; Houston, J.B. Prediction of in vivo drug-drug interactions from in vitro data: Factors affecting prototypic drug-drug interactions involving CYP2C9, CYP2D6 and CYP3A4. Clin. Pharmacol. 2006, 45, 1035–1050. [Google Scholar] [CrossRef]
- Richter, T.; Murdter, T.E.; Heinkele, G.; Pleiss, J.; Tatzel, S.; Schwab, M.; Eichelbaum, M.; Zanger, U.M. Potent mechanism-based inhibition of human CYP2B6 by clopidogrel and ticlopidine. J. Pharmacol. Exp. Ther. 2004, 308, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Palacharla, R.C.; Molgara, P.; Panthangi, H.R.; Boggavarapu, R.K.; Manoharan, A.K.; Ponnamaneni, R.K.; Ajjala, D.R.; Nirogi, R. Methoxsalen as an in vitro phenotyping tool in comparison with 1-aminobenzotriazole. Xenobiotica 2019, 49, 169–176. [Google Scholar] [CrossRef]
- Stresser, D.M.; Broudy, M.I.; Ho, T.; Cargill, C.E.; Blanchard, A.P.; Sharma, R.; Dandeneau, A.A.; Goodwin, J.J.; Turner, S.D.; Erve, J.C.; et al. Highly selective inhibition of human CYP3Aa in vitro by azamulin and evidence that inhibition is irreversible. Drug Metab. Dispos. 2004, 32, 105–112. [Google Scholar] [CrossRef]
- Bjornsson, T.D.; Callaghan, J.T.; Einolf, H.J.; Fischer, V.; Gan, L.; Grimm, S.; Kao, J.; King, S.P.; Miwa, G.; Ni, L.; et al. The conduct of in vitro and in vivo drug-drug interaction studies: A Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab. Dispos. 2003, 31, 815–832. [Google Scholar] [CrossRef]
- Lee, H.; Heo, J.K.; Lee, G.H.; Park, S.Y.; Jang, S.N.; Kim, H.J.; Kwon, M.J.; Song, I.S.; Liu, K.H. Ginsenoside rc is a new selective ugt1a9 inhibitor in human liver microsomes and recombinant human ugt isoforms. Drug Metab. Dispos. 2019, 47, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, X.; Lin, D.; Xu, D.; Li, S.; Huang, J.; Weng, S.; Lin, Z.; Zheng, Y.; Yao, H.; et al. Proliposomes for oral delivery of total biflavonoids extract from Selaginella doederleinii: Formulation development, optimization, and in vitro-in vivo characterization. Int. J. Nanomed. 2019, 14, 6691–6706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Wang, X.; Zou, Y.; Chen, W.; Wang, G.; Yao, W.; Shi, P.; Li, S.; Lin, S.; Lin, X.; et al. Simultaneous quantification of five biflavonoids in rat plasma by LC-ESI-MS/MS and its application to a comparatively pharmacokinetic study of Selaginella doederleinii Hieron extract in rats. J. Pharm. Biomed. Anal. 2018, 149, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wu, J.; Xie, H.; Liu, M.; Takau, I.; Zhang, H.; Xiong, Y.; Xia, C. Inhibitory effect of hesperetin and naringenin on human udp-glucuronosyltransferase enzymes: Implications for herb-drug interactions. Biol. Pharm. Bull. 2016, 39, 2052–2059. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.S.; Neuvonen, M.; Wen, X.; Backman, J.T.; Neuvonen, P.J. Gemfibrozil inhibits CYP2C8-mediated cerivastatin metabolism in human liver microsomes. Drug Metab. Dispos. 2002, 30, 1352–1356. [Google Scholar] [CrossRef] [Green Version]
- Vaclavikova, R.; Horsky, S.; Simek, P.; Gut, I. Paclitaxel metabolism in rat and human liver microsomes is inhibited by phenolic antioxidants. Naunyn. Schmiedebergs Arch. Pharmacol. 2003, 368, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, S.J.; Clarke, S.E.; Chenery, R.J. Characterization of the cytochrome P450 enzymes involved in the in vitro metabolism of rosiglitazone. Br. J. Clin. Pharmacol. 1999, 48, 424–432. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P450 Enzyme | Substrates | Concentration (μM) | Metabolites | SRM Transition (m/z) | Polarity | Collision Energy (eV) |
|---|---|---|---|---|---|---|
| 1A2 | Phenacetin | 100 | Acetaminophen | 152 > 110 | ESI+ | 25 |
| 2A6 | Coumarin | 5 | 7-Hydroxycoumarin | 163 > 107 | ESI+ | 17 |
| 2B6 | Bupropion | 50 | 6-Hydroxybupropion | 256 > 238 | ESI+ | 20 |
| 2C8 | Amodiaquine | 1 | N-Desethylamodiaquine | 328 > 283 | ESI+ | 17 |
| Rosiglitazone | 5 | p-Hydroxyrosiglitazone | 374 > 151 | ESI+ | 17 | |
| 2C9 | Tolbutamide | 100 | 4-Hydroxytolbutamide | 287 > 89 | ESI+ | 60 |
| Diclofenac | 10 | 4-Hydroxydiclofenac | 312 > 231 | ESI+ | 15 | |
| 2C19 | Omeprazole | 20 | 5-Hydroxyomeprazole | 362 > 214 | ESI+ | 10 |
| 2D6 | Dextromethorphan | 5 | Dextrorphan | 258 > 157 | ESI+ | 35 |
| 2E1 | Chlorzoxazone | 50 | 6-Hydroxychlorzoxazone | 184 > 120 | ESI− | 18 |
| 3A | Midazolam | 5 | 1′-Hydroxymidazolam | 342 > 203 | ESI+ | 25 |
| IS | Trimipramine | 0.007 | 295 > 100 | ESI+ | 17 |
| UGT Enzyme | Substrates | Concentration (μM) | Metabolites | SRM Transition (m/z) | Polarity | Collision Energy (eV) |
|---|---|---|---|---|---|---|
| 1A1 | SN-38 | 0.5 | SN-38 glucuronide | 569 > 393 | ESI+ | 30 |
| 1A3 | Chenodeoxycholic acid (CDCA) | 2 | CDCA-24 glucuronide | 567 > 391 | ESI− | 20 |
| 1A4 | Trifluoperazine (TFP) | 0.5 | TFP N-glucuronide | 584 > 408 | ESI+ | 30 |
| 1A6 | N-Acetylserotonin (N-SER) | 1 | N-SER glucuronide | 395 > 219 | ESI+ | 10 |
| 1A9 | Mycophenolic acid (MPA) | 0.2 | MPA 7-O-glucuronide | 495 > 319 | ESI− | 25 |
| 2B7 | Naloxone (NX) | 0.2 | NX 3-glucuronide | 504 > 310 | ESI+ | 30 |
| IS | Estrone glucuronide | 0.25 | 445 > 269 | ESI− | 35 |
| P450 Enzyme | Substrate | IC50 (µM) | |||||
|---|---|---|---|---|---|---|---|
| Selamaris-Cina A | Amento-Flavone | Robusta-Flavone | Cupressu-Flavone | Taiwania-Flavone | Montelukast | ||
| 1A2 | Phenacetin | 7.4 | 4.4 | 4.5 | 5.9 | 6.8 | >50 |
| 2A6 | Coumarin | 11.6 | 11.9 | 11.8 | >20 | 10.6 | >50 |
| 2B6 | Bupropion | 5.3 | 7.1 | 5.7 | 6.7 | 6.4 | >50 |
| 2C8 | Amodiaquine | 0.019 | 0.084 | 0.083 | 0.083 | 0.12 | 0.52 |
| 2C9 | Diclofenac | 0.047 | 0.15 | 0.15 | 0.21 | 0.20 | 9.73 |
| 2C19 | Omeprazole | 13.3 | 3.4 | 6.4 | 3.0 | 5.0 | >50 |
| 2D6 | Dextromethorphan | 10.6 | 2.6 | 2.2 | 2.7 | 3.2 | >50 |
| 2E1 | Chlorzoxazone | >20 | 3.3 | 2.9 | 2.3 | 6.0 | >50 |
| 3A | Midazolam | 2.7 | 1.3 | 1.2 | 1.5 | 1.2 | >50 |
| P450 Enzyme | Substrate | Inhibitor | Ki (µM) a | Mode of Inhibition |
|---|---|---|---|---|
| CYP2C8 | Amodiaquine | Selamariscina A | 0.018 ± 0.002 | Noncompetitive |
| Amentoflavone | 0.083 ± 0.009 | Noncompetitive | ||
| Robustaflavone | 0.084 ± 0.016 | Noncompetitive | ||
| Cupressuflavone | 0.103 ± 0.017 | Noncompetitive | ||
| Taiwaniaflavone | 0.142 ± 0.026 | Noncompetitive |
| P450 Enzyme | Substrate | Ki (µM) a | Mode of Inhibition |
|---|---|---|---|
| 1A2 | Phenacetin | 3.1 ± 0.6 | Competitive |
| 2B6 | Bupropion | 7.9 ± 1.1 | Noncompetitive |
| 2C8 | Amodiaquine | 0.018 ± 0.002 | Noncompetitive |
| Rosiglitazone | 0.010 ± 0.003 | Noncompetitive, partial | |
| 2C9 | Diclofenac | 0.032 ± 0.007 | Competitive |
| Tolbutamide | 0.065 ± 0.01 | Noncompetitive | |
| 3A | Midazolam | 4.5 ± 0.5 | Noncompetitive |
| UGT Enzyme | Substrate | IC50 (µM) a |
|---|---|---|
| 1A1 | SN-38 * | 1.7 ± 0.5 |
| 1A3 | Chenodeoxycholic acid | >50 |
| 1A4 | Trifluoperazine | 7.7 ± 1.9 |
| 1A6 | N-Acetylserotonin | 46.1 ± 11.7 |
| 1A9 | Mycophenolic acid | 40.4 ± 11.1 |
| 2B7 | Naloxone | >50 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.-Y.; Nguyen, P.-H.; Kim, G.; Jang, S.-N.; Lee, G.-H.; Phuc, N.M.; Wu, Z.; Liu, K.-H. Strong and Selective Inhibitory Effects of the Biflavonoid Selamariscina A against CYP2C8 and CYP2C9 Enzyme Activities in Human Liver Microsomes. Pharmaceutics 2020, 12, 343. https://doi.org/10.3390/pharmaceutics12040343
Park S-Y, Nguyen P-H, Kim G, Jang S-N, Lee G-H, Phuc NM, Wu Z, Liu K-H. Strong and Selective Inhibitory Effects of the Biflavonoid Selamariscina A against CYP2C8 and CYP2C9 Enzyme Activities in Human Liver Microsomes. Pharmaceutics. 2020; 12(4):343. https://doi.org/10.3390/pharmaceutics12040343
Chicago/Turabian StylePark, So-Young, Phi-Hung Nguyen, Gahyun Kim, Su-Nyeong Jang, Ga-Hyun Lee, Nguyen Minh Phuc, Zhexue Wu, and Kwang-Hyeon Liu. 2020. "Strong and Selective Inhibitory Effects of the Biflavonoid Selamariscina A against CYP2C8 and CYP2C9 Enzyme Activities in Human Liver Microsomes" Pharmaceutics 12, no. 4: 343. https://doi.org/10.3390/pharmaceutics12040343
APA StylePark, S. -Y., Nguyen, P. -H., Kim, G., Jang, S. -N., Lee, G. -H., Phuc, N. M., Wu, Z., & Liu, K. -H. (2020). Strong and Selective Inhibitory Effects of the Biflavonoid Selamariscina A against CYP2C8 and CYP2C9 Enzyme Activities in Human Liver Microsomes. Pharmaceutics, 12(4), 343. https://doi.org/10.3390/pharmaceutics12040343