Avoiding the Pitfalls of siRNA Delivery to the Retinal Pigment Epithelium with Physiologically Relevant Cell Models

 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. siRNA Delivery Systems
2.2.1. Polyplexes
2.2.2. Lipoplexes
2.2.3. Lipidoid-siRNA Complexes
2.2.4. Commercial Carriers
2.3. Dynamic Light Scattering (DLS) and Nanoparticle Tracking Analysis (NTA)
2.4. Retinal Pigment Epithelium (RPE) Cell Cultures
2.4.1. Human Retinal Pigment Epithelial Cells (ARPE-19)
2.4.2. Primary Porcine RPE (pRPE)
2.4.3. Human Embryonic Stem Cell-Derived RPE (hESC-RPE)
2.5. Transepithelial Electrical Resistance Measurement (TEER)
2.6. Cytotoxicity
2.7. Gene Silencing
2.7.1. Interleukin 6 (IL-6) Protein Knockdown
2.7.2. Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) and Hypoxanthine Phosphoribosyltransferase 1 (HPRT1) MRNA Knockdown
2.8. Cell Uptake
2.9. Intracellular Distribution
2.10. Statistical Analysis
3. Results
3.1. Particle Size and Polydispersity Index (PdI)
3.2. Cytotoxicity
3.3. Knockdown Efficacy in RPE Cell Models
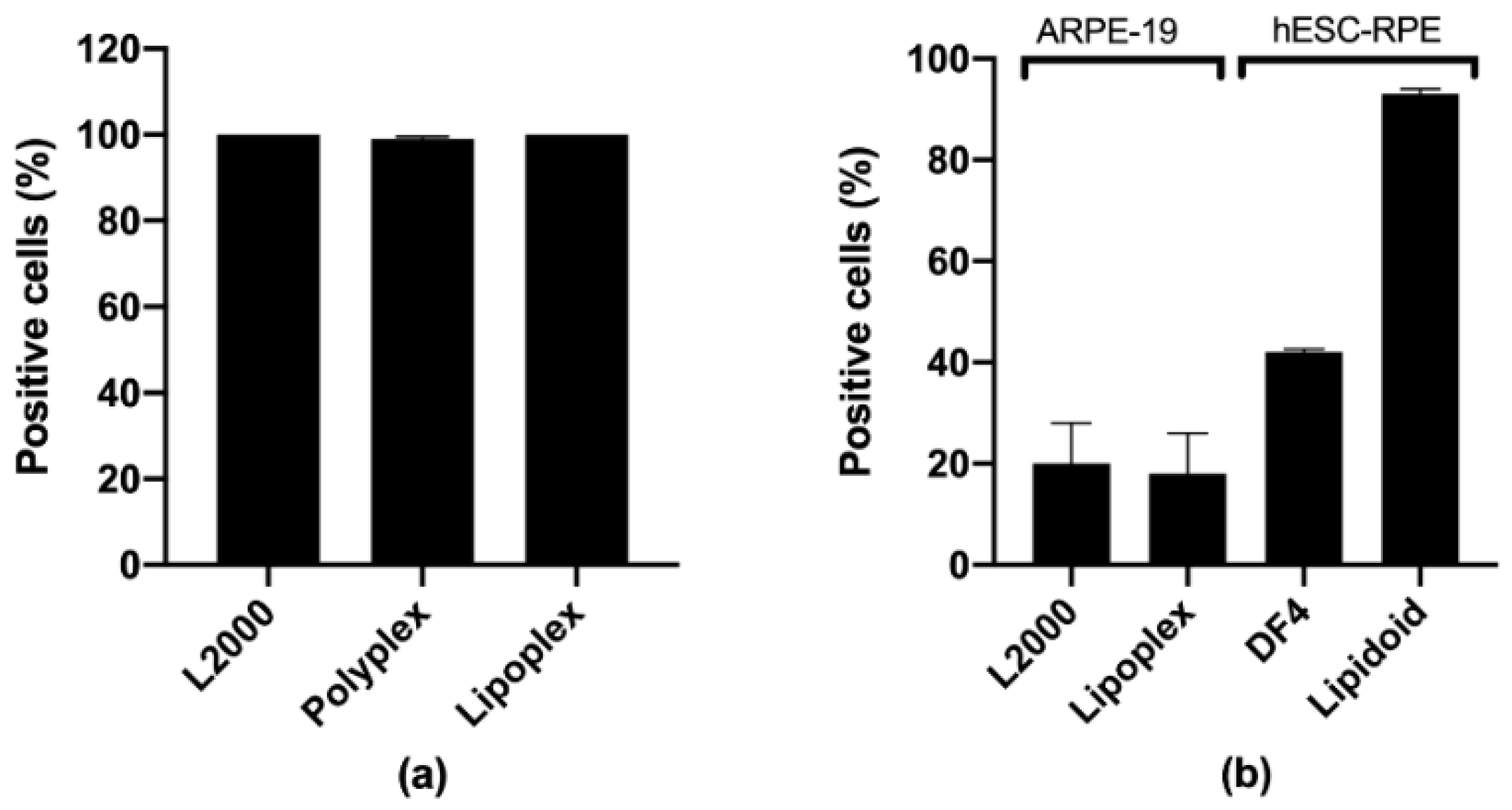
3.3.1. Human ARPE-19
3.3.2. Primary Porcine RPE (pRPE)
3.3.3. Human Embryonic Stem Cell-derived RPE (hESC-RPE)
3.4. Cell Uptake
3.5. Intracellular Distribution in hESC-RPE
3.5.1. 2-Week Matured hESC-RPE
3.5.2. 4-Week Matured hESC-RPE
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Penfold, P.L.; Killingsworth, M.C.; Sarks, S.H. An ultrastructural study of the role of leucocytes and fibroblasts in the breakdown of bruch’s membrane. Aust. J. Ophthalmol. 1984, 12, 23–31. [Google Scholar] [PubMed]
- Penfold, P.L.; Killingsworth, M.C.; Sarks, S.H. Senile macular degeneration: The involvement of immunocompetent cells. Graefes Arch. Clin. Exp. Ophthalmol. 1985, 223, 69–76. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Hollyfield, J.G.; Bonilha, V.L.; Rayborn, M.E.; Yang, X.; Shadrach, K.G.; Lu, L.; Ufret, R.L.; Salomon, R.G.; Perez, V.L. Oxidative damage—Induced inflammation initiates age-related macular degeneration. Nat. Med. 2008, 14, 194–198. [Google Scholar] [CrossRef]
- Perez, V.L.; Caspi, R.R. Immune mechanisms in inflammatory and degenerative eye disease. Trends Immunol. 2015, 36, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell. Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef] [Green Version]
- Copland, D.A.; Theodoropoulou, S.; Liu, J.; Dick, A.D. A perspective of AMD through the eyes of immunology. Invest. Ophthalmol. Vis. Sci. 2018, 59, AMD83–AMD92. [Google Scholar] [CrossRef] [Green Version]
- Joussen, A.M.; Poulaki, V.; Le, M.L.; Koizumi, K.; Esser, C.; Janicki, H.; Schraermeyer, U.; Kociok, N.; Fauser, S.; Kirchhof, B.; et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004, 18, 1450–1452. [Google Scholar] [CrossRef]
- Funatsu, H.; Yamashita, H.; Noma, H.; Mimura, T.; Nakamura, S.; Sakata, K.; Hori, S. Aqueous humor levels of cytokines are related to vitreous levels and progression of diabetic retinopathy in diabetic patients. Graefes Arch. Clin. Exp. Ophthalmol. 2005, 243, 3–8. [Google Scholar] [CrossRef]
- Tang, J.; Kern, T.S. Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef] [Green Version]
- Rübsam, A.; Parikh, S.; Fort, P.E. Role of inflammation in diabetic retinopathy. Int. J. Mol. Sci. 2018, 19, 942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vohra, R.; Tsai, J.C.; Kolko, M. The role of inflammation in the pathogenesis of glaucoma. Surv. Ophthalmol. 2013, 58, 311–320. [Google Scholar] [CrossRef] [PubMed]
- del Amo, E.M.; Rimpelä, A.-K.; Heikkinen, E.; Kari, O.K.; Ramsay, E.; Lajunen, T.; Schmitt, M.; Pelkonen, L.; Bhattacharya, M.; Richardson, D.; et al. Pharmacokinetic aspects of retinal drug delivery. Prog. Retin. Eye Res. 2017, 57, 134–185. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.F.; Wolfensberger, T.J. The Retinal Pigment Epithelium: Function and Disease; Oxford University Press: New York, NY, USA, 1998; ISBN 978-0-19-510956-6. [Google Scholar]
- Xu, H.; Chen, M.; Forrester, J.V. Para-inflammation in the aging retina. Prog. Retin. Eye Res. 2009, 28, 348–368. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar] [CrossRef]
- Guzman-Aranguez, A.; Loma, P.; Pintor, J. Small-interfering RNAs (siRNAs) as a promising tool for ocular therapy. Br. J. Pharmacol. 2013, 170, 730–747. [Google Scholar] [CrossRef] [Green Version]
- Setten, R.L.; Rossi, J.J.; Han, S. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- Hornung, V.; Guenthner-Biller, M.; Bourquin, C.; Ablasser, A.; Schlee, M.; Uematsu, S.; Noronha, A.; Manoharan, M.; Akira, S.; de Fougerolles, A.; et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 2005, 11, 263–270. [Google Scholar] [CrossRef]
- Caillaud, M.; El Madani, M.; Massaad-Massade, L. Small interfering RNA from the lab discovery to patients’ recovery. J. Control. Release 2020, 321, 616–628. [Google Scholar] [CrossRef]
- Chen, C.-W.; Yeh, M.-K.; Shiau, C.-Y.; Chiang, C.-H.; Lu, D.-W. Efficient downregulation of VEGF in retinal pigment epithelial cells by integrin ligand-labeled liposome-mediated siRNA delivery. Int. J. Nanomed. 2013, 8, 2613–2627. [Google Scholar]
- Liu, X.; Jann, J.; Xavier, C.; Wu, H. Glutaredoxin 1 (Grx1) protects human retinal pigment epithelial cells from oxidative damage by preventing AKT glutathionylation. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2821–2832. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, Q.; Tang, M.; Zhang, J.; Fan, W. Complement factor H expressed by retinal pigment epithelium cells can suppress neovascularization of human umbilical vein endothelial cells: An in vitro study. PLoS ONE 2015, 10, e0129945. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Yang, W.; Gao, Z.; Ruan, X.; Zhang, Y. Up-regulation of P-gp via NF-κB activation confers protection against oxidative damage in the retinal pigment epithelium cells. Exp. Eye Res. 2019, 181, 367–373. [Google Scholar] [CrossRef]
- Osipova, O.; Sharoyko, V.; Zashikhina, N.; Zakharova, N.; Tennikova, T.; Urtti, A.; Korzhikova-Vlakh, E. Amphiphilic polypeptides for VEGF siRNA delivery into retinal epithelial cells. Pharmaceutics 2020, 12, 39. [Google Scholar] [CrossRef] [Green Version]
- Hellinen, L.; Hongisto, H.; Ramsay, E.; Kaarniranta, K.; Vellonen, K.-S.; Skottman, H.; Ruponen, M. Drug flux across RPE cell models: The hunt for an appropriate outer blood–retinal barrier model for use in early drug discovery. Pharmaceutics 2020, 12, 176. [Google Scholar] [CrossRef] [Green Version]
- Subia, B.; Reinisalo, M.; Dey, N.; Tavakoli, S.; Subrizi, A.; Ganguli, M.; Ruponen, M. Nucleic acid delivery to differentiated retinal pigment epithelial cells using cell-penetrating peptide as a carrier. Eur. J. Pharm. Biopharm. 2019, 140, 91–99. [Google Scholar] [CrossRef]
- Fasbender, A.; Marshall, J.; Moninger, T.O.; Grunst, T.; Cheng, S.; Welsh, M.J. Effect of co-lipids in enhancing cationic lipid-mediated gene transfer in vitro and in vivo. Gene Ther. 1997, 4, 716–725. [Google Scholar] [CrossRef]
- Uduehi, A.N.; Moss, S.H.; Nuttall, J.; Pouton, C.W. Cationic lipid-mediated transfection of differentiated Caco-2 cells: A filter culture model of gene delivery to a polarized epithelium. Pharm. Res. 1999, 16, 1805–1811. [Google Scholar] [CrossRef]
- Toropainen, E.; Hornof, M.; Kaarniranta, K.; Johansson, P.; Urtti, A. Corneal epithelium as a platform for secretion of transgene products after transfection with liposomal gene eyedrops. J. Gene Med. 2007, 9, 208–216. [Google Scholar] [CrossRef]
- Stukenkemper, T.; Dose, A.; Gonzalez, M.C.; Groenen, A.J.J.; Hehir, S.; Andrés-Guerrero, V.; Vanrell, R.H.; Cameron, N.R. Block copolypeptide nanoparticles for the delivery of ocular therapeutics. Macromol. Biosci. 2015, 15, 138–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruponen, M.; Ylä-Herttuala, S.; Urtti, A. Interactions of polymeric and liposomal gene delivery systems with extracellular glycosaminoglycans: Physicochemical and transfection studies. Biochim. Biophys. Acta BBA–Biomembr. 1999, 1415, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Mannermaa, E.; Rönkkö, S.; Ruponen, M.; Reinisalo, M.; Urtti, A. Long-lasting secretion of transgene product from differentiated and filter-grown retinal pigment epithelial cells after nonviral gene transfer. Curr. Eye Res. 2005, 30, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Love, K.T.; Mahon, K.P.; Levins, C.G.; Whitehead, K.A.; Querbes, W.; Dorkin, J.R.; Qin, J.; Cantley, W.; Qin, L.L.; Racie, T.; et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. USA 2010, 107, 1864–1869. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Yang, C.; Thomsen, J.S.; Dagnæs-Hansen, F.; Jakobsen, M.; Brüel, A.; Deleuran, B.; Kjems, J. Lipidoid-siRNA nanoparticle-mediated IL-1β gene silencing for systemic arthritis therapy in a mouse model. Mol. Ther. 2019, 27, 1424–1435. [Google Scholar] [CrossRef]
- Skottman, H. Derivation and characterization of three new human embryonic stem cell lines in Finland. In Vitro Cell. Dev. Biol. Anim. 2010, 46, 206–209. [Google Scholar] [CrossRef]
- Subrizi, A.; Hiidenmaa, H.; Ilmarinen, T.; Nymark, S.; Dubruel, P.; Uusitalo, H.; Yliperttula, M.; Urtti, A.; Skottman, H. Generation of hESC-derived retinal pigment epithelium on biopolymer coated polyimide membranes. Biomaterials 2012, 33, 8047–8054. [Google Scholar] [CrossRef]
- Vaajasaari, H.; Ilmarinen, T.; Juuti-Uusitalo, K.; Rajala, K.; Onnela, N.; Narkilahti, S.; Suuronen, R.; Hyttinen, J.; Uusitalo, H.; Skottman, H. Toward the defined and xeno-free differentiation of functional human pluripotent stem cell-derived retinal pigment epithelial cells. Mol. Vis. 2011, 17, 558–575. [Google Scholar]
- Leung, K.W.; Barnstable, C.J.; Tombran-Tink, J. Bacterial endotoxin activates retinal pigment epithelial cells and induces their degeneration through IL-6 and IL-8 autocrine signaling. Mol. Immunol. 2009, 46, 1374–1386. [Google Scholar] [CrossRef]
- Fraley, R.; Straubinger, R.M.; Rule, G.; Springer, E.L.; Papahadjopoulos, D. Liposome-mediated delivery of deoxyribonucleic acid to cells: Enhanced efficiency of delivery by changes in lipid composition and incubation conditions. Biochemistry 1981, 20, 6978–6987. [Google Scholar] [CrossRef]
- Hinze, C.; Boucrot, E. Endocytosis in proliferating, quiescent and terminally differentiated cells. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cryan, S.-A.; O’ Driscoll, C.M. Mechanistic studies on nonviral gene delivery to the intestine using in vitro differentiated cell culture models and an in vivo rat intestinal loop. Pharm. Res. 2003, 20, 569–575. [Google Scholar] [CrossRef] [PubMed]
- O’ Neill, M.J.; Guo, J.; Byrne, C.; Darcy, R.; O’ Driscoll, C.M. Mechanistic studies on the uptake and intracellular trafficking of novel cyclodextrin transfection complexes by intestinal epithelial cells. Int. J. Pharm. 2011, 413, 174–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilina, P.; Partti, S.; Niklander, J.; Ruponen, M.; Lou, Y.-R.; Yliperttula, M. Effect of differentiation on endocytic profiles of endothelial and epithelial cell culture models. Exp. Cell Res. 2015, 332, 89–101. [Google Scholar] [CrossRef]
- Foerg, C.; Ziegler, U.; Fernandez-Carneado, J.; Giralt, E.; Merkle, H.P. Differentiation restricted endocytosis of cell penetrating peptides in MDCK cells corresponds with activities of Rho-GTPases. Pharm. Res. 2007, 24, 628–642. [Google Scholar] [CrossRef] [Green Version]
- Matsui, H.; Johnson, L.G.; Randell, S.H.; Boucher, R.C. Loss of binding and entry of liposome-DNA complexes decreases transfection efficiency in differentiated airway epithelial cells. J. Biol. Chem. 1997, 272, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Boulton, M.E. Studying melanin and lipofuscin in RPE cell culture models. Exp. Eye Res. 2014, 126, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Verrey, F. Dégénérescence pigmentaire de la rétine d’origine médicamenteuse. Ophthalmologica 1956, 131, 296–303. (In French) [Google Scholar] [CrossRef]
- Goar, E.L.; Fletcher, M.C. Toxic chorioretinopathy following the use of NP 207. Am. J. Ophthalmol. 1957, 44, 603–608. [Google Scholar] [CrossRef]
- Hobbs, H.E.; Sorsby, A.; Freedman, A. Retinopathy following chloroquine therapy. Lancet Lond. Engl. 1959, 2, 478–480. [Google Scholar] [CrossRef]
- Jakubiak, P.; Reutlinger, M.; Mattei, P.; Schuler, F.; Urtti, A.; Alvarez-Sánchez, R. Understanding molecular drivers of melanin binding to support rational design of small molecule ophthalmic drugs. J. Med. Chem. 2018, 61, 10106–10115. [Google Scholar] [CrossRef]
- Rimpelä, A.-K.; Reinisalo, M.; Hellinen, L.; Grazhdankin, E.; Kidron, H.; Urtti, A.; del Amo, E.M. Implications of melanin binding in ocular drug delivery. Adv. Drug Deliv. Rev. 2018, 126, 23–43. [Google Scholar] [CrossRef]
- Pitkänen, L.; Ranta, V.-P.; Moilanen, H.; Urtti, A. Binding of betaxolol, metoprolol and oligonucleotides to synthetic and bovine ocular melanin, and prediction of drug binding to melanin in human choroid-retinal pigment epithelium. Pharm. Res. 2007, 24, 2063–2070. [Google Scholar] [CrossRef]
- Geng, J.; Yuan, P.; Shao, C.; Yu, S.-B.; Zhou, B.; Zhou, P.; Chen, X.-D. Bacterial melanin interacts with double-stranded DNA with high affinity and may inhibit cell metabolism in vivo. Arch. Microbiol. 2010, 192, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Schraermeyer, U.; Stieve, H. A newly discovered pathway of melanin formation in cultured retinal pigment epithelium of cattle. Cell Tissue Res. 1994, 276, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Schraermeyer, U.; Peters, S.; Thumann, G.; Kociok, N.; Heimann, K. Melanin granules of retinal pigment epithelium are connected with the lysosomal degradation pathway. Exp. Eye Res. 1999, 68, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Delevoye, C.; Marks, M.S.; Raposo, G. Lysosome-related organelles as functional adaptations of the endolysosomal system. Curr. Opin. Cell Biol. 2019, 59, 147–158. [Google Scholar] [CrossRef]
- Lu, J.J.; Langer, R.; Chen, J. A novel mechanism is involved in cationic lipid-mediated functional siRNA delivery. Mol. Pharm. 2009, 6, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Zelphati, O.; Szoka, F.C. Mechanism of oligonucleotide release from cationic liposomes. Proc. Natl. Acad. Sci. USA 1996, 93, 11493–11498. [Google Scholar] [CrossRef] [Green Version]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stöter, M.; et al. Image-based analysis of lipid nanoparticle–mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carrier | Size, DLS (nm) | PdI | Size, NTA (nm) |
|---|---|---|---|
| Polyplex | 46.4 ± 14 | 0.20 | n/a |
| Lipoplex | 216 ± 27 | 0.40 ± 0.05 | n/a |
| Lipidoid | 166 ± 18 | 0.21 ± 0.03 | 191 ± 62 |
| DharmaFect 4 | n/a | n/a | 281 ± 160 |
| Metafectene PRO | n/a | n/a | 160 ± 88 |
| Lipofectamine 2000 | n/a | n/a | n/a |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramsay, E.; Raviña, M.; Sarkhel, S.; Hehir, S.; Cameron, N.R.; Ilmarinen, T.; Skottman, H.; Kjems, J.; Urtti, A.; Ruponen, M.; et al. Avoiding the Pitfalls of siRNA Delivery to the Retinal Pigment Epithelium with Physiologically Relevant Cell Models. Pharmaceutics 2020, 12, 667. https://doi.org/10.3390/pharmaceutics12070667
Ramsay E, Raviña M, Sarkhel S, Hehir S, Cameron NR, Ilmarinen T, Skottman H, Kjems J, Urtti A, Ruponen M, et al. Avoiding the Pitfalls of siRNA Delivery to the Retinal Pigment Epithelium with Physiologically Relevant Cell Models. Pharmaceutics. 2020; 12(7):667. https://doi.org/10.3390/pharmaceutics12070667
Chicago/Turabian StyleRamsay, Eva, Manuela Raviña, Sanjay Sarkhel, Sarah Hehir, Neil R. Cameron, Tanja Ilmarinen, Heli Skottman, Jørgen Kjems, Arto Urtti, Marika Ruponen, and et al. 2020. "Avoiding the Pitfalls of siRNA Delivery to the Retinal Pigment Epithelium with Physiologically Relevant Cell Models" Pharmaceutics 12, no. 7: 667. https://doi.org/10.3390/pharmaceutics12070667
APA StyleRamsay, E., Raviña, M., Sarkhel, S., Hehir, S., Cameron, N. R., Ilmarinen, T., Skottman, H., Kjems, J., Urtti, A., Ruponen, M., & Subrizi, A. (2020). Avoiding the Pitfalls of siRNA Delivery to the Retinal Pigment Epithelium with Physiologically Relevant Cell Models. Pharmaceutics, 12(7), 667. https://doi.org/10.3390/pharmaceutics12070667