NLRP3 Inflammasome and Allergic Contact Dermatitis: A Connection to Demystify

 , ,
, ,  , ,
, ,  and
and 
Abstract
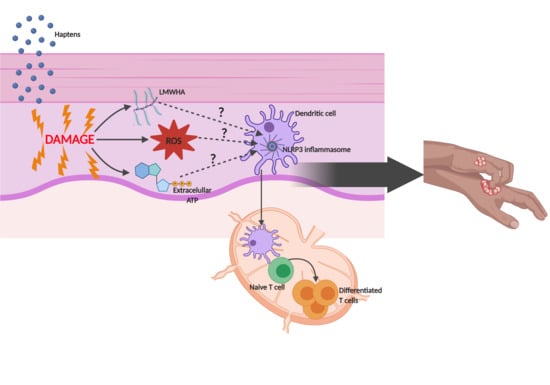
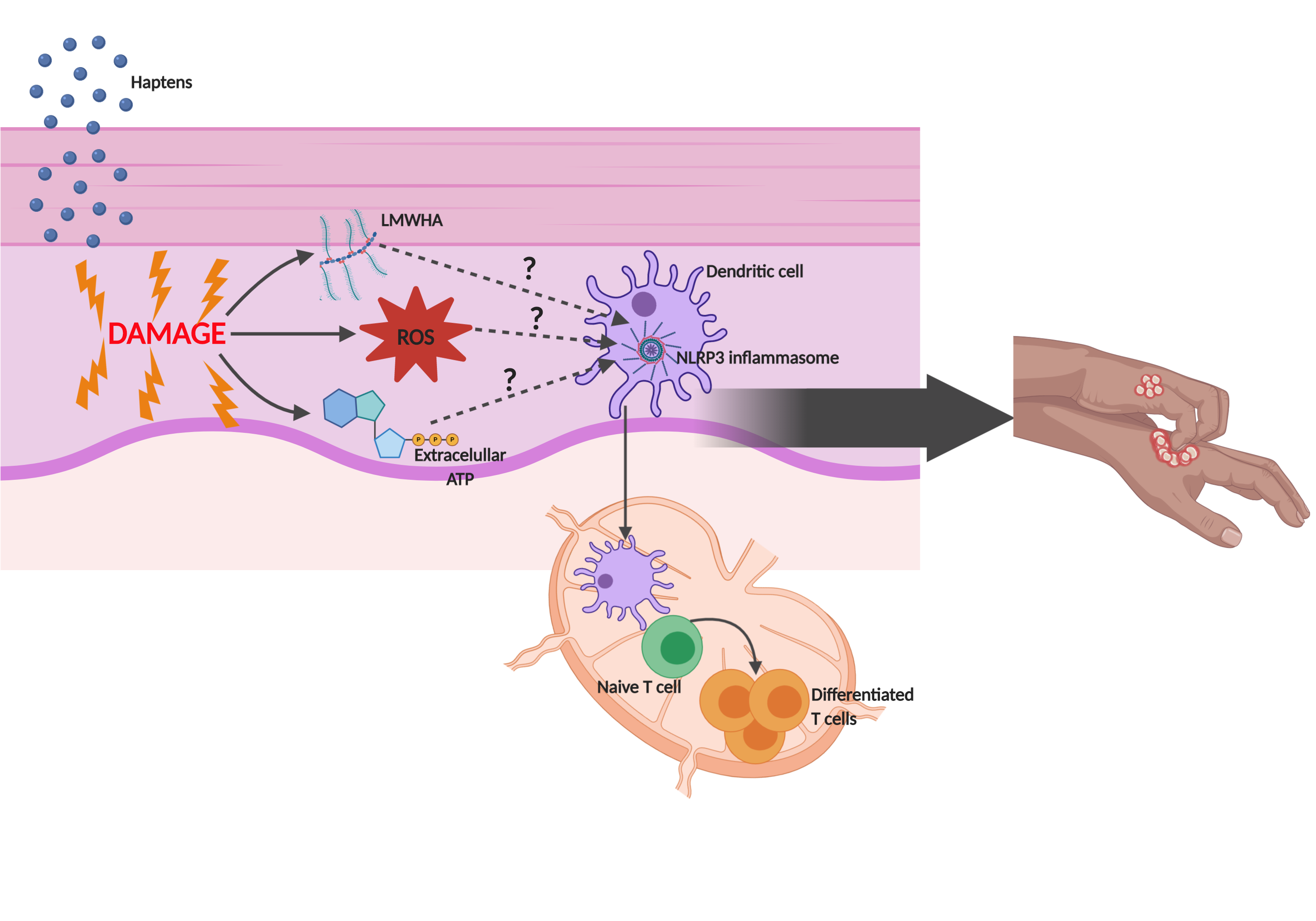
:
1. Introduction
2. Skin Sensitizers and Danger Signals
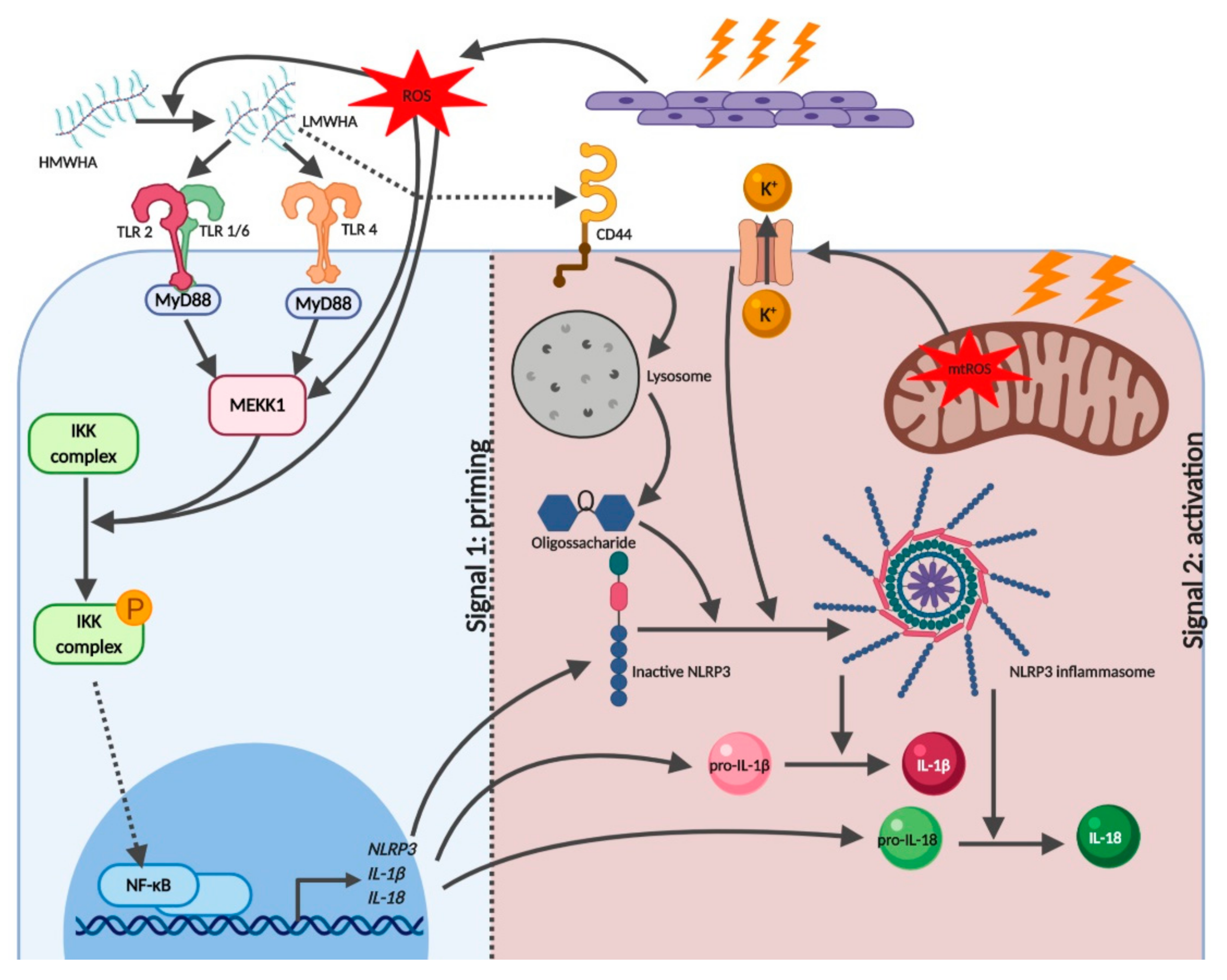
2.1. Reactive Oxygen Species
2.2. Low Molecular Weight Hyaluronic Acid
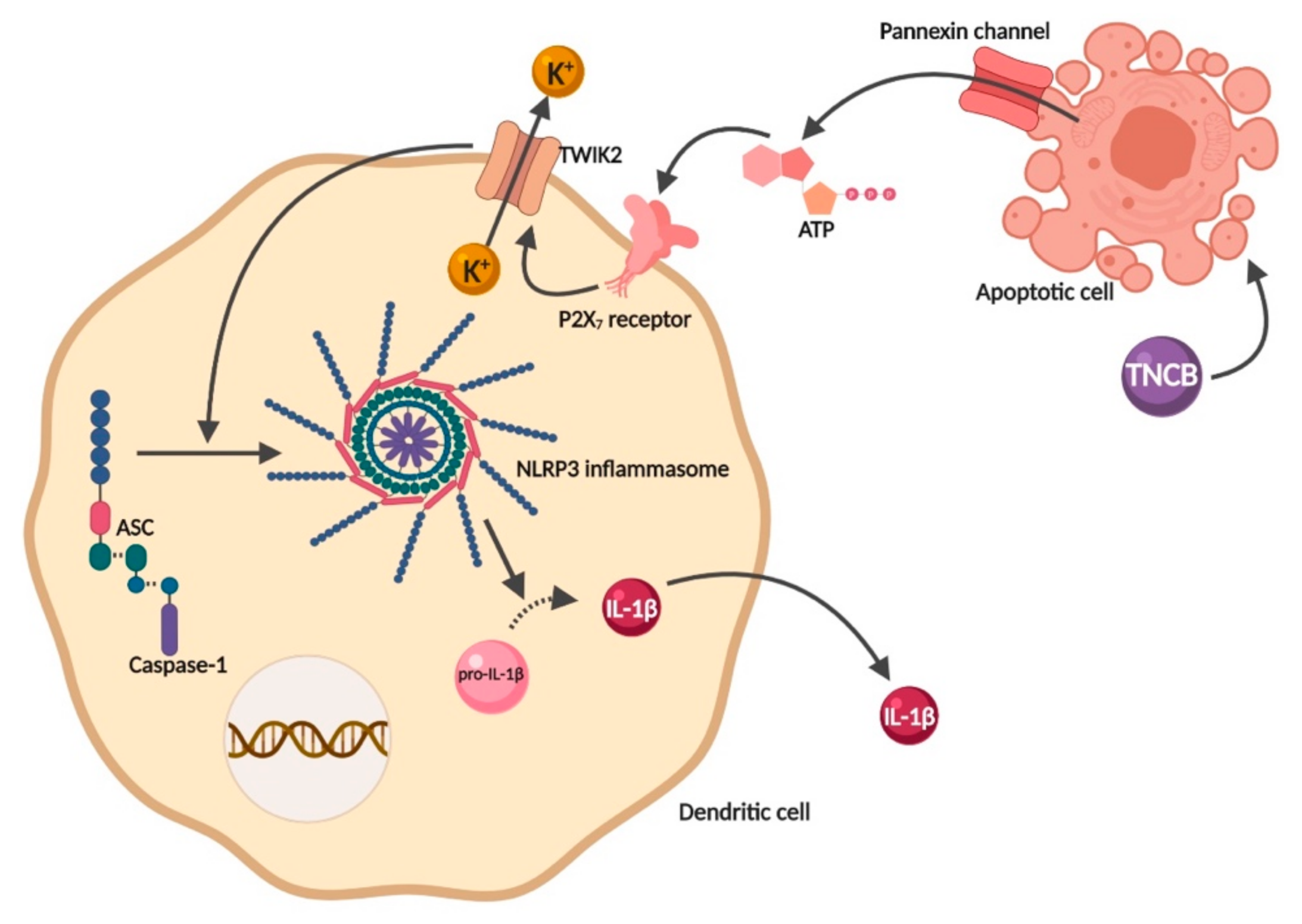
2.3. Adenosine Triphosphate
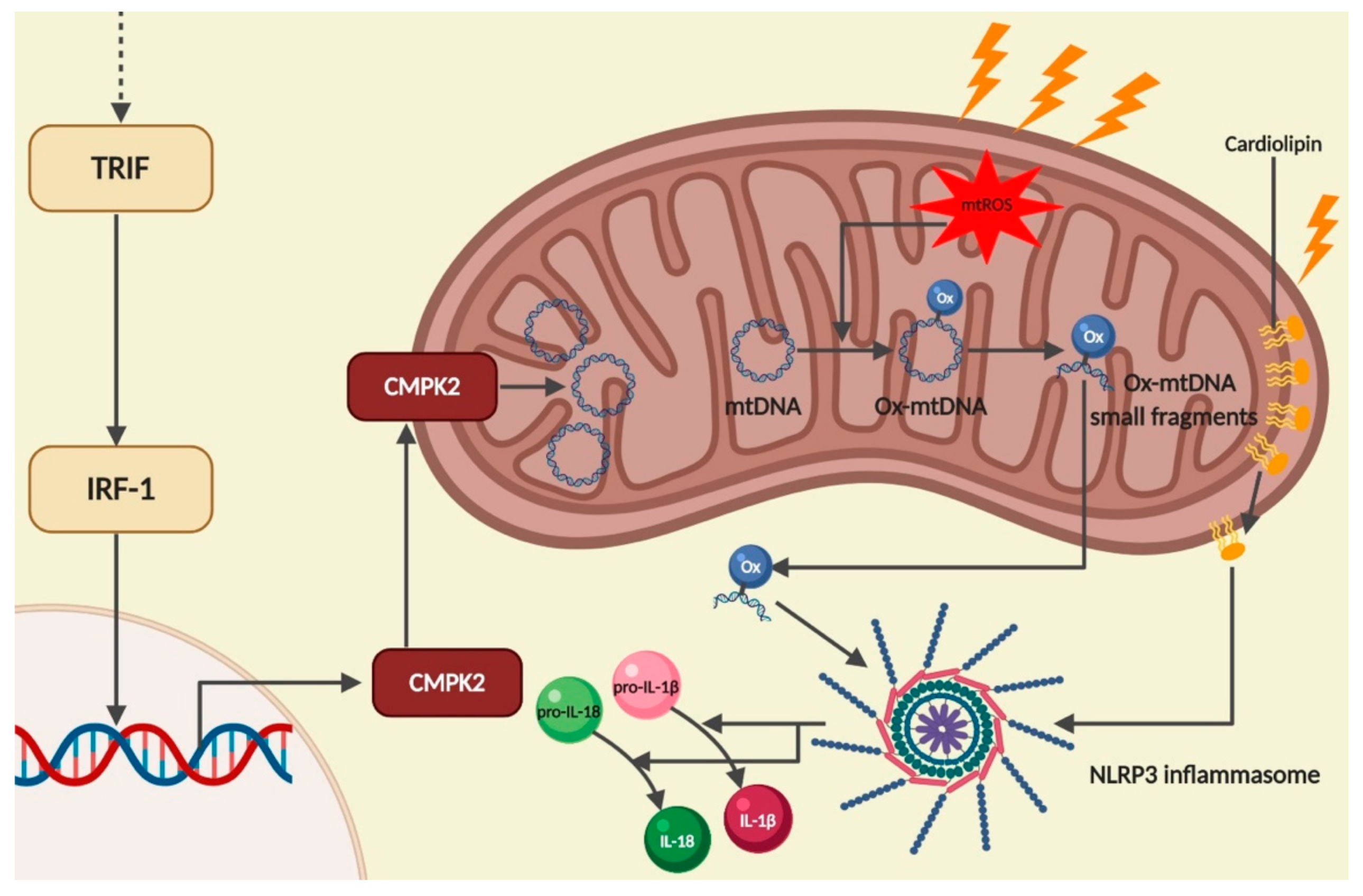
2.4. Mitochondrial DNA
2.5. Cardiolipin
2.6. Uric Acid
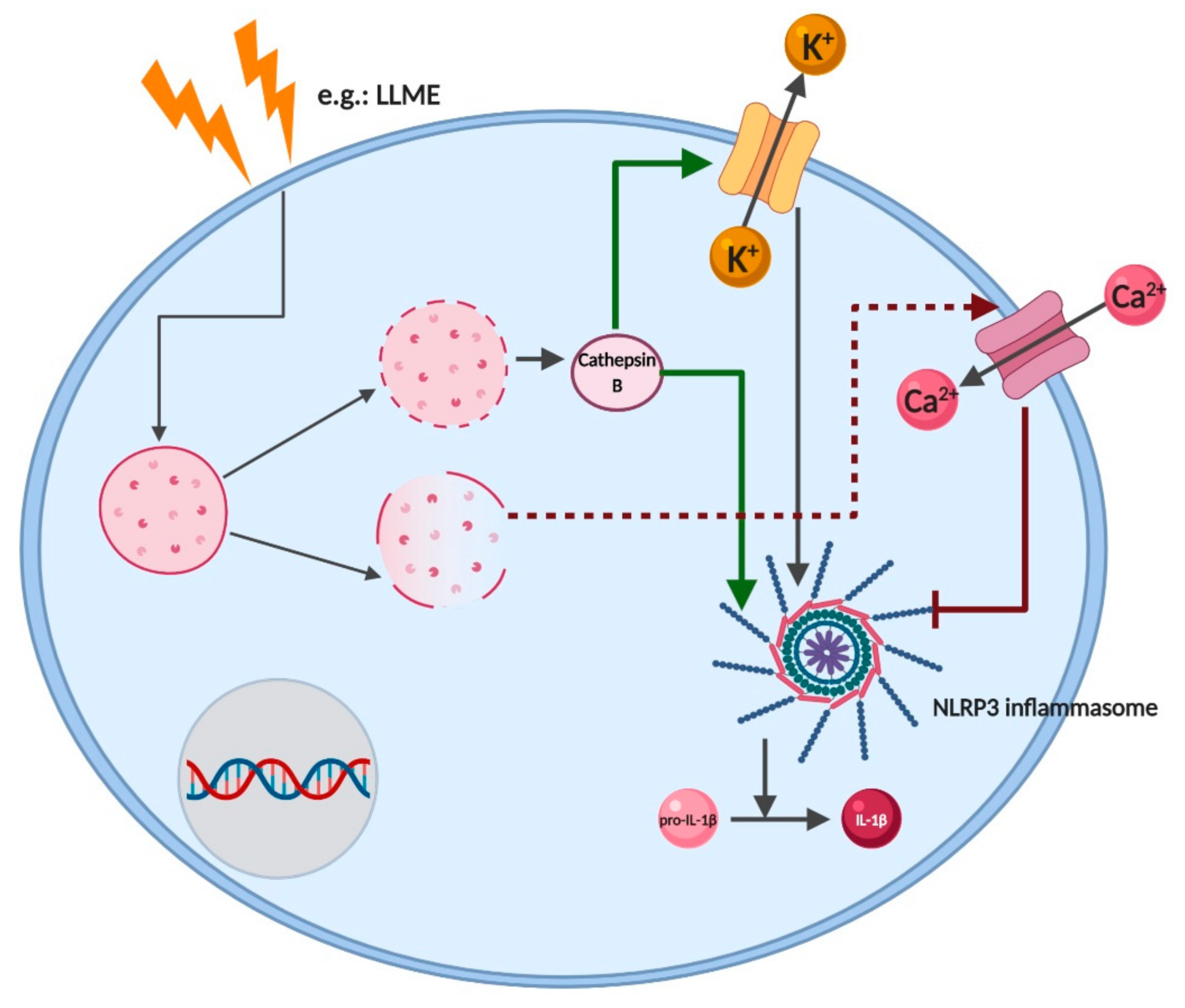
2.7. Cathepsins
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Esser, P.R.; Martin, S.F. Pathomechanisms of Contact Sensitization. Curr. Allergy Asthma Rep. 2017, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Kostner, L.; Anzengruber, F.; Guillod, C.; Recher, M.; Schmid-Grendelmeier, P.; Navarini, A. Allergic Contact Dermatitis. Immunol. Allergy Clin. N. Am. 2017, 37, 141–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.F. Immunological mechanisms in allergic contact dermatitis. Curr. Opin. Allergy Clin. Immunol. 2015, 15, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.H.; Igyarto, B.Z.; Gaspari, A.A. Early immune events in the induction of allergic contact dermatitis. Nat. Rev. Immunol. 2012, 12, 114–124. [Google Scholar] [CrossRef]
- Ainscough, J.; Gerberick, G.F.; Dearman, R.J.; Kimber, I. Danger, intracellular signaling, and the orchestration of dendritic cell function in skin sensitization. J. Immunotoxicol. 2012, 10, 223–234. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.F. Contact dermatitis: From pathomechanisms to immunotoxicology. Exp. Dermatol. 2012, 21, 382–389. [Google Scholar] [CrossRef]
- Schaefer, L. Complexity of Danger: The Diverse Nature of Damage-associated Molecular Patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.F. Allergic contact dermatitis: Xenoinflammation of the skin. Curr. Opin. Immunol. 2012, 24, 720–729. [Google Scholar] [CrossRef]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Johnston, G.A.; Mustapa, M.M.; Slack, J.; Coulson, I.; English, J.; Bourke, J.; McHenry, P.; Gibbon, K.; Buckley, D.; Leslie, T.; et al. British Association of Dermatologists’ guidelines for the management of contact dermatitis . Br. J. Dermatol. 2017, 176, 317–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natsuaki, Y.; Egawa, G.; Nakamizo, S.; Ono, S.; Hanakawa, S.; Okada, T.; Kusuba, N.; Otsuka, A.; Kitoh, A.; Honda, T.; et al. Perivascular leukocyte clusters are essential for efficient activation of effector T cells in the skin. Nat. Immunol. 2014, 15, 1064–1069. [Google Scholar] [CrossRef] [PubMed]
- Tuckermann, J.P.; Kleiman, A.; Moriggl, R.; Spanbroek, R.; Neumann, A.; Illing, A.; Clausen, B.E.; Stride, B.; Förster, I.; Habenicht, A.J.R.; et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J. Clin. Investig. 2007, 117, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Meguro, K.; Nakagomi, D.; Nakajima, H. Roles of alternatively activated M2 macrophages in allergic contact dermatitis. Allergol. Int. 2017, 66, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2019, 20, 95–112. [Google Scholar] [CrossRef]
- Martin, S.F.; Dudda, J.C.; Bachtanian, E.; Lembo, A.; Liller, S.; Duürr, C.; Heimesaat, M.M.; Bereswill, S.; Fejer, G.; Vassileva, R.; et al. Toll-like receptor and IL-12 signaling control susceptibility to contact hypersensitivity. J. Exp. Med. 2008, 205, 2151–2162. [Google Scholar] [CrossRef]
- Schmidt, M.; Raghavan, B.; Müller, V.; Vogl, T.; Fejer, G.; Tchaptchet, S.; Keck, S.; Kalis, C.; Nielsen, P.J.; Galanos, C.; et al. Crucial role for human Toll-like receptor 4 in the development of contact allergy to nickel. Nat. Immunol. 2010, 11, 814–819. [Google Scholar] [CrossRef]
- Watanabe, H.; Gaide, O.; Petrilli, V.; Martinon, F.; Contassot, E.; Roques, S.; Kummer, J.A.; Tschopp, J.; French, L.E. Activation of the IL-1β-Processing Inflammasome Is Involved in Contact Hypersensitivity. J. Investig. Dermatol. 2007, 127, 1956–1963. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95. [Google Scholar] [CrossRef] [PubMed]
- De Sá, D.C.; Neto, C.F. Inflammasomes and dermatology. An. Bras. Dermatol. 2016, 91, 566–578. [Google Scholar] [CrossRef]
- Tang, L.; Zhou, F. Inflammasomes in Common Immune-Related Skin Diseases. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Chipinda, I.; Hettick, J.M.; Siegel, P.D. Haptenation: Chemical Reactivity and Protein Binding. J. Allergy 2011, 2011, 839682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fyhrquist, N.T.; Lehto, E.; Lauerma, A. New findings in allergic contact dermatitis. Curr. Opin. Allergy Clin. Immunol. 2014, 14, 430–435. [Google Scholar] [CrossRef]
- Kermani, F.; Flint, M.S.; Hotchkiss, S.A. Induction and Localization of Cutaneous Interleukin-1β mRNA during Contact Sensitization. Toxicol. Appl. Pharmacol. 2000, 169, 231–237. [Google Scholar] [CrossRef]
- Wang, B.-J.; Chiu, H.-W.; Lee, Y.-L.; Li, C.-Y.; Wang, Y.-J.; Kuo, H.-C. Pterostilbene Attenuates Hexavalent Chromium-Induced Allergic Contact Dermatitis by Preventing Cell Apoptosis and Inhibiting IL-1β-Related NLRP3 Inflammasome Activation. J. Clin. Med. 2018, 7, 489. [Google Scholar] [CrossRef] [Green Version]
- Cumberbatch, M.; Dearman, R.J.; Kimber, I. Langerhans cells require signals from both tumour necrosis factor-α and interleukin-1β for migration. Immunol. 1997, 92, 388–395. [Google Scholar] [CrossRef]
- Nishibu, A.; Ward, B.; Boes, M.; Takashima, A. Roles for IL-1 and TNFα in dynamic behavioral responses of Langerhans cells to topical hapten application. J. Dermatol. Sci. 2007, 45, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Byamba, D.; Kim, T.-G.; Kim, D.H.; Je, J.H.; Lee, M.G. The Roles of Reactive Oxygen Species Produced by Contact Allergens and Irritants in Monocyte-derived Dendritic Cells. Ann. Dermatol. 2010, 22, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, I.; Silva, A.; Martins, J.D.; Neves, B.M.; Cruz, M.T. Nature and kinetics of redox imbalance triggered by respiratory and skin chemical sensitizers on the human monocytic cell line THP-1. Redox Biol. 2018, 16, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Youn, G.S.; Lee, K.-W.; Choi, S.Y.; Park, J.-S. Overexpression of HDAC6 induces pro-inflammatory responses by regulating ROS-MAPK-NF-κB/AP-1 signaling pathways in macrophages. Free. Radic. Biol. Med. 2016, 97, 14–23. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Yen, W.-C.; Wu, Y.-H.; Wu, C.-C.; Lin, H.-R.; Stern, A.; Chen, S.-H.; Shu, J.-C.; Chiu, D.T.-Y. Impaired inflammasome activation and bacterial clearance in G6PD deficiency due to defective NOX/p38 MAPK/AP-1 redox signaling. Redox Biol. 2020, 28, 101363. [Google Scholar] [CrossRef] [PubMed]
- Shelnutt, S.R.; Goad, P.; Belsito, D.V. Dermatological Toxicity of Hexavalent Chromium. Crit. Rev. Toxicol. 2007, 37, 375–387. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.; Cronin, M.T.D. Metals, Toxicity and Oxidative Stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [Green Version]
- Adam, C.; Wohlfarth, J.; Haußmann, M.; Sennefelder, H.; Rodin, A.; Maler, M.; Martin, S.F.; Goebeler, M.; Schmidt, M. Allergy-Inducing Chromium Compounds Trigger Potent Innate Immune Stimulation Via ROS-Dependent Inflammasome Activation. J. Investig. Dermatol. 2017, 137, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.F.; Rustemeyer, T.; Thyssen, J.P. Recent advances in understanding and managing contact dermatitis. F1000Research 2018, 7, 810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Liu, H.; Jian, Z.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L.; et al. Nickel induces inflammatory activation via NF-κB, MAPKs, IRF3 and NLRP3 inflammasome signaling pathways in macrophages. Aging 2019, 11, 11659–11672. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhong, F. Nickel Induces Interleukin-1β Secretion via the NLRP3–ASC–Caspase-1 Pathway. Inflammation 2013, 37, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Kowol, C.; Heffeter, P.; Miklos, W.; Gille, L.; Trondl, R.; Cappellacci, L.; Berger, W.; Keppler, B.K. Mechanisms underlying reductant-induced reactive oxygen species formation by anticancer copper(II) compounds. JBIC J. Biol. Inorg. Chem. 2011, 17, 409–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Lin, E.; Johansen, M.J.; Madden, T.; Felix, E.; Martirosyan, K.S.; Frank, S.J. Reactive Oxygen Species Generation in Human Cells by a Novel Magnetic Resonance Imaging Contrast Agent. J. Toxicol. 2018, 2018, 6362426. [Google Scholar] [CrossRef] [Green Version]
- Park, E.-J.; Park, K. Induction of reactive oxygen species and apoptosis in BEAS-2B cells by mercuric chloride. Toxicol. Vitr. 2007, 21, 789–794. [Google Scholar] [CrossRef]
- Lee, S.R. Critical Role of Zinc as Either an Antioxidant or a Prooxidant in Cellular Systems. Oxidative Med. Cell. Longev. 2018, 2018, 9156285. [Google Scholar] [CrossRef] [Green Version]
- Salwowska, N.M.; Bebenek, K.A.; Żądło, D.A.; Wcisło-Dziadecka, D. Physiochemical properties and application of hyaluronic acid: A systematic review. J. Cosmet. Dermatol. 2016, 15, 520–526. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Keen, M.A. Hyaluronic Acid in Dermatology. Skin 2017, 15, 441–448. [Google Scholar]
- Stern, R.; Asari, A.A.; Sugahara, K.N. Hyaluronan fragments: An information-rich system. Eur. J. Cell Biol. 2006, 85, 699–715. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Lee, C.-H.; Dedaj, R.; Zhao, H.; Mrabat, H.; Shiedlin, A.; Syrkina, O.L.; Huang, P.-M.; Garg, H.G.; Hales, C.A.; et al. High-molecular-weight hyaluronan—A possible new treatment for sepsis-induced lung injury: A preclinical study in mechanically ventilated rats. Crit. Care 2008, 12, R102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esser, P.R.; Wölfle, U.; Dürr, C.; Von Loewenich, F.D.; Schempp, C.M.; Freudenberg, M.A.; Jakob, T.; Martin, S.F. Contact Sensitizers Induce Skin Inflammation via ROS Production and Hyaluronic Acid Degradation. PLoS ONE 2012, 7, e41340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavasi, R.-M.; Berdiaki, A.; Spyridaki, I.; Papoutsidakis, A.; Corsini, E.; Tsatsakis, A.; Tzanakakis, G.N.; Nikitovic, D.; Dragana, N. Contact allergen (PPD and DNCB)-induced keratinocyte sensitization is partly mediated through a low molecular weight hyaluronan (LMWHA)/TLR4/NF-κB signaling axis. Toxicol. Appl. Pharmacol. 2019, 377, 114632. [Google Scholar] [CrossRef]
- Nikitovic, D.; Berdiaki, A.; Galbiati, V.; Kavasi, R.-M.; Papale, A.; Tsatsakis, A.; Tzanakakis, G.N.; Corsini, E. Hyaluronan regulates chemical allergen-induced IL-18 production in human keratinocytes. Toxicol. Lett. 2015, 232, 89–97. [Google Scholar] [CrossRef]
- Heo, J.H.; Heo, Y.; Lee, H.J.; Kim, M.; Shin, H.-Y. Topical anti-inflammatory and anti-oxidative effects of porcine placenta extracts on 2,4-dinitrochlorobenzene-induced contact dermatitis. BMC Complement. Altern. Med. 2018, 18, 331. [Google Scholar] [CrossRef]
- Muto, J.; Morioka, Y.; Yamasaki, K.; Kim, M.; Garcia, A.; Carlin, A.F.; Varki, A.; Gallo, R.L. Hyaluronan digestion controls DC migration from the skin. J. Clin. Investig. 2014, 124, 1309–1319. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, K.; Muto, J.; Taylor, K.R.; Cogen, A.L.; Audish, D.; Bertin, J.; Grant, E.P.; Coyle, A.J.; Misaghi, A.; Hoffman, H.M.; et al. NLRP3/Cryopyrin Is Necessary for Interleukin-1β (IL-1β) Release in Response to Hyaluronan, an Endogenous Trigger of Inflammation in Response to Injury. J. Biol. Chem. 2009, 284, 12762–12771. [Google Scholar] [CrossRef] [Green Version]
- Brown, T.A.; Bouchard, T.; John, T.S.; Wayner, E.; Carter, W.G. Human keratinocytes express a new CD44 core protein (CD44E) as a heparan-sulfate intrinsic membrane proteoglycan with additional exons. J. Cell Biol. 1991, 113, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Patel, S. Danger-Associated Molecular Patterns (DAMPs): The Derivatives and Triggers of Inflammation. Curr. Allergy Asthma Rep. 2018, 18, 63. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, K.; Useliene, J.; Ring, S.; Kage, P.; Jendrossek, V.; Robson, S.C.; Bylaite-Bucinskiene, M.; Steinbrink, K.; Enk, A.H. Down-Regulation of CD62L Shedding in T Cells by CD39+ Regulatory T Cells Leads to Defective Sensitization in Contact Hypersensitivity Reactions. J. Investig. Dermatol. 2016, 137, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, A.; Xiong, S.; Ye, Z.; Malireddi, R.K.S.; Kometani, S.; Mittal, M.; Kanneganti, T.-D.; Rehman, J.; Malik, A.B.; Zhong, M.; et al. The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation. Immunity 2018, 49, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, F.C.; Esser, P.R.; Müller, T.; Ganesan, J.; Pellegatti, P.; Simon, M.M.; Zeiser, R.; Idzko, M.; Jakob, T.; Martin, S.F. Lack of the purinergic receptor P2X7 results in resistance to contact hypersensitivity. J. Exp. Med. 2010, 207, 2609–2619. [Google Scholar] [CrossRef] [Green Version]
- Martins, J.D.; Silva, A.; Ferreira, I.; Gonçalo, M.; Custódio, J.; Lopes, M.C.; Domingues, M.R.M.; Neves, B.M.; Cruz, M.T. Adenosine diphosphate involvement in THP-1 maturation triggered by the contact allergen 1-fluoro-2,4-dinitrobenzene. Toxicol. Res. 2016, 5, 1512–1521. [Google Scholar] [CrossRef] [Green Version]
- Neuberger, A.; Ring, S.; Silva-Vilches, C.; Schrader, J.; Enk, A.; Mahnke, K. Expression of CD73 slows down migration of skin dendritic cells, affecting the sensitization phase of contact hypersensitivity reactions in mice. J. Dermatol. Sci. 2017, 87, 292–299. [Google Scholar] [CrossRef] [Green Version]
- Danquah, W.; Meyer-Schwesinger, C.; Rissiek, B.; Pinto, C.; Serracant-Prat, A.; Amadi, M.; Iacenda, D.; Knop, J.-H.; Hammel, A.; Bergmann, P.; et al. Nanobodies that block gating of the P2X7 ion channel ameliorate inflammation. Sci. Transl. Med. 2016, 8, 366ra162. [Google Scholar] [CrossRef]
- Banoth, B.; Cassel, S.L. Mitochondria in innate immune signaling. Transl. Res. 2018, 202, 52–68. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2010, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Liang, S.; Sánchez-López, E.; He, F.; Shalapour, S.; Lin, X.-J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.D.; Maciel, E.; Silva, A.; Ferreira, I.; Ricardo, F.; Domingues, P.; Neves, B.M.; Domingues, M.R.; Cruz, M.T. Phospholipidomic Profile Variation on THP-1 Cells Exposed to Skin or Respiratory Sensitizers and Respiratory Irritant. J. Cell. Physiol. 2016, 231, 2639–2651. [Google Scholar] [CrossRef] [PubMed]
- Elliott, E.I.; Miller, A.N.; Banoth, B.; Iyer, S.S.; Stotland, A.; Weiss, J.; Gottlieb, R.A.; Sutterwala, F.S.; Cassel, S.L. Cutting Edge: Mitochondrial Assembly of the NLRP3 Inflammasome Complex Is Initiated at Priming. J. Immunol. 2018, 200, 3047–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiuolo, J.; Oppedisano, F.; Gratteri, S.; Muscoli, C.; Mollace, V. Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 2016, 213, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Evans, J.E.; Rock, K.L. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 2003, 425, 516–521. [Google Scholar] [CrossRef]
- Yu, L.; Wang, L.; Chen, S. Endogenous toll-like receptor ligands and their biological significance. J. Cell. Mol. Med. 2010, 14, 2592–2603. [Google Scholar] [CrossRef] [Green Version]
- So, A.; Thorens, B. Uric acid transport and disease. J. Clin. Investig. 2010, 120, 1791–1799. [Google Scholar] [CrossRef] [Green Version]
- Katsnelson, M.A.; Lozada-Soto, K.M.; Russo, H.M.; Miller, B.A.; Dubyak, G.R. NLRP3 inflammasome signaling is activated by low-level lysosome disruption but inhibited by extensive lysosome disruption: Roles for K+ efflux and Ca2+ influx. Am. J. Physiol. Physiol. 2016, 311, C83–C100. [Google Scholar] [CrossRef] [Green Version]
- Braga, T.T.; Forni, M.F.; Correa-Costa, M.; Ramos, R.N.; Barbuto, J.A.M.; Branco, P.; Castoldi, A.; Hiyane, M.I.; Davanso, M.R.; Latz, E.; et al. Soluble Uric Acid Activates the NLRP3 Inflammasome. Sci. Rep. 2017, 7, 39884. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Inoue, H.; Nakayama, H.; Kanno, R.; Kanno, M. The Endogenous Danger Signal Uric Acid Augments Contact Hypersensitivity Responses in Mice. Pathobiology 2007, 74, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Conus, S.; Simon, H.-U. Cathepsins: Key modulators of cell death and inflammatory responses. Biochem. Pharmacol. 2008, 76, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Adair, B.; Reinheckel, T. Specialized roles for cysteine cathepsins in health and disease. J. Clin. Investig. 2010, 120, 3421–3431. [Google Scholar] [CrossRef] [Green Version]
- Conus, S.; Simon, H.-U. Cathepsins and their involvement in immune responses. Swiss Med. Wkly. 2010, 140. [Google Scholar] [CrossRef]
- Hsing, L.C.; Rudensky, A.Y. The lysosomal cysteine proteases in MHC class II antigen presentation. Immunol. Rev. 2005, 207, 229–241. [Google Scholar] [CrossRef]
- Driessen, C.; Lennon-Duménil, A.-M.; Ploegh, H.L. Individual cathepsins degrade immune complexes internalized by antigen-presenting cells via Fcγ receptors. Eur. J. Immunol. 2001, 31, 1592–1601. [Google Scholar] [CrossRef]
- Fiebiger, E.; Meraner, P.; Weber, E.; Fang, I.-F.; Stingl, G.; Ploegh, H.L.; Maurer, D. Cytokines Regulate Proteolysis in Major Histocompatibility Complex Class II–Dependent Antigen Presentation by Dendritic Cells. J. Exp. Med. 2001, 193, 881–892. [Google Scholar] [CrossRef]
- Schwenck, J.; Maurer, A.; Fehrenbacher, B.; Mehling, R.; Knopf, P.; Mucha, N.; Haupt, D.; Fuchs, K.; Griessinger, C.M.; Bukala, D.; et al. Cysteine-type cathepsins promote the effector phase of acute cutaneous delayed-type hypersensitivity reactions. Theranostics 2019, 9, 3903–3917. [Google Scholar] [CrossRef]
- Orlowski, G.M.; Colbert, J.D.; Sharma, S.; Bogyo, M.; Robertson, S.A.; Rock, K.L. Multiple Cathepsins Promote Pro-IL-1β Synthesis and NLRP3-Mediated IL-1β Activation. J. Immunol. 2015, 195, 1685–1697. [Google Scholar] [CrossRef]
- Dostert, C.; Petrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate Immune Activation Through Nalp3 Inflammasome Sensing of Asbestos and Silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Núñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]







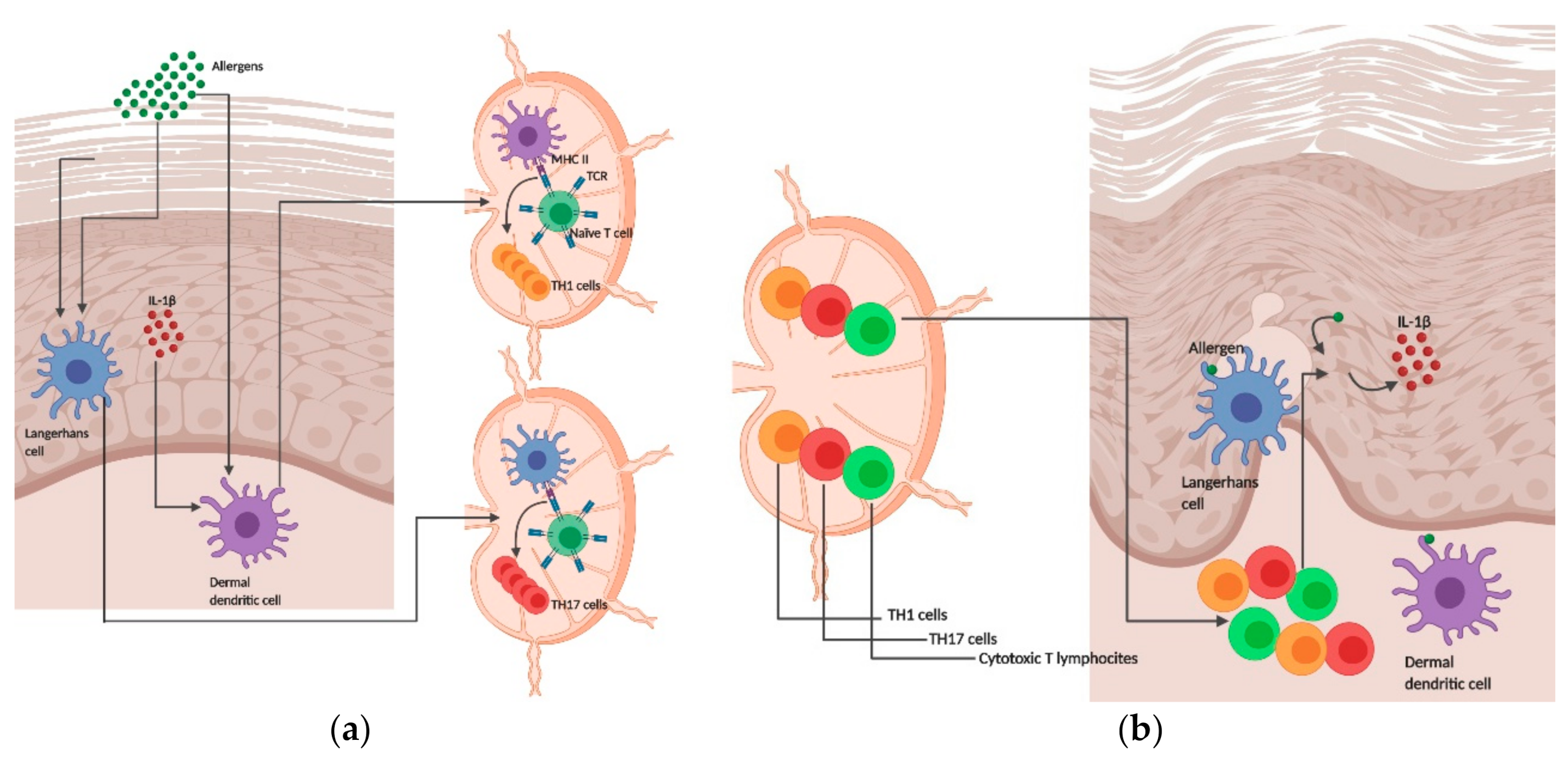
{kind=link}
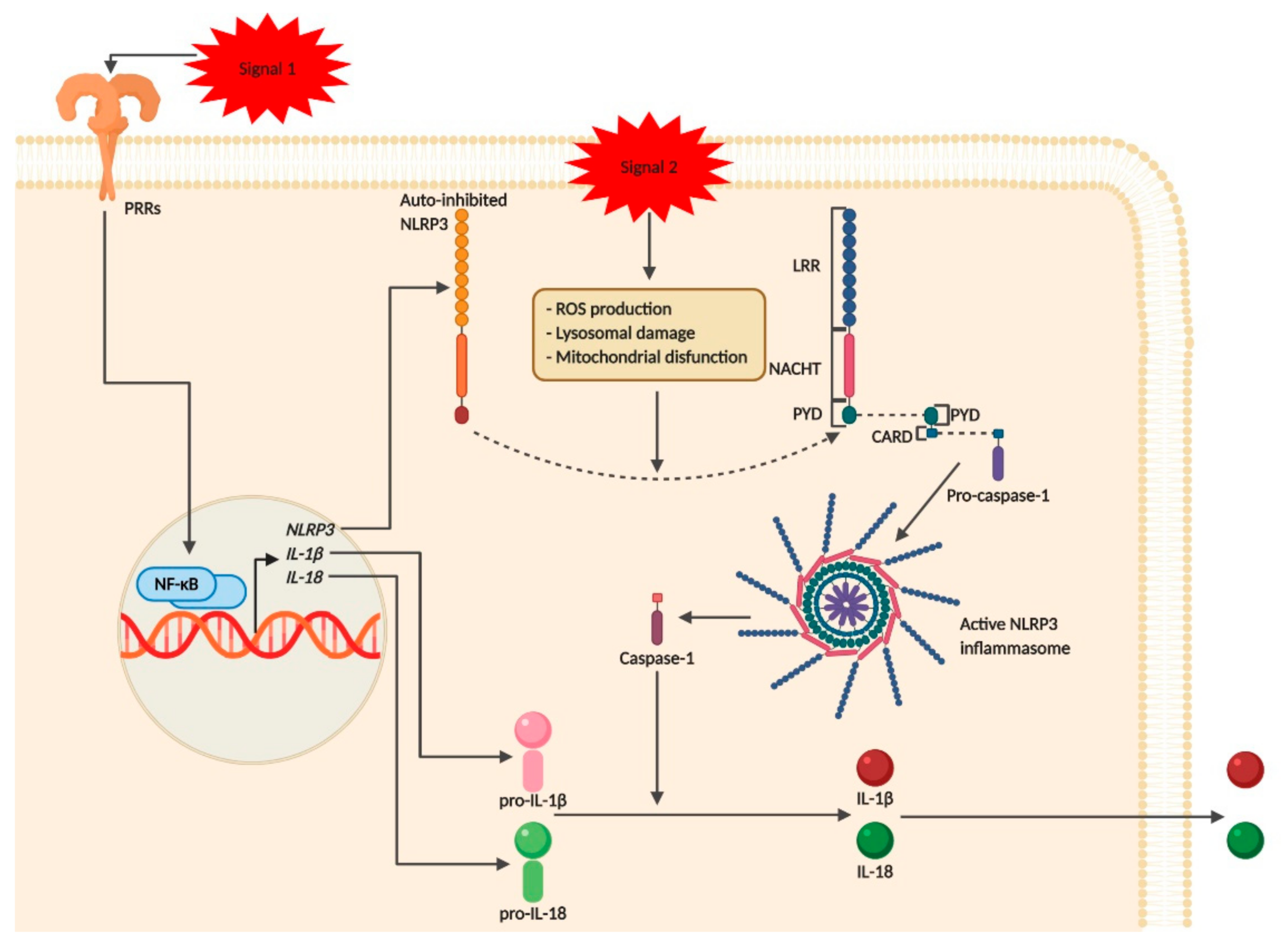
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Priming Phase | Activation Phase | ||
|---|---|---|---|
| DAMPs | Allergens | DAMPs | Allergens |
| ROS | DNFB, Cr (VI) | mtROS | Cr (VI) Ni |
| LMWHA | PPD, DNCB | ATP | TNCB |
| (ox-)mtDNA | Ni | ||
| Cardiolipin | DNFB | ||
| Uric Acid | TNCB | ||
| Cathepsins | TNCB | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sebastião, A.I.; Ferreira, I.; Brites, G.; Silva, A.; Neves, B.M.; Teresa Cruz, M. NLRP3 Inflammasome and Allergic Contact Dermatitis: A Connection to Demystify. Pharmaceutics 2020, 12, 867. https://doi.org/10.3390/pharmaceutics12090867
Sebastião AI, Ferreira I, Brites G, Silva A, Neves BM, Teresa Cruz M. NLRP3 Inflammasome and Allergic Contact Dermatitis: A Connection to Demystify. Pharmaceutics. 2020; 12(9):867. https://doi.org/10.3390/pharmaceutics12090867
Chicago/Turabian StyleSebastião, Ana Isabel, Isabel Ferreira, Gonçalo Brites, Ana Silva, Bruno Miguel Neves, and Maria Teresa Cruz. 2020. "NLRP3 Inflammasome and Allergic Contact Dermatitis: A Connection to Demystify" Pharmaceutics 12, no. 9: 867. https://doi.org/10.3390/pharmaceutics12090867
APA StyleSebastião, A. I., Ferreira, I., Brites, G., Silva, A., Neves, B. M., & Teresa Cruz, M. (2020). NLRP3 Inflammasome and Allergic Contact Dermatitis: A Connection to Demystify. Pharmaceutics, 12(9), 867. https://doi.org/10.3390/pharmaceutics12090867