Design and Characterization of Phosphatidylcholine-Based Solid Dispersions of Aprepitant for Enhanced Solubility and Dissolution



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of PC-Based Solid Dispersions
2.3. Instrument Characterization of Solid Dispersions
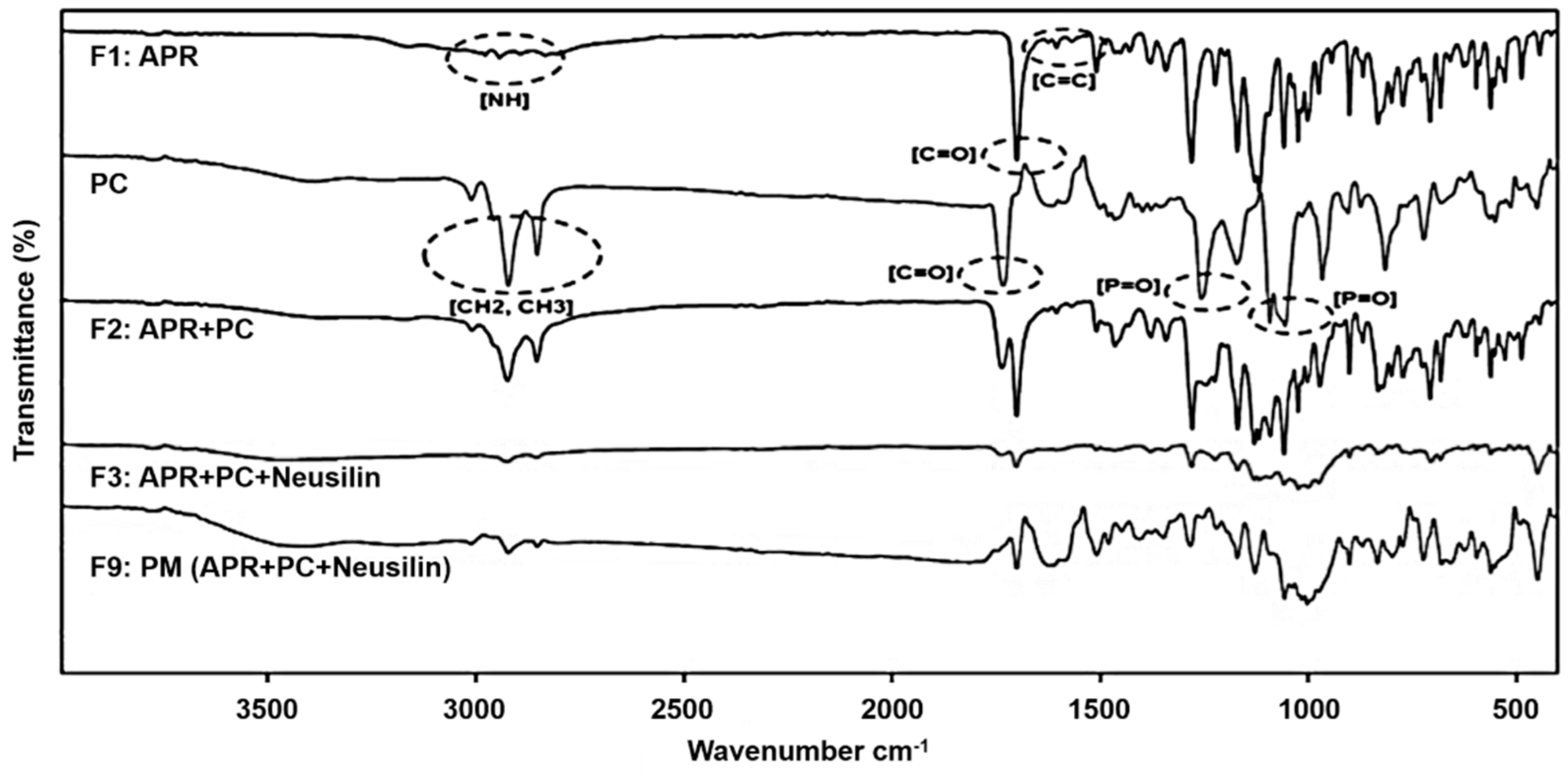
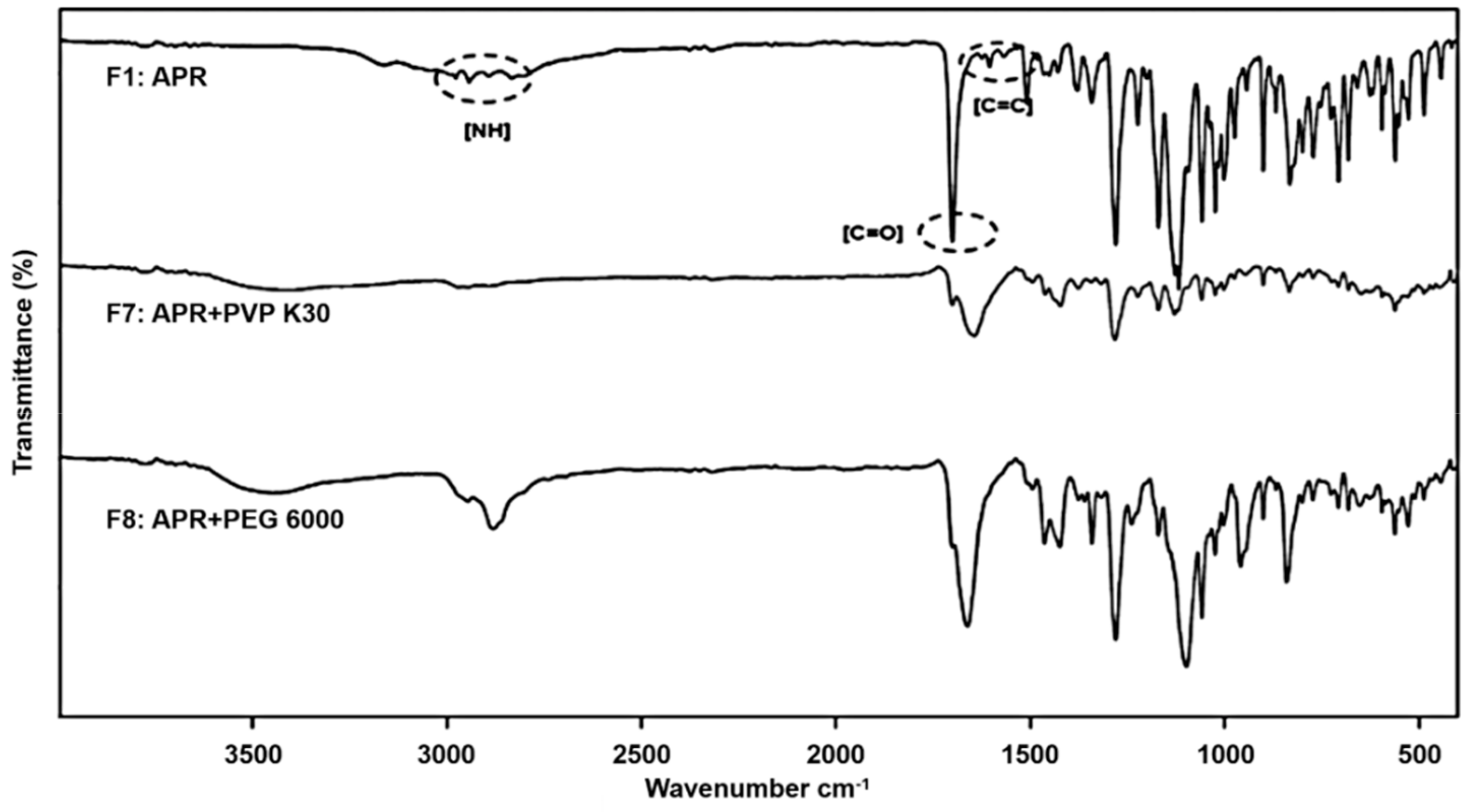
2.3.1. Fourier Transform Infrared Spectroscopy (FTIR)
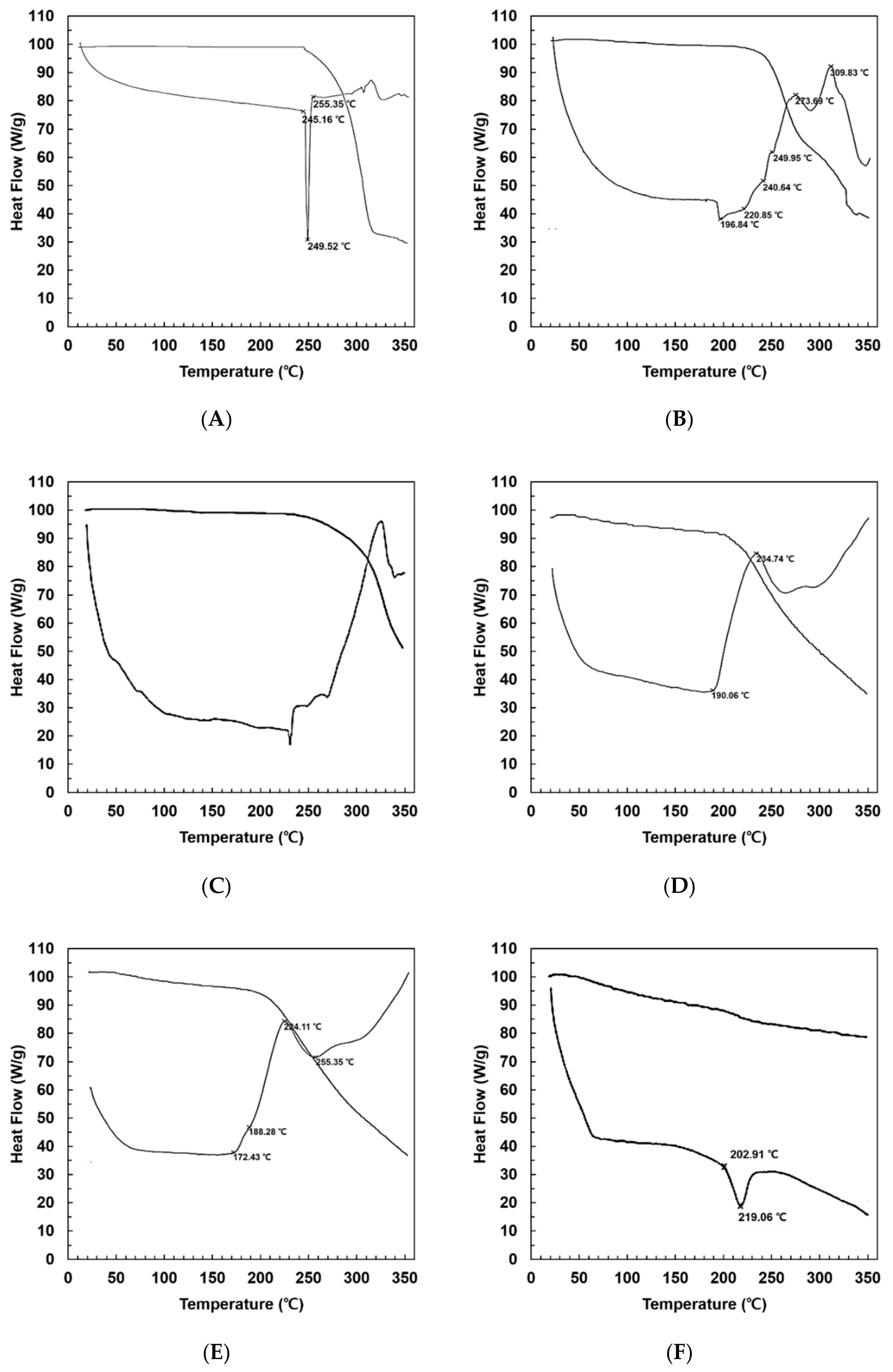
2.3.2. Differential Scanning Calorimetry (DSC)
2.3.3. Morphological Evaluation
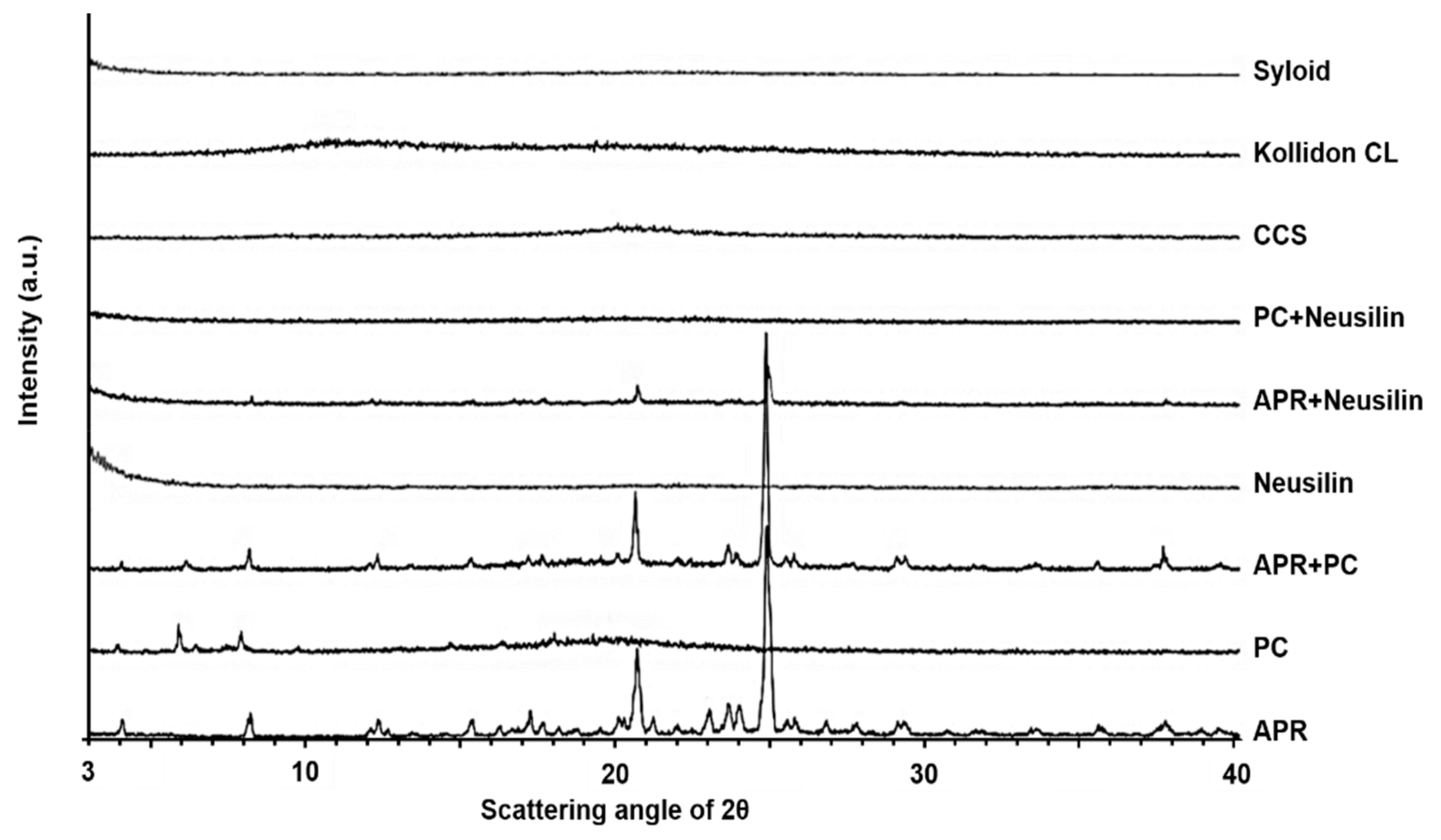
2.3.4. X-ray Powder Diffraction (XRPD)
2.4. Evaluation of Physical Properties of Powderized Solid Dispersions
2.4.1. Measurement of Powder Density
2.4.2. Measurement of Compressibility
2.4.3. Measurement of Flow Properties
2.5. Evaluation of Solubility and Dissolution Rate
2.5.1. Evaluation of Solubility in Water
2.5.2. Drug Dissolution Study
2.6. HPLC Analysis
2.7. Statistical Analysis
3. Results and Discussion
3.1. Instrument Characterization of PC-Based Dispersion of APR
3.1.1. Fourier Transform Infrared Spectroscopy
3.1.2. Differential Scanning Calorimetry
3.1.3. Morphological Evaluation
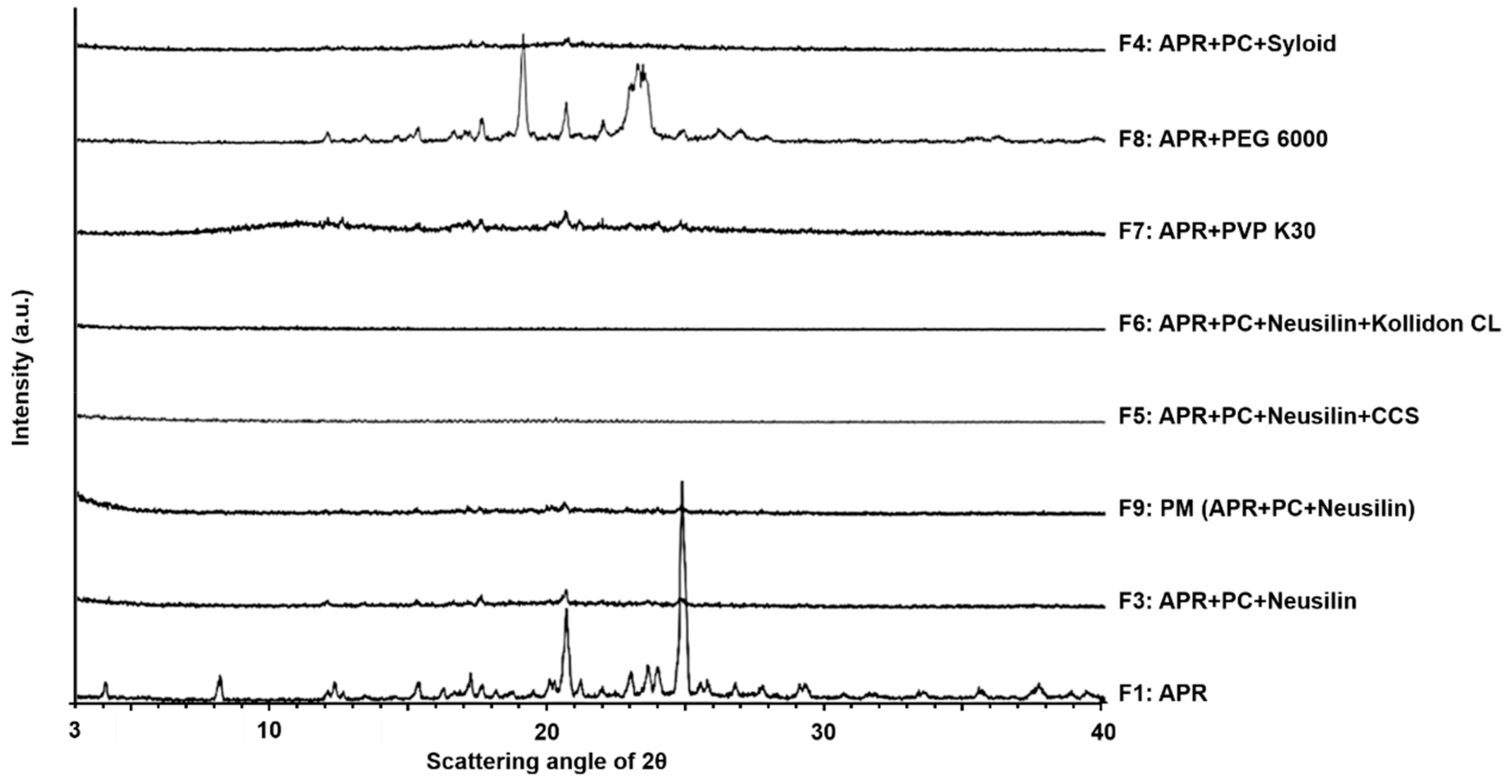
3.1.4. X-ray Powder Diffraction
3.2. Evaluation of Physical Properties of Powderized Solid Dispersions
3.2.1. Measurement of Powder Density and Compressibility
3.2.2. Measurement of Flow Properties
3.3. Evaluation of Solubility and Dissolution Rate
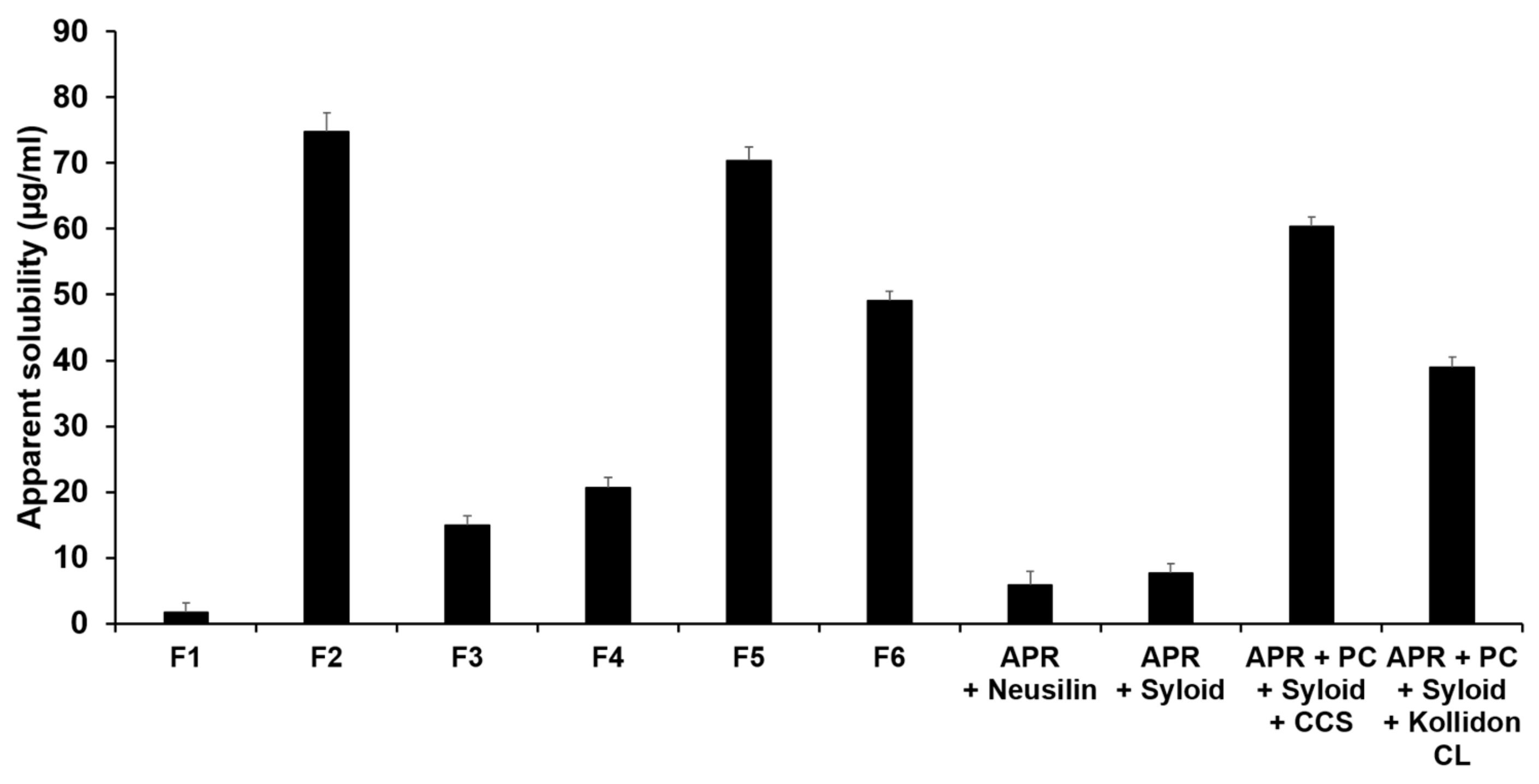
3.3.1. Evaluation of Solubility in Water
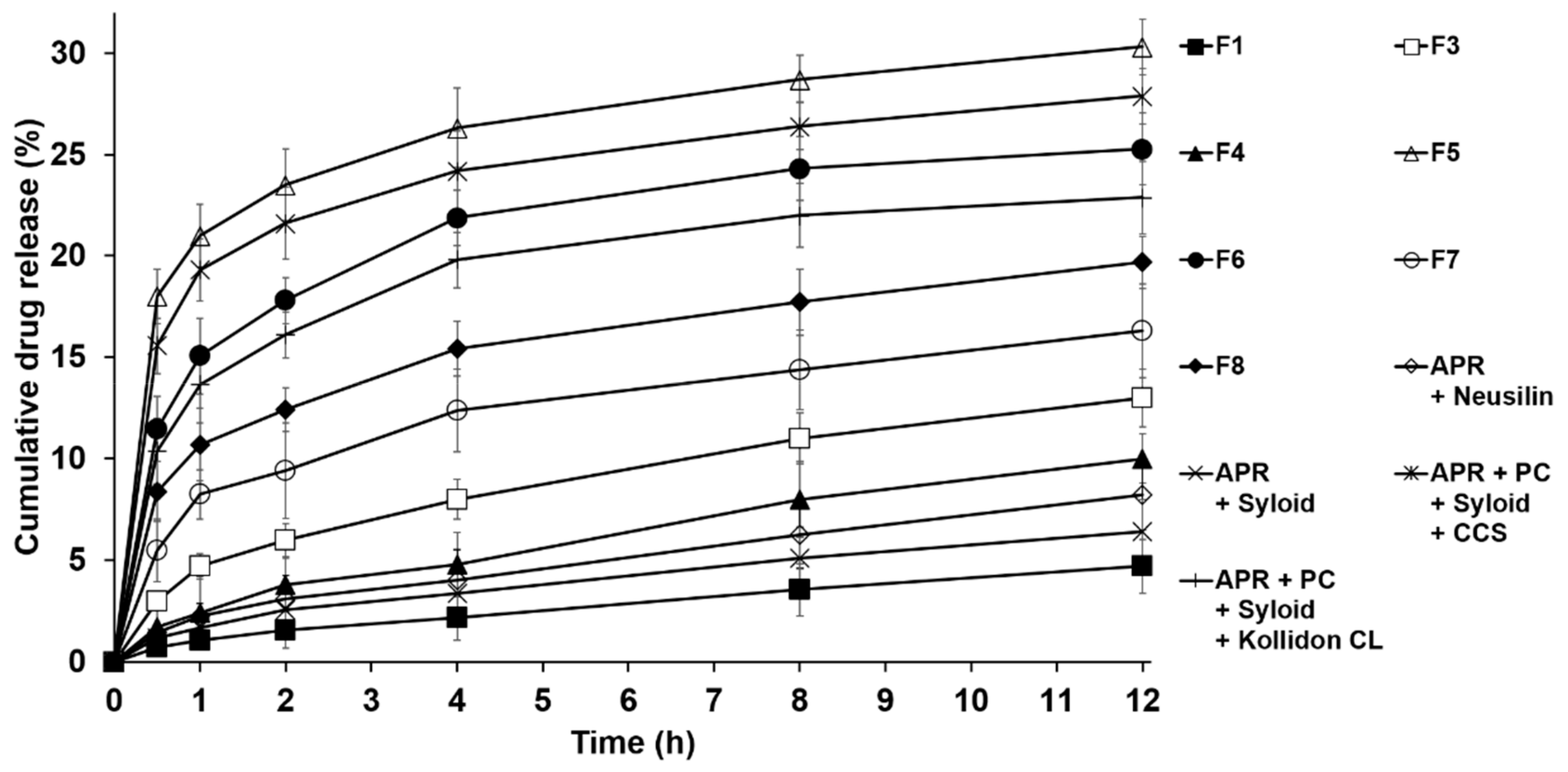
3.3.2. Drug Dissolution Studies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singh, G.; Pai, R.S. Trans-resveratrol self-nano-emulsifying drug delivery system (SNEDDS) with enhanced bioavailability potential: Optimization, pharmacokinetics and in situ single pass intestinal perfusion (SPIP) studies. Drug Deliv. 2015, 22, 522–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, D.; Busciglio, I.; Burton, D.; Halawi, H.; Oduyebo, I.; Rhoten, D.; Ryks, M.; Harmsen, W.S.; Camilleri, M. Effects of NK1 receptors on gastric motor functions and satiation in healthy humans: Results from a controlled trial with the NK1 antagonist aprepitant. Am. J. Physiol. Gastrointest Liver Physiol. 2017, 313, G505–G510. [Google Scholar] [CrossRef]
- Ohanyan, T.; Schoepke, N.; Eirefelt, S.; Hoey, G.; Koopmann, W.; Hawro, T.; Maurer, M.; Metz, M. Role of substance P and its receptor neurokinin 1 in chronic prurigo: A randomized, proof-of-concept, controlled trial with topical aprepitant. Acta Derm. Venereol. 2018, 98, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Zhou, Y.; Wei, P.; Li, M.; Chen, G. Preparation and pharmacokinetic study of aprepitant–sulfobutyl ether-β-cyclodextrin complex. AAPS PharmSciTech 2014, 15, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olver, I.; Shelukar, S.; Thompson, K.C. Nanomedicines in the treatment of emesis during chemotherapy: Focus on aprepitant. Int. J. Nanomed. 2007, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zou, M.; Piao, H.; Liu, Y.; Tang, B.; Gao, Y.; Ma, N.; Cheng, G. Characterization and pharmacokinetic study of aprepitant solid dispersions with soluplus®. Molecules 2015, 20, 11345–11356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanaki, S.; Eleftheriou, R.M.; Barmpalexis, P.; Kostoglou, M.; Karavas, E.; Bikiaris, D. Evaluation of dissolution enhancement of aprepitant drug in ternary pharmaceutical solid dispersions with Soluplus® and Poloxamer 188 prepared by melt mixing. Sci 2019, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Loper, A.; Landis, E.; Hettrick, L.; Novak, L.; Lynn, K.; Chen, C.; Thompson, K.; Higgins, R.; Batra, U. The role of biopharmaceutics in the development of a clinical nanoparticle formulation of MK-0869: A Beagle dog model predicts improved bioavailability and diminished food effect on absorption in human. Int. J. Pharm. 2004, 285, 135–146. [Google Scholar] [CrossRef]
- Angi, R.; Solymosi, T.; Ötvös, Z.; Ordasi, B.; Glavinas, H.; Filipcsei, G.; Heltovics, G.; Darvas, F. Novel continuous flow technology for the development of a nanostructured aprepitant formulation with improved pharmacokinetic properties. Eur. J. Pharm. Biopharm. 2014, 86, 361–368. [Google Scholar] [CrossRef]
- Ridhurkar, D.N.; Ansari, K.A.; Kumar, D.; Kaul, N.S.; Krishnamurthy, T.; Dhawan, S.; Pillai, R. Inclusion complex of aprepitant with cyclodextrin: Evaluation of physico-chemical and pharmacokinetic properties. Drug Dev. Ind. Pharm. 2013, 39, 1783–1792. [Google Scholar] [CrossRef]
- Zhang, X.; Xing, H.; Zhao, Y.; Ma, Z. Pharmaceutical dispersion techniques for dissolution and bioavailability enhancement of poorly water-soluble drugs. Pharmaceutics 2018, 10, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugano, K.; Terada, K. Rate-and extent-limiting factors of oral drug absorption: Theory and applications. J. Pharm. Sci. 2015, 104, 2777–2788. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Okuda, T.; Lu, X.-Y.; Chan, H.-K. Amorphous powders for inhalation drug delivery. Adv. Drug Deliv. Rev. 2016, 100, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Kanaujia, P.; Poovizhi, P.; Ng, W.; Tan, R. Amorphous formulations for dissolution and bioavailability enhancement of poorly soluble APIs. Powder Technol. 2015, 285, 2–15. [Google Scholar] [CrossRef]
- Park, J.H.; Yan, Y.D.; Chi, S.C.; Hwang, D.H.; Shanmugam, S.; Lyoo, W.S.; Woo, J.S.; Yong, C.S.; Choi, H.G. Preparation and evaluation of Cremophor-free paclitaxel solid dispersion by a supercritical antisolvent process. J. Pharm. Pharmacol. 2011, 63, 491–499. [Google Scholar] [CrossRef]
- Pouton, C.W. Lipid formulations for oral administration of drugs: Non-emulsifying, self-emulsifying and ‘self-microemulsifying’drug delivery systems. Eur. J. Pharm. Sci. 2000, 11, S93–S98. [Google Scholar] [CrossRef]
- Sanka, K.; Suda, D.; Bakshi, V. Optimization of solid-self nanoemulsifying drug delivery system for solubility and release profile of clonazepam using simplex lattice design. J. Drug Deliv. Sci. Technol. 2016, 33, 114–124. [Google Scholar] [CrossRef]
- Prajapat, M.D.; Patel, N.J.; Bariya, A.; Patel, S.S.; Butani, S.B. Formulation and evaluation of self-emulsifying drug delivery system for nimodipine, a BCS class II drug. J. Drug Deliv. Sci. Technol. 2017, 39, 59. [Google Scholar] [CrossRef]
- Wang, X.; Li, L.; Huo, W.; Hou, L.; Zhao, Z.; Li, W. Characterization and stability of tanshinone IIA solid dispersions with hydroxyapatite. Materials 2013, 6, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Eloy, J.O.; Marchetti, J.M. Solid dispersions containing ursolic acid in Poloxamer 407 and PEG 6000: A comparative study of fusion and solvent methods. Powder Technol. 2014, 253, 98–106. [Google Scholar] [CrossRef]
- Loh, Z.H.; Samanta, A.K.; Heng, P.W.S. Overview of milling techniques for improving the solubility of poorly water-soluble drugs. Asian J. Pharm. Sci. 2015, 10, 255–274. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Gupta, S.K. Pharmaceutical solid dispersion technology: A strategy to improve dissolution of poorly water-soluble drugs. Recent Pat. Drug Deliv. Formul. 2013, 7, 111–121. [Google Scholar] [CrossRef]
- Dhirendra, K.; Lewis, S.; Udupa, N.; Atin, K. Solid dispersions: A review. Pak. J. Pharm. Sci. 2009, 22, 234–246. [Google Scholar]
- Sosnik, A.; Seremeta, K.P. Advantages and challenges of the spray-drying technology for the production of pure drug particles and drug-loaded polymeric carriers. J. Colloid Interface Sci. 2015, 223, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Narvekar, M.; Xue, H.Y.; Wong, H.L. A novel hybrid delivery system: Polymer-oil nanostructured carrier for controlled delivery of highly lipophilic drug all-trans-retinoic acid (ATRA). Int. J. Pharm. 2012, 436, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Taylor, L.S. Dissolution performance of high drug loading celecoxib amorphous solid dispersions formulated with polymer combinations. Pharm. Res. 2016, 33, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Taylor, L.S. Tailoring supersaturation from amorphous solid dispersions. J. Control. Release 2018, 279, 114–125. [Google Scholar] [CrossRef]
- Dos Santos, K.M.; Barbosa, R.d.M.; Vargas, F.G.A.; de Azevedo, E.P.; Lins, A.C.d.S.; Camara, C.A.; Aragão, C.F.; Moura, T.F.; Raffin, F.N. Development of solid dispersions of β-lapachone in PEG and PVP by solvent evaporation method. Drug Dev. Ind. Pharm. 2018, 44, 750–756. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, K. Solid dispersion-strategy to enhance solubility and dissolution of poorly water soluble drugs. Univers. J. Pharm. Res. 2017, 2, 54–59. [Google Scholar] [CrossRef]
- Mir, K.B.; Khan, N.A. Solid dispersion: Overview of the technology. Int. J. Pharm. Sci. Rev. Res. 2017, 8, 2378–2387. [Google Scholar]
- Inugala, S.; Eedara, B.B.; Sunkavalli, S.; Dhurke, R.; Kandadi, P.; Jukanti, R.; Bandari, S. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) of darunavir for improved dissolution and oral bioavailability: In vitro and in vivo evaluation. Eur. J. Pharm. Sci. 2015, 74, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Khalid, N.; Sarfraz, M.; Arafat, M.; Akhtar, M.; Löbenberg, R.; Rehman, N.U. Nano-sized droplets of self-emulsifying system for enhancing oral bioavailability of chemotherapeutic agent VP-16 in rats: A nano lipid carrier for BCS class IV drugs. J. Pharm. Pharm. Sci. 2018, 21, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Aydin, F.; Chu, X.; Greenstein, J.; Dutt, M. A review on phospholipid vesicles flowing through channels. MRS Commun. 2018, 8, 718–726. [Google Scholar] [CrossRef]
- Semalty, A. Cyclodextrin and phospholipid complexation in solubility and dissolution enhancement: A critical and meta-analysis. Expert. Opin. Drug Deliv. 2014, 11, 1255–1272. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Zhang, T.; Wang, C.; Huang, Z.; Luo, X.; Deng, Y. A review on phospholipids and their main applications in drug delivery systems. Asian J. Pharm. Sci. 2015, 10, 81–98. [Google Scholar] [CrossRef]
- Hussain, M.D.; Saxena, V.; Brausch, J.F.; Talukder, R.M. Ibuprofen–phospholipid solid dispersions: Improved dissolution and gastric tolerance. Int. J. Pharm. 2012, 422, 290–294. [Google Scholar] [CrossRef]
- Ickenstein, L.M.; Sandström, M.C.; Mayer, L.D.; Edwards, K. Effects of phospholipid hydrolysis on the aggregate structure in DPPC/DSPE-PEG2000 liposome preparations after gel to liquid crystalline phase transition. BBA-Biomembranes 2006, 1758, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, H.; Nagira, S.; Yamamoto, H.; Kawashima, Y. Solid dispersion particles of tolbutamide prepared with fine silica particles by the spray-drying method. Powder Technol. 2004, 141, 187–195. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Y.; Deng, J.; Jia, X.; Zhou, J.; Lv, H. Solid dispersion of berberine–phospholipid complex/TPGS 1000/SiO2: Preparation, characterization and in vivo studies. Int. J. Pharm. 2014, 465, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Jeganath, S.; Shafiq, K.M.; Mahesh, P.; Kumar, S.S. Formulation and evaluation of non-effervescent floating tablets of linagliptin using low-density carriers. Drug Invent. Today 2018, 10, 322–329. [Google Scholar]
- Rajurkar, V.; Sunil, N.; Ghawate, V. Tablet formulation and enhancement of aqueous solubility of efavirenz by solvent evaporation Co-Crystal technique. Med. Chem. 2015, 2, 2161-0444.10000. [Google Scholar] [CrossRef] [Green Version]
- Vadher, A.H.; Parikh, J.R.; Parikh, R.H.; Solanki, A.B. Preparation and characterization of co-grinded mixtures of aceclofenac and Neusilin US 2 for dissolution enhancement of aceclofenac. AAPS PharmSciTech 2009, 10, 606–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vojinović, T.; Medarević, D.; Vranić, E.; Potpara, Z.; Krstić, M.; Djuriš, J.; Ibrić, S. Development of ternary solid dispersions with hydrophilic polymer and surface adsorbent for improving dissolution rate of carbamazepine. Saudi. Pharm. J. 2018, 26, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Fan, N.; Wang, X.; Li, C.; Sun, M.; Wang, J.; Fu, Q.; He, Z. Interfacial interaction track of amorphous solid dispersions established by water-soluble polymer and indometacin. Eur. J. Pharm. Sci. 2017, 106, 244–253. [Google Scholar] [CrossRef]
- Keating, A.V.; Soto, J.; Tuleu, C.; Forbes, C.; Zhao, M.; Craig, D.Q. Solid state characterisation and taste masking efficiency evaluation of polymer based extrudates of isoniazid for paediatric administration. Int. J. Pharm. 2018, 536, 536–546. [Google Scholar] [CrossRef]
- Sharma, D. Solubility enhancement strategies for poorly water-soluble drugs in solid dispersions: A review. Afr. J. Pharm. Pharmacol. 2016, 1. [Google Scholar] [CrossRef]
- Angelo, J.M.; Lenhoff, A.M. Determinants of protein elution rates from preparative ion-exchange adsorbents. J. Chromatogr. A 2016, 1440, 94–104. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.; Dudhedia, M.; Deng, W.; Shepard, K.; Zhong, L.; Povilaitis, E.; Zimny, E. Development of tablet formulation of amorphous solid dispersions prepared by hot melt extrusion using quality by design approach. AAPS PharmSciTech 2016, 17, 214–232. [Google Scholar] [CrossRef] [Green Version]
- Schachter, D.M.; Xiong, J.; Tirol, G.C. Solid state NMR perspective of drug–polymer solid solutions: A model system based on poly (ethylene oxide). Int. J. Pharm. 2004, 281, 89–101. [Google Scholar] [CrossRef]
- Shalaev, E.; Wu, K.; Shamblin, S.; Krzyzaniak, J.F.; Descamps, M. Crystalline mesophases: Structure, mobility, and pharmaceutical properties. Adv. Drug Deliv. Rev. 2016, 100, 194–211. [Google Scholar] [CrossRef]
- Azad, M.; Moreno, J.; Davé, R. Stable and fast-dissolving amorphous drug composites preparation via impregnation of Neusilin® UFL2. J. Pharm. Sci. 2018, 107, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Indulkar, A.S.; Waters, J.E.; Mo, H.; Gao, Y.; Raina, S.A.; Zhang, G.G.; Taylor, L.S. Origin of nanodroplet formation upon dissolution of an amorphous solid dispersion: A mechanistic isotope scrambling study. J. Pharm. Sci. 2017, 106, 1998–2008. [Google Scholar] [CrossRef] [PubMed]
- The United States Pharmacopeial Convention. <1174> Powder Flow; US Pharmacopoeia: Rockville, MD, USA, 2012; Volume 18. [Google Scholar]
- Mura, P.; Valleri, M.; Fabianelli, E.; Maestrelli, F.; Cirri, M. Characterization and evaluation of different mesoporous silica kinds as carriers for the development of effective oral dosage forms of glibenclamide. Int. J. Pharm. 2019, 563, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.-F.; Wang, L.-Y.; Tian, Y.-J.; Zhou, Z.-X.; Tang, J.-B.; Liu, X.-R.; Jiang, H.-P.; Shen, Y.-Q. Enhanced water solubility, antioxidant activity, and oral absorption of hesperetin by D-α-tocopheryl polyethylene glycol 1000 succinate and phosphatidylcholine. J. Zhejiang Univ. Sci. B 2019, 20, 273–281. [Google Scholar] [CrossRef]
- Hespeler, D.; El Nomeiri, S.; Kaltenbach, J.; Müller, R.H. Nanoporous smartPearls for dermal application–identification of optimal silica types and a scalable production process as prerequisites for marketed products. Beilstein J. Nanotechnol. 2019, 10, 1666–1678. [Google Scholar] [CrossRef]
- Müller, R.H.; Hespeler, D.; Jin, N.; Pyo, S.M. smartPearls–Novel physically stable amorphous delivery system for poorly soluble dermal actives. Int. J. Pharm. 2019, 555, 314–321. [Google Scholar] [CrossRef]
- Alwadei, M.; Kazi, M.; Alanazi, F.K. Novel oral dosage regimen based on self-nanoemulsifying drug delivery systems for codelivery of phytochemicals–Curcumin and thymoquinone. Saudi Pharm. J. 2019, 27, 866–876. [Google Scholar] [CrossRef]
- Seljak, K.B.; Ilić, I.G.; Gašperlin, M.; Pobirk, A.Z. Self-microemulsifying tablets prepared by direct compression for improved resveratrol delivery. Int. J. Pharm. 2018, 548, 263–275. [Google Scholar] [CrossRef]
- Matji, A.; Carvajal, L.; Conde, F.; Peña, M.A.; Donato, N.; Serrano, D.R.; Torrado, J.J. Effect of the characteristics of raw material ibuprofen on roller compaction and dissolution. J. Drug Deliv. Sci. Technol. 2017, 42, 237–244. [Google Scholar] [CrossRef]
- Zaheer, K.; Langguth, P. Formulation strategy towards minimizing viscosity mediated negative food effect on disintegration and dissolution of immediate release tablets. Drug Dev. Ind. Pharm. 2018, 44, 444–451. [Google Scholar] [CrossRef]
- Bahmani, A.; Saaidpour, S.; Rostami, A. A simple, robust and efficient computational method for n-octanol/water partition coefficients of substituted aromatic drugs. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Barmpalexis, P.; Syllignaki, P.; Kachrimanis, K. A study of water uptake by selected superdisintegrants from the sub-molecular to the particulate level. Pharm. Dev. Technol. 2018, 23, 476–487. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| APR (mg) | PC (mg) | Adsorbent | Disintegrant | Polymeric Carriers | Control | ||||
|---|---|---|---|---|---|---|---|---|---|
| Neusilin® (mg) | Syloid® (mg) | CCS (mg) | Kollidon® CL (mg) | PVP K30 (mg) | PEG 6000 (mg) | ||||
| F1 | 100 | ||||||||
| F2 | 100 | 100 | |||||||
| F3 | 100 | 100 | 200 | ||||||
| F4 | 100 | 100 | 200 | ||||||
| F5 | 100 | 100 | 200 | 50 | |||||
| F6 | 100 | 100 | 200 | 50 | |||||
| F7 | 100 | 300 | |||||||
| F8 | 100 | 300 | |||||||
| F9 | 100 | 100 | 200 | PM * | |||||
| NH (cm−1) | C=C (cm−1) | C=O (cm−1) | |
|---|---|---|---|
| APR | 2943, 2893, 2834 | 1605 | 1700 |
| F2 | 3009, 2924, 2853 | - | 1700 |
| F3 | 2925, 2854 | - | 1702 |
| F9 | - | - | 1700 |
| CH2, CH3 (cm−1) | C=O (ester) (cm−1) | P=O (cm−1) | |
|---|---|---|---|
| PC | 2922, 2822 | 1733 | 1257, 1092, 1055 |
| F2 | - | 1736 | 1091, 1058 |
| F3 | - | 1739 | 1091, 1058 |
| F9 | 2922, 2822 | - | 1057 |
| NH (cm−1) | C=C (cm−1) | C=O (cm−1) | |
|---|---|---|---|
| APR | 2943, 2893, 2834 | 1605 | 1700 |
| F7 | 2972 | - | 1700 |
| F8 | 2946, 2882 | - | 1662 |
| Bulk Density (g/mL) | Tapped Density (g/mL) | Carr’s Index (%) | Hausner Ratio | |
|---|---|---|---|---|
| F3 | 0.155 ± 0.001 | 0.180 ± 0.001 | 13.864 ± 0.096 | 1.161 ± 0.001 |
| F4 | 0.172 ± 0.001 | 0.201 ± 0.001 | 14.405 ± 0.089 | 1.168 ± 0.001 |
| F5 | 0.171 ± 0.004 | 0.193 ± 0.005 | 11.395 ± 0.164 | 1.129 ± 0.002 |
| F6 | 0.166 ± 0.002 | 0.189 ± 0.003 | 12.500 ± 0.143 | 1.143 ± 0.002 |
| Angle of Repose (θ) | Flow Rate (g/s) | |
|---|---|---|
| F3 | 33.81 ± 0.43 | 4.57 ± 0.25 |
| F4 | 34.71 ± 0.89 | 4.03 ± 0.32 |
| F5 | 31.54 ± 0.32 | 6.47 ± 0.30 |
| F6 | 32.17 ± 0.43 | 5.21 ± 0.31 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeo, S.; An, J.; Park, C.; Kim, D.; Lee, J. Design and Characterization of Phosphatidylcholine-Based Solid Dispersions of Aprepitant for Enhanced Solubility and Dissolution. Pharmaceutics 2020, 12, 407. https://doi.org/10.3390/pharmaceutics12050407
Yeo S, An J, Park C, Kim D, Lee J. Design and Characterization of Phosphatidylcholine-Based Solid Dispersions of Aprepitant for Enhanced Solubility and Dissolution. Pharmaceutics. 2020; 12(5):407. https://doi.org/10.3390/pharmaceutics12050407
Chicago/Turabian StyleYeo, Sooho, Jieun An, Changhee Park, Dohyun Kim, and Jaehwi Lee. 2020. "Design and Characterization of Phosphatidylcholine-Based Solid Dispersions of Aprepitant for Enhanced Solubility and Dissolution" Pharmaceutics 12, no. 5: 407. https://doi.org/10.3390/pharmaceutics12050407
APA StyleYeo, S., An, J., Park, C., Kim, D., & Lee, J. (2020). Design and Characterization of Phosphatidylcholine-Based Solid Dispersions of Aprepitant for Enhanced Solubility and Dissolution. Pharmaceutics, 12(5), 407. https://doi.org/10.3390/pharmaceutics12050407