Investigating the Potential of Transmucosal Delivery of Febuxostat from Oral Lyophilized Tablets Loaded with a Self-Nanoemulsifying Delivery System

 ,
, 

Abstract
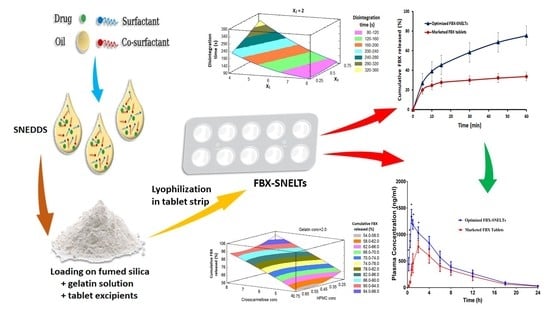
:
1. Introduction
2. Materials and Methods
2.1. Materials
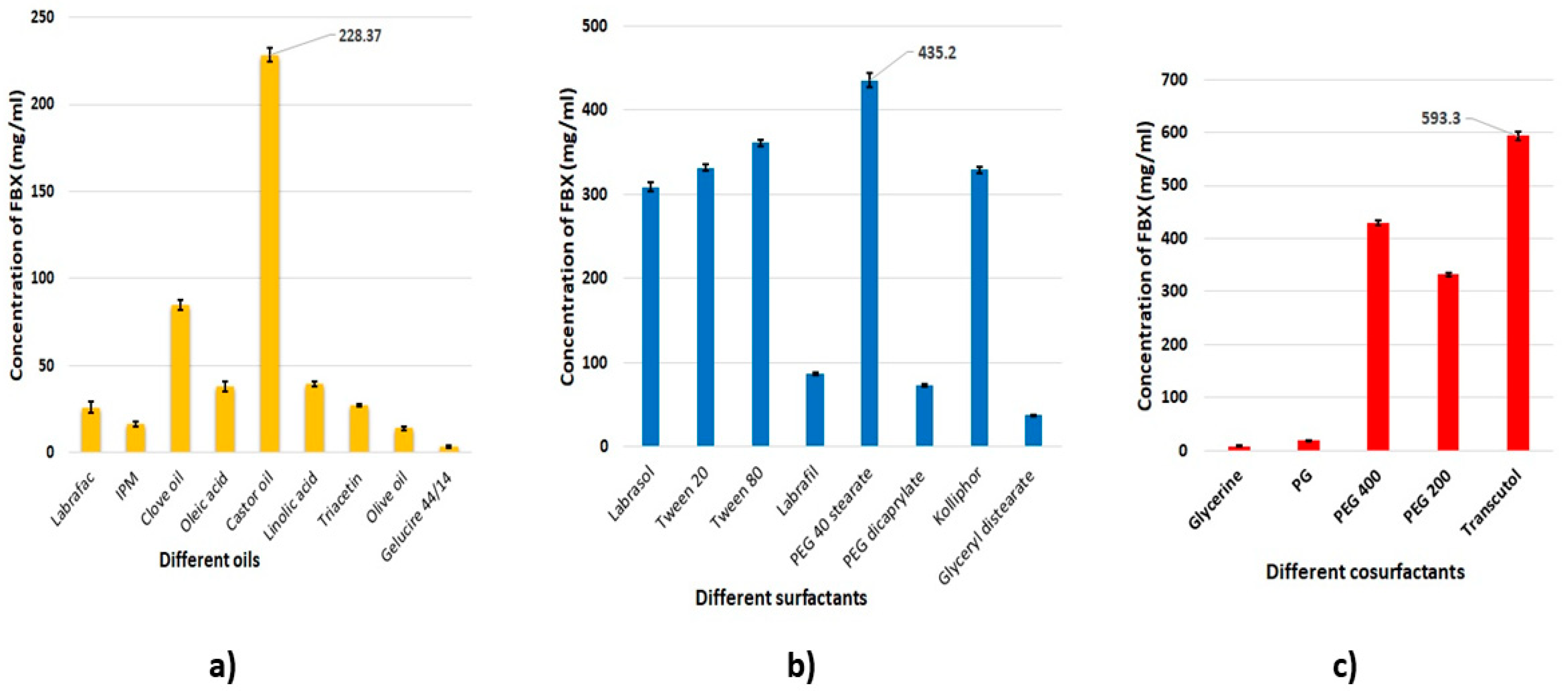
2.2. Solubility Studies of FBX in Different Self-Nanoemulsion Components
2.3. Construction of Pseudo-Ternary Phase Diagram
2.4. Formulation of FBX-Loaded SNEDS according to the Mixture Design
2.5. Evaluation of the FBX-NE Formulations
2.5.1. Visual Inspection for Emulsification Ability
2.5.2. Globule Size Determination
2.5.3. Thermodynamic Stability Studies
2.5.4. Morphology of NE
2.6. Preparation of FBX-SNELTs
2.7. Optimization of FBX-SNELTs
2.8. In Vivo Pharmacokinetic Studies
3. Results and Discussion
3.1. Solubility Studies
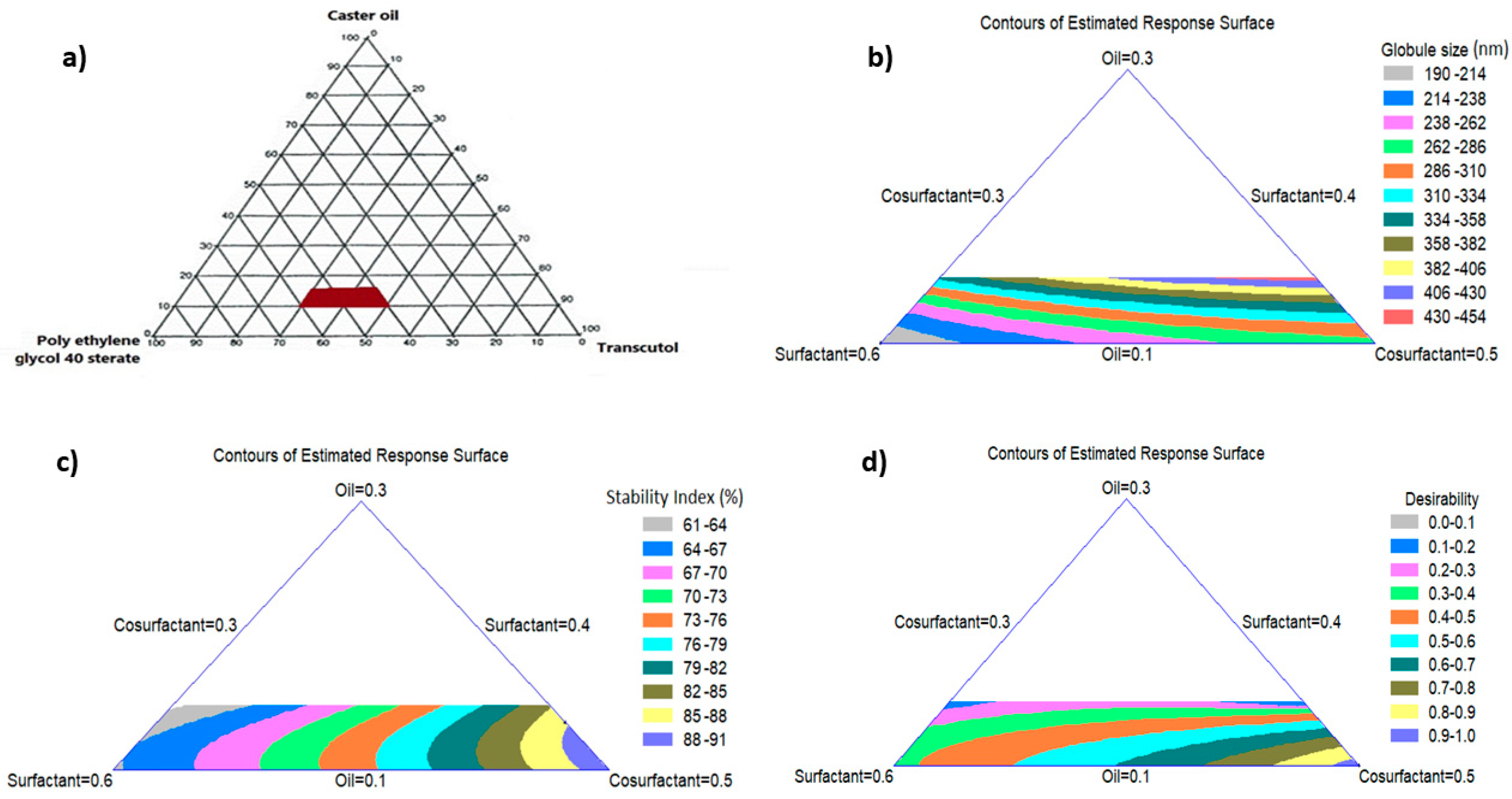
3.2. Construction of Pseudo-Ternary Phase Diagram
3.3. Optimization of FBX-NE Formulations
3.3.1. Effect of NE Components on the Globule Size
3.3.2. Effect of NE Components on the Stability Index
3.3.3. Multiple Response Optimization Using the Desirability Function
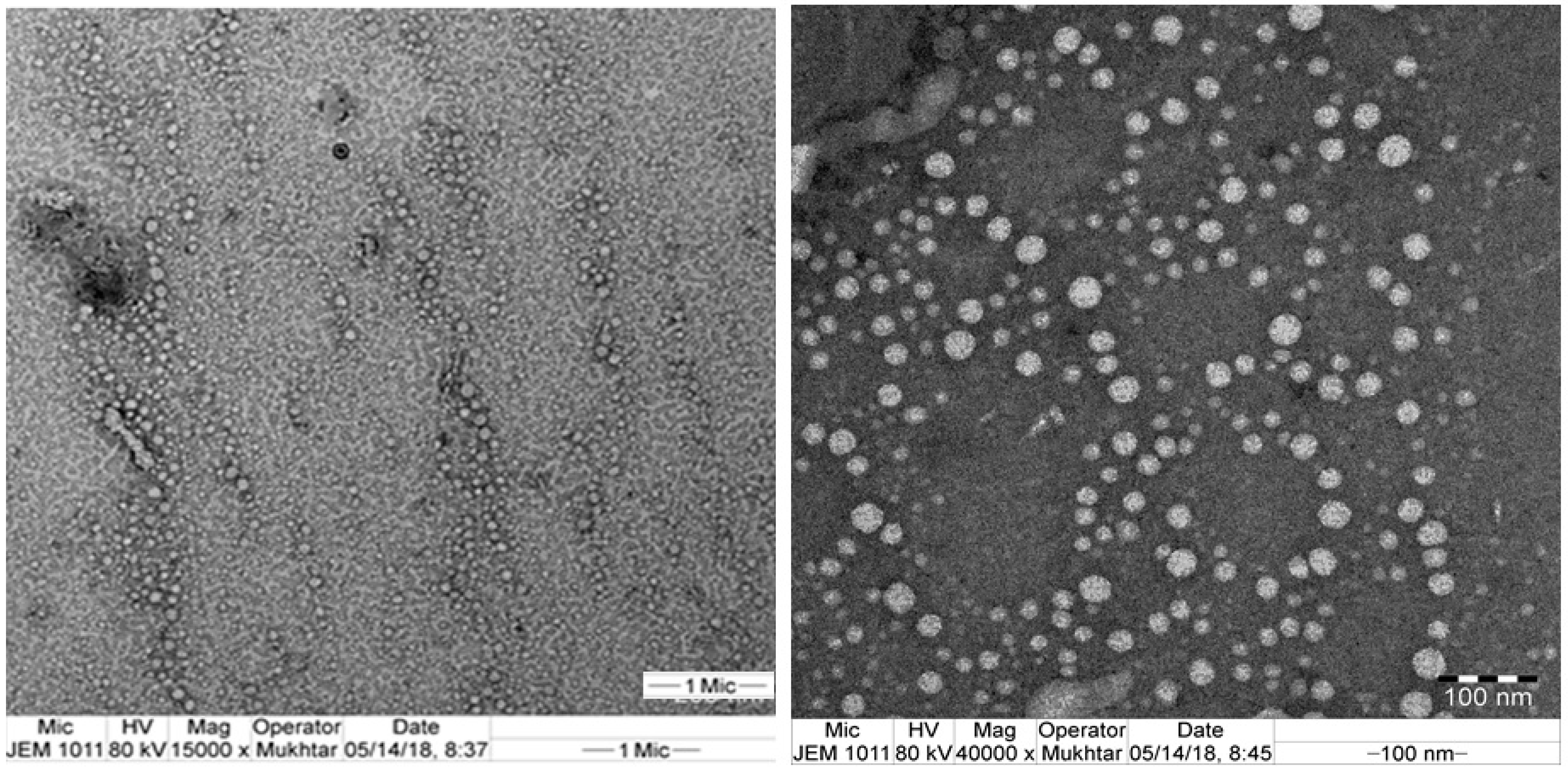
3.4. Morphological Examination Using TEM
3.5. Formulation of FBX-SNELTs
3.6. Evaluation of the Prepared FBX-SNELTs
3.7. In Vitro Disintegration Study
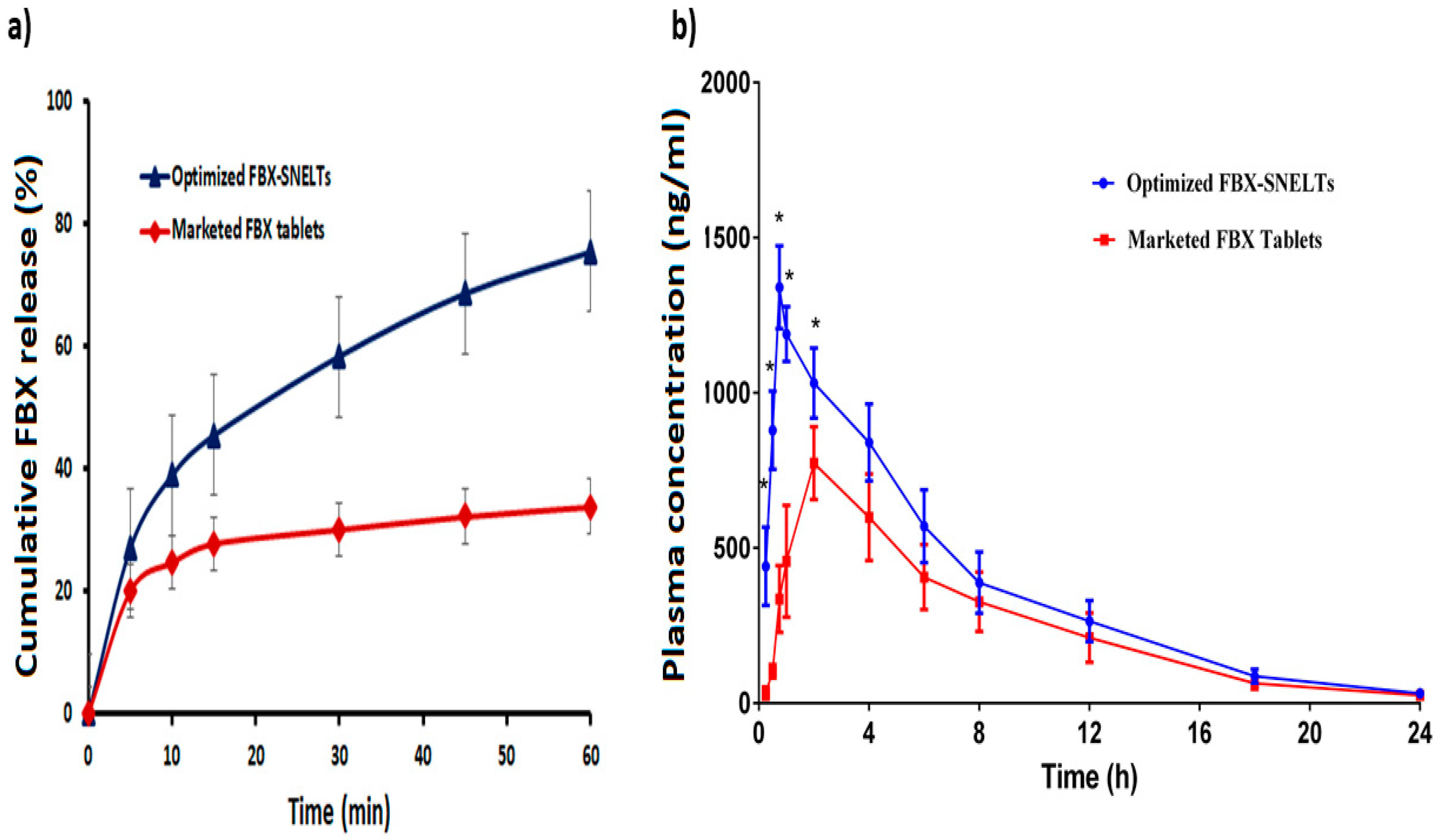
3.8. In Vitro Dissolution Study
3.9. Response Surface Methodology for Optimization of FBX-SNELTs
3.9.1. Influence of the Independent Variables on Tablet Disintegration (Y1)
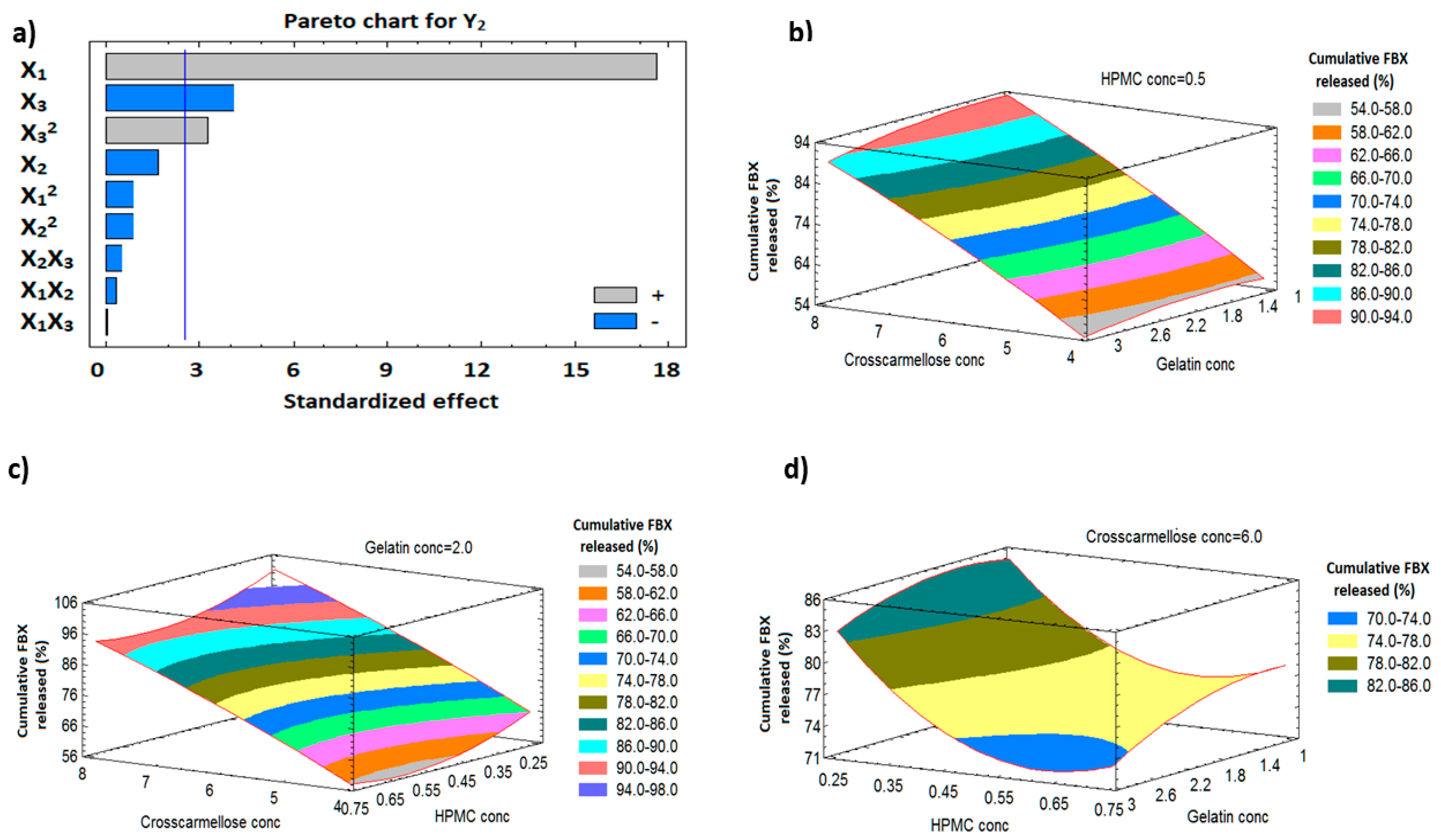
3.9.2. Influence of Independent Variables on Cumulative FBX Release (Y2)
3.9.3. Optimization
3.10. In Vivo Pharmacokinetic Study in Healthy Human Volunteers
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perez-Ruiz, F.; Dalbeth, N.; Bardin, T. A Review of Uric Acid, Crystal Deposition Disease, and Gout. Adv. Ther. 2014, 32, 31–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altan, A.; Shiozawa, A.; Bancroft, T.; Singh, J.A. A real-world study of switching from allopurinol to febuxostat in a health plan database. J. Clin. Rheumatol. 2015, 21, 411–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Yang, H.; Guo, Y.; Wei, F.; Yang, X.; Li, D.; Li, M.; Xu, W.; Li, W.; Sun, L.; et al. Comparative efficacy and safety of urate-lowering therapy for the treatment of hyperuricemia: A systematic review and network meta-analysis. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, D.; Fitzgerald, J.D.; Khanna, P.P.; Bae, S.; Singh, M.K.; Neogi, T.; Pillinger, M.H.; Merill, J.; Lee, S.; Prakash, S.; et al. American college of rheumatology guidelines for management of gout. part 1: Systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. 2012, 64, 1431–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisht, M.; Bist, S. Febuxostat: A novel agent for management of Hyperuricemia in gout. Indian J. Pharm. Sci. 2012, 73, 597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, M.A.; Schumacher, H.R.; Wortmann, R.L.; MacDonald, P.A.; Eustace, D.; Palo, W.A.; Streit, J.; Joseph-Ridge, N. Febuxostat Compared with Allopurinol in Patients with Hyperuricemia and Gout. N. Engl. J. Med. 2005, 353, 2450–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudhe, P.B.; Chavare, P.D.; Shelke, P.S. Spectrophotometric Determination of Febuxostat from Bulk and Tablet Dosage form by Area under Curve Method. Int. J. ChemTech Res. 2017, 10, 183–189. [Google Scholar]
- Kumar, K.K.; Srinivas, L.; Kishore, V.S.; Basha, S.N. Formulation and Evaluation of Poorly Soluble Febuxostat Orodispersable Tablet. AjaddCoUk 2014, 2, 191–202. [Google Scholar]
- Satalkar, P.; Elger, B.S.; Hunziker, P.; Shaw, D. Challenges of clinical translation in nanomedicine: A qualitative study. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 893–900. [Google Scholar] [CrossRef] [Green Version]
- El-Say, K.M.; El-Sawy, H.S. Polymeric nanoparticles: Promising platform for drug delivery. Int. J. Pharm. 2017, 528, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.V.R.; Yajurvedi, K.; Shao, J. Self-nanoemulsifying drug delivery system (SNEDDS) for oral delivery of protein drugs: III. In vivo oral absorption study. Int. J. Pharm. 2008, 362, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Abdelghani, G.M.; Nouh, A.T. Self-nanoemulsifying drug-delivery systems for potentiated anti-inflammatory activity of diacerein. Int. J. Nanomed. 2018, 13, 6585–6602. [Google Scholar]
- Al-Subaie, M.M.; Hosny, K.M.; El-Say, K.M.; Ahmed, T.A.; Aljaeid, B.M. Utilization of nanotechnology to enhance percutaneous absorption of acyclovir in the treatment of herpes simplex viral infections. Int. J. Nanomed. 2015, 10, 3973–3985. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, O.A.A.; Badr-Eldin, S.M.; Tawfik, M.K.; Ahmed, T.A.; El-Say, K.M.; Badr, J.M. Design and Optimization of Self-Nanoemulsifying Delivery System to Enhance Quercetin Hepatoprotective Activity in Paracetamol-Induced Hepatotoxicity. J. Pharm. Sci. 2014, 103, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, K.; Raghavan, C.V.; Selvan, N.T.; Prasad, R.H.; Abdu, S. Self nanoemulsifying drug delivery system (SNEDDS) of Rosuvastatin calcium: Design, formulation, bioavailability and pharmacokinetic evaluation. Colloids Surf. B Biointerfaces 2013, 112, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Villar, A.M.S.; Naveros, B.C.; Campmany, A.C.C.; Trenchs, M.A.; Rocabert, C.B.; Bellowa, L.H. Design and optimization of self-nanoemulsifying drug delivery systems (SNEDDS) for enhanced dissolution of gemfibrozil. Int. J. Pharm. 2012, 431, 161–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Sharma, U.S. Liposomes in drug delivery: Progress and limitations. Int. J. Pharm. 1997, 154, 123–140. [Google Scholar] [CrossRef]
- MuÈller, R.H.; MaÈder, K.; Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery - a review of the state of the art. Eur. J. Pharm. Biopharm. 2000, 50, 161–177. [Google Scholar] [CrossRef]
- Pouton, C.W. Lipid formulations for oral administration of drugs: Non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur. J. Pharm. Sci. 2000, 11, S93–S98. [Google Scholar] [CrossRef]
- Tan, A.; Rao, S.; Prestidge, C.A. Transforming lipid-based oral drug delivery systems into solid dosage forms: An overview of solid carriers, physicochemical properties, and biopharmaceutical performance. Pharm. Res. 2013, 30, 2993–3017. [Google Scholar] [CrossRef]
- Mahmoud, E.A.; Bendas, E.R.; Mohamed, M.I. Preparation and evaluation of self-nanoemulsifying tablets of carvedilol. AAPS Pharmscitech 2009, 10, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, T.A.; El-Say, K.M.; Hosny, K.M.; Aljaeid, B.M. Development of optimized self-nanoemulsifying lyophilized tablets (SNELTs) to improve finasteride clinical pharmacokinetic behavior. Drug Dev. Ind. Pharm. 2018, 44, 652–661. [Google Scholar] [CrossRef] [PubMed]
- El-Say, K.M.; Ahmed, T.A.; Ahmed, O.A.A.; Hosny, K.M.; Abd-Allah, F.I. Self-Nanoemulsifying Lyophilized Tablets for Flash Oral Transmucosal Delivery of Vitamin K: Development and Clinical Evaluation. J. Pharm. Sci. 2017, 106, 2447–2456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, Y.G.; Kim, D.W.; Yousaf, A.M.; Park, J.H.; Chang, P.S.; Baek, H.H.; Lim, S.J.; Kim, J.O.; Yong, C.S.; Choi, H.G. Solid self-nanoemulsifying drug delivery system (SNEDDS) for enhanced oral bioavailability of poorly water-soluble tacrolimus: Physicochemical characterisation and pharmacokinetics. J. Microencapsul. 2015, 32, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Inugala, S.; Eedara, B.B.; Sunkavalli, S.; Dhurke, R.; Kandadi, P.; Jukanti, R.; Bandari, S. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) of darunavir for improved dissolution and oral bioavailability: In vitro and in vivo evaluation. Eur. J. Pharm. Sci. 2015, 74, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Oh, D.H.; Oh, Y.K.; Yong, C.S.; Choi, H.G. Effects of solid carriers on the crystalline properties, dissolution and bioavailability of flurbiprofen in solid self-nanoemulsifying drug delivery system (solid SNEDDS). Eur. J. Pharm. Biopharm. 2012, 80, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Nasr, A.; Gardouh, A.; Ghorab, M. Novel solid self-nanoemulsifying drug delivery system (S-SNEDDS) for oral delivery of olmesartan medoxomil: Design, formulation, pharmacokinetic and bioavailability evaluation. Pharmaceutics 2016, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Mohd, A.B.; Sanka, K.; Bandi, S.; Diwan, P.V.; Shastri, N. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) for oral delivery of glimepiride: Development and antidiabetic activity in albino rabbits. Drug Deliv. 2015, 22, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Sastry, S.V.; Nyshadham, J.R.; Fix, J.A. Recent technological advances in oral drug delivery—A review. Pharm. Sci. Technolo. Today 2000, 3, 138–145. [Google Scholar] [CrossRef]
- El-Nesr, O.H.; Yahiya, S.A.; El-Gazayerly, O.N. Effect of formulation design and freeze-drying on properties of fluconazole multilamellar liposomes. Saudi Pharm. J. 2010, 18, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, J.M.; González, C.; Maestro, A.; Solè, I.; Pey, C.M.; Nolla, J. Nano-emulsions: New applications and optimization of their preparation. Curr. Opin. Colloid Interface Sci. 2008, 13, 245–251. [Google Scholar] [CrossRef]
- Lawrence, M.J.J.; Rees, G.D. Microemulsion-based media as novel drug delivery systems. Adv. Drug Deliv. Rev. 2000, 45, 89–121. [Google Scholar] [CrossRef]
- Khosravan, R.; Grabowski, B.A.; Wu, J.T.; Joseph-Ridge, N.; Vernillet, L. Pharmacokinetics, pharmacodynamics and safety of febuxostat, a non-purine selective inhibitor of xanthine oxidase, in a dose escalation study in healthy subjects. Clin. Pharmacokinet. 2006, 45, 821–841. [Google Scholar] [CrossRef]
- Boby, J.G. A Review on Self Emulsifying Nanoemulsion. Open Access J. Pharm. Res. 2017, 1, 1–17. [Google Scholar] [CrossRef]
- Mensah, M.B.; Awudza, J.A.M.; Brien, P.O. Castor oil: A suitable green source of capping agent for nanoparticle syntheses and facile surface functionalization. Royal Soc. Open Sci. 2018, 5, 180824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidts, T.; Dobler, D.; Nissing, C.; Runkel, F. Influence of hydrophilic surfactants on the properties of multiple W/O/W emulsions. J. Colloid Interface Sci. 2009, 338, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Chime, S.A.; Kenechukwu, F.C.; Attama, A.A. Nanoemulsions—Advances in Formulation, Characterization and Applications in Drug Delivery. In Nanotechnology and Nanomaterials “Application of Nanotechnology in Drug Delivery”; Sezer, A.D., Ed.; InTech: Rijeka, Croatia, 2014; Chapter 3; pp. 77–126. [Google Scholar]
- Dapčević Hadnadev, T.; Dokić, P.; Krstonošić, V.; Hadnadev, M. Influence of oil phase concentration on droplet size distribution and stability of oil-in-water emulsions. Eur. J. Lipid Sci. Technol. 2013, 115, 313–321. [Google Scholar] [CrossRef]
- Aboul-einien, M. Design and in-Vitro Evaluation of Olanzapine- Loaded Self Nanoemulsifying Drug Delivery System. Int. J. Ind. Pharm. Life Sci. 2012, 2, 12–32. [Google Scholar]
- Resende, K.X.; Corrêa, M.A.; Oliveira, A.G.D.; Scarpa, M.V. Effect of cosurfactant on the supramolecular structure and physicochemical properties of non-ionic biocompatible microemulsions. Revista Brasileira de Ciências Farmacêuticas 2008, 44, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Ma, X.H.; Yao, G.L.; Zhang, W.T.; Zhao, Y. Microemulsion-based anthocyanin systems: Effect of surfactants, cosurfactants, and its stability. Int. J. Food Prop. 2018, 21, 1152–1165. [Google Scholar] [CrossRef]
- Teagarden, D.L.; Baker, D.S.; Baheti, A.; Kumar, L.; Bansal, A.K. Excipients used in lyophilization of small molecules. J. Excip. Food Chem. 2010, 1, 41–54. [Google Scholar]
- Darkwah, J. Characterisation of Freeze Dried Amino Acids and Gelatin Based Rapidly Disintegrating Tablets, De Montfort University, 2011. Available online: https://www.dora.dmu.ac.uk/handle/2086/6018 (accessed on 9 June 2019).
- Zhang, X.; Cresswell, M. Silica-Based Amorphous Drug Delivery Systems. In Inorganic Controlled Release Technology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 93–137. ISBN 9780080999913. [Google Scholar]
- Hesari, Z.; Shafiee, A.; Hooshfar, S.; Mobarra, N.; Mortazavic, S.A. Formulation and taste masking of ranitidine orally disintegrating tablet. Iran. J. Pharm. Res. 2016, 15, 677–686. [Google Scholar] [PubMed]
- Marais, A.F.; Song, M.; de Villiers, M.M. Effect of compression force, humidity and disintegrant concentration on the disintegration and dissolution of directly compressed furosemide tablets using croscarmellose sodium as disintegrant. Trop. J. Pharm. Res. 2003, 2, 125–135. [Google Scholar]
- AlHusban, F.; Perrie, Y.; Mohammed, A. Preparation, Optimisation and Characterisation of Lyophilised Rapid Disintegrating Tablets Based on Gelatin and Saccharide. Curr. Drug Deliv. 2010, 7, 65–75. [Google Scholar] [CrossRef]
- Dave, V.; Yadav, R.B.; Ahuja, R.; Yadav, S. Formulation design and optimization of novel fast dissolving tablet of chlorpheniramine maleate by using lyophilization techniques. Bull. Fac. Pharmacy Cairo Univ. 2017, 55, 31–39. [Google Scholar] [CrossRef]
- Liew, K.B.; Peh, K.K. Investigation on the effect of polymer and starch on the tablet properties of lyophilized orally disintegrating tablet. Arch. Pharm. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Aboul-Einien, M. In vitro and in vivo evaluation of a fast-disintegrating lyophilized dry emulsion tablet containing griseofulvin. Eur. J. Pharm. Sci. 2007, 32, 58–68. [Google Scholar] [CrossRef]
- Elkordy, A.A.; Tan, X.N.; Essa, E.A. Spironolactone release from liquisolid formulations prepared with CapryolTM 90, Solutol® HS-15 and Kollicoat® SR 30 D as non-volatile liquid vehicles. Eur. J. Pharm. Biopharm. 2013, 83, 203–223. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.M.; Er, P.X.H.; Liew, C.V.; Heng, P.W.S. Functionality of Disintegrants and Their Mixtures in Enabling Fast Disintegration of Tablets by a Quality by Design Approach. AAPS PharmSciTech 2014, 15, 1093–1104. [Google Scholar] [CrossRef] [Green Version]
- Audu-peter, J.D.; Ibrahim, M.A. Interactions of binder, disintegrant and compression pressure in tablets ii: Effect of the differences in their levels on friability, hardness and disintegration time. J. Pharm. Allied Sci. 2016, 11, 2133–2141. [Google Scholar]
- Widjaja, B.; Setyawan, D.; Moechtar, J. Development of piroxicam orally disintegrating tablets by freeze drying method. Int. J. Pharm. Pharm. Sci. 2013, 5, 795–798. [Google Scholar]
- Tanuwijaya, J. Karsono the effects of crospovidone and croscarmellose sodium as superdisintegrants on the characteristics of piroxicam nanoparticles ODT (orally disintegrating tablet). Int. J. PharmTech Res. 2013, 5, 1590–1597. [Google Scholar]
- Nazmi, M.; Ashraful Islam, S.M.; Bhuiyan, M.A.; Reza, M.S. Effect of superdisintegrants and their mode of incorporation on disintegration time and release profile of carbamazepine from immediate release tablet. J. Appl. Pharm. Sci. 2013, 3, 80–84. [Google Scholar] [CrossRef]
- Arzani, G.; Haeri, A.; Daeihamed, M.; Bakhtiari-Kaboutaraki, H.; Dadashzadeh, S. Niosomal carriers enhance oral bioavailability of carvedilol: Effects of bile salt-enriched vesicles and carrier surface charge. Int. J. Nanomed. 2015, 10, 4797–4813. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Level | |
|---|---|---|
| Low | High | |
| Oil percentage (X1) | 10 | 15 |
| Surfactant percentage (X2) | 40 | 60 |
| Co-surfactant percentage (X3) | 30 | 50 |
| Formula Code | Mixture Components | Dependent Responses | |||
|---|---|---|---|---|---|
| X1 (%) | X2 (%) | X3 (%) | Y1 (nm) | Y2 (%) | |
| NE-1 | 10 | 60 | 30 | 202.2 | 61 |
| NE-2 | 10 | 40 | 50 | 175.7 | 91 |
| NE-3 | 15 | 55 | 30 | 355.7 | 59 |
| NE-4 | 15 | 40 | 45 | 452.8 | 85 |
| NE-5 | 11.25 | 54.375 | 34.375 | 210.7 | 72 |
| NE-6 | 11.25 | 44.375 | 44.375 | 288.7 | 80 |
| NE-7 | 13.75 | 51.875 | 34.375 | 366.9 | 70 |
| NE-8 | 13.75 | 44.375 | 41.875 | 389.3 | 78 |
| NE-9 | 10 | 50 | 40 | 256.3 | 75 |
| NE-10 | 12.5 | 57.5 | 30 | 232.7 | 63 |
| NE-11 | 12.5 | 40 | 47.5 | 347.5 | 89 |
| NE-12 | 15 | 47.5 | 37.5 | 401.3 | 69 |
| NE-13 | 12.5 | 48.75 | 38.75 | 328.9 | 73 |
| NE-14 | 10 | 60 | 30 | 197.5 | 65 |
| Formula Code | X1 | X2 | X3 |
|---|---|---|---|
| SNELT-1 | 6.0 | 1.0 | 0.75 |
| SNELT-2 | 6.0 | 2.0 | 0.5 |
| SNELT-3 | 8.0 | 1.0 | 0.5 |
| SNELT-4 | 8.0 | 2.0 | 0.75 |
| SNELT-5 | 4.0 | 1.0 | 0.5 |
| SNELT-6 | 6.0 | 2.0 | 0.5 |
| SNELT-7 | 6.0 | 1.0 | 0.25 |
| SNELT-8 | 8.0 | 3.0 | 0.5 |
| SNELT-9 | 4.0 | 2.0 | 0.75 |
| SNELT-10 | 4.0 | 2.0 | 0.25 |
| SNELT-11 | 6.0 | 3.0 | 0.25 |
| SNELT-12 | 8.0 | 2.0 | 0.25 |
| SNELT-13 | 6.0 | 3.0 | 0.75 |
| SNELT-14 | 6.0 | 2.0 | 0.5 |
| SNELT-15 | 4.0 | 3.0 | 0.5 |
| Factors | Disintegration Time (Y1), min | Cumulative Release after 60 min (Y2), % | ||||
|---|---|---|---|---|---|---|
| Estimate | F-Ratio | p-Value | Estimate | F-Ratio | p-Value | |
| X1 | −190.0 | 2166.00 | 0.0001 * | 35.83 | 311.63 | 0.0001 * |
| X2 | 35.0 | 73.50 | 0.0004 * | −3.58 | 3.10 | 0.1384 |
| X3 | 65.0 | 253.50 | 0.0001 * | −8.5 | 17.54 | 0.0086 * |
| X1X1 | 50.83 | 71.56 | 0.0004 * | −2.83 | 0.90 | 0.3864 |
| X1X2 | 5.0 | 0.75 | 0.4261 | −0.9 | 0.10 | 0.7665 |
| X1X3 | −55.0 | 90.75 | 0.0002 * | 0.05 | 0.00 | 0.9868 |
| X2X2 | 10.83 | 3.25 | 0.1313 | −2.83 | 0.90 | 0.3864 |
| X2X3 | 15.0 | 6.75 | 0.0484 * | −1.35 | 0.22 | 0.6579 |
| X3X3 | 10.83 | 3.25 | 0.1313 | 9.72 | 10.58 | 0.0226 * |
| R2 | 99.81 | 98.58 | ||||
| Adj. R2 | 99.48 | 96.01 | ||||
| SEE | 5.77 | 2.87 | ||||
| MAE | 2.44 | 1.23 | ||||
| PK Parameters | Optimized FBX-SNELTs | Marketed FBX Tablets |
|---|---|---|
| Cmax (ng/mL) | 1340.0 ± 134.0 | 773.5 ± 117.6 |
| Tmax (min) | 45.0 ± 0.0 | 120.0 ± 0.0 |
| t1/2 (h) | 4.0 ± 0.27 | 4.28 ± 0.50 |
| AUC0–t (ng/mL h) | 8885.9 ± 1578.3 | 6069.9 ± 1640.0 |
| AUC0–inf (ng/mL h) | 9068.6 ± 1590.0 | 6230.7 ± 1715.7 |
| AUMC0–inf (ng/mL h2) | 60,175.0 ± 12,212.0 | 46,481.8 ± 15,071.3 |
| Kel (h−1) | 0.173 ± 0.01 | 0.165 ± 0.02 |
| MRT (h) | 6.61 ± 0.19 | 7.39 ± 0.39 |
| Relative BA (%) | 146.4 | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Amodi, Y.A.; Hosny, K.M.; Alharbi, W.S.; Safo, M.K.; El-Say, K.M. Investigating the Potential of Transmucosal Delivery of Febuxostat from Oral Lyophilized Tablets Loaded with a Self-Nanoemulsifying Delivery System. Pharmaceutics 2020, 12, 534. https://doi.org/10.3390/pharmaceutics12060534
Al-Amodi YA, Hosny KM, Alharbi WS, Safo MK, El-Say KM. Investigating the Potential of Transmucosal Delivery of Febuxostat from Oral Lyophilized Tablets Loaded with a Self-Nanoemulsifying Delivery System. Pharmaceutics. 2020; 12(6):534. https://doi.org/10.3390/pharmaceutics12060534
Chicago/Turabian StyleAl-Amodi, Yasir A., Khaled M Hosny, Waleed S. Alharbi, Martin K. Safo, and Khalid M El-Say. 2020. "Investigating the Potential of Transmucosal Delivery of Febuxostat from Oral Lyophilized Tablets Loaded with a Self-Nanoemulsifying Delivery System" Pharmaceutics 12, no. 6: 534. https://doi.org/10.3390/pharmaceutics12060534
APA StyleAl-Amodi, Y. A., Hosny, K. M., Alharbi, W. S., Safo, M. K., & El-Say, K. M. (2020). Investigating the Potential of Transmucosal Delivery of Febuxostat from Oral Lyophilized Tablets Loaded with a Self-Nanoemulsifying Delivery System. Pharmaceutics, 12(6), 534. https://doi.org/10.3390/pharmaceutics12060534