
Elucidating the Innate Immunological Effects of Mild Magnetic Hyperthermia on U87 Human Glioblastoma Cells: An In Vitro Study

 ,
, 
 ,
,
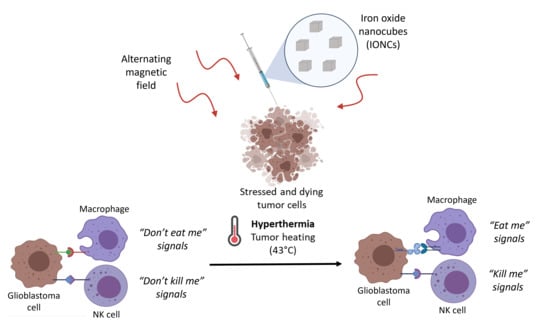
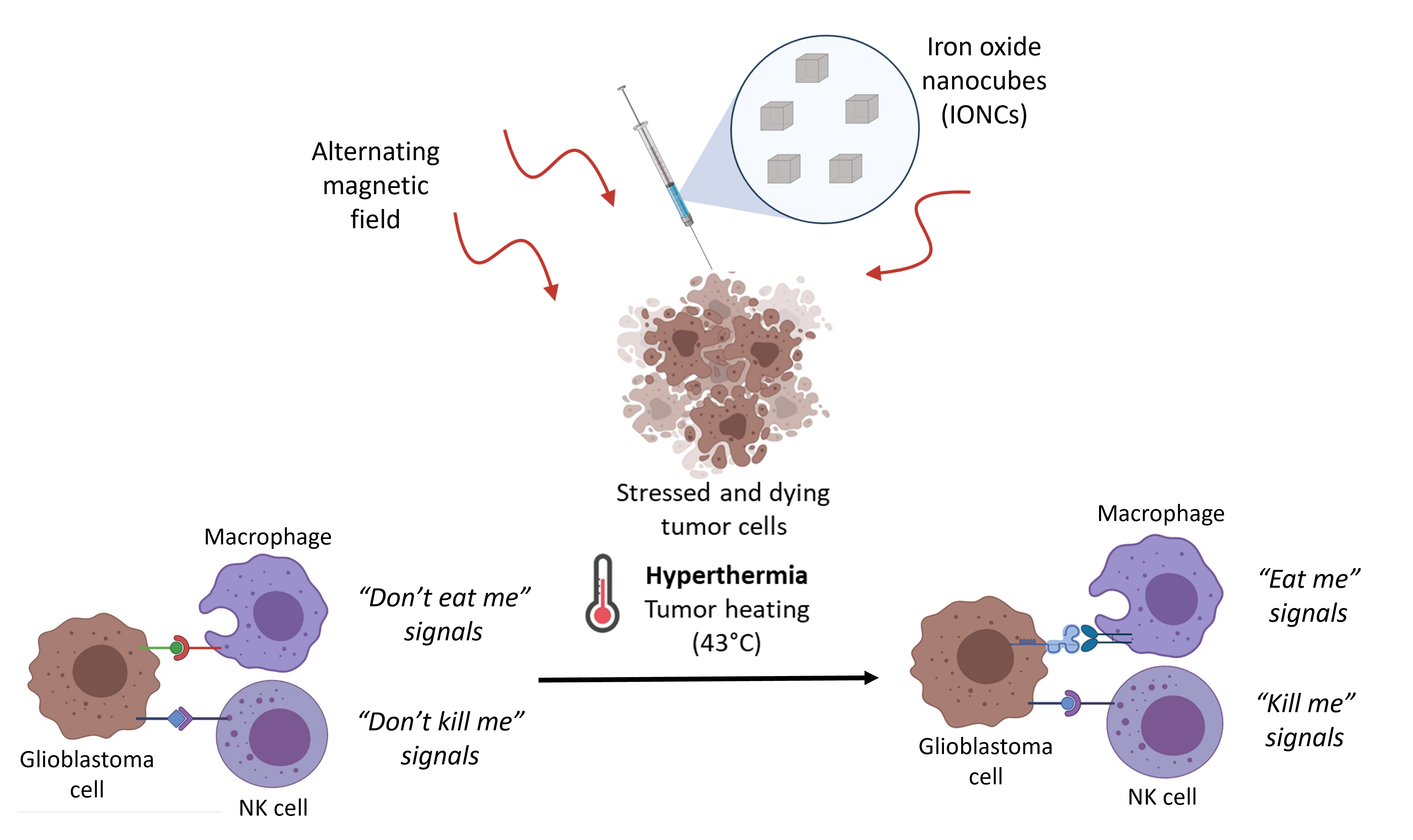
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
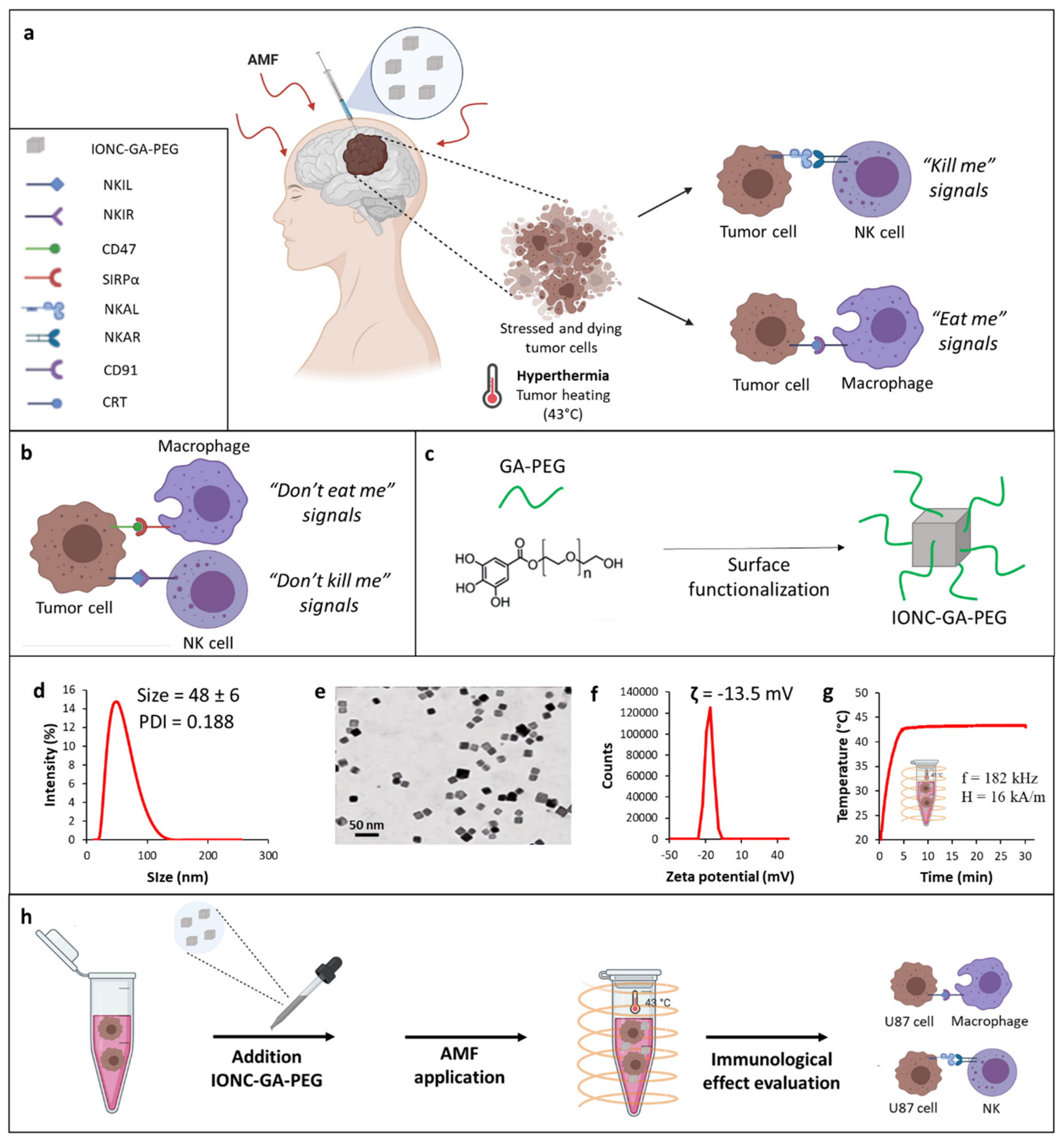
2.1. Synthesis and Characterization of GA-PEG-Coated IONCs
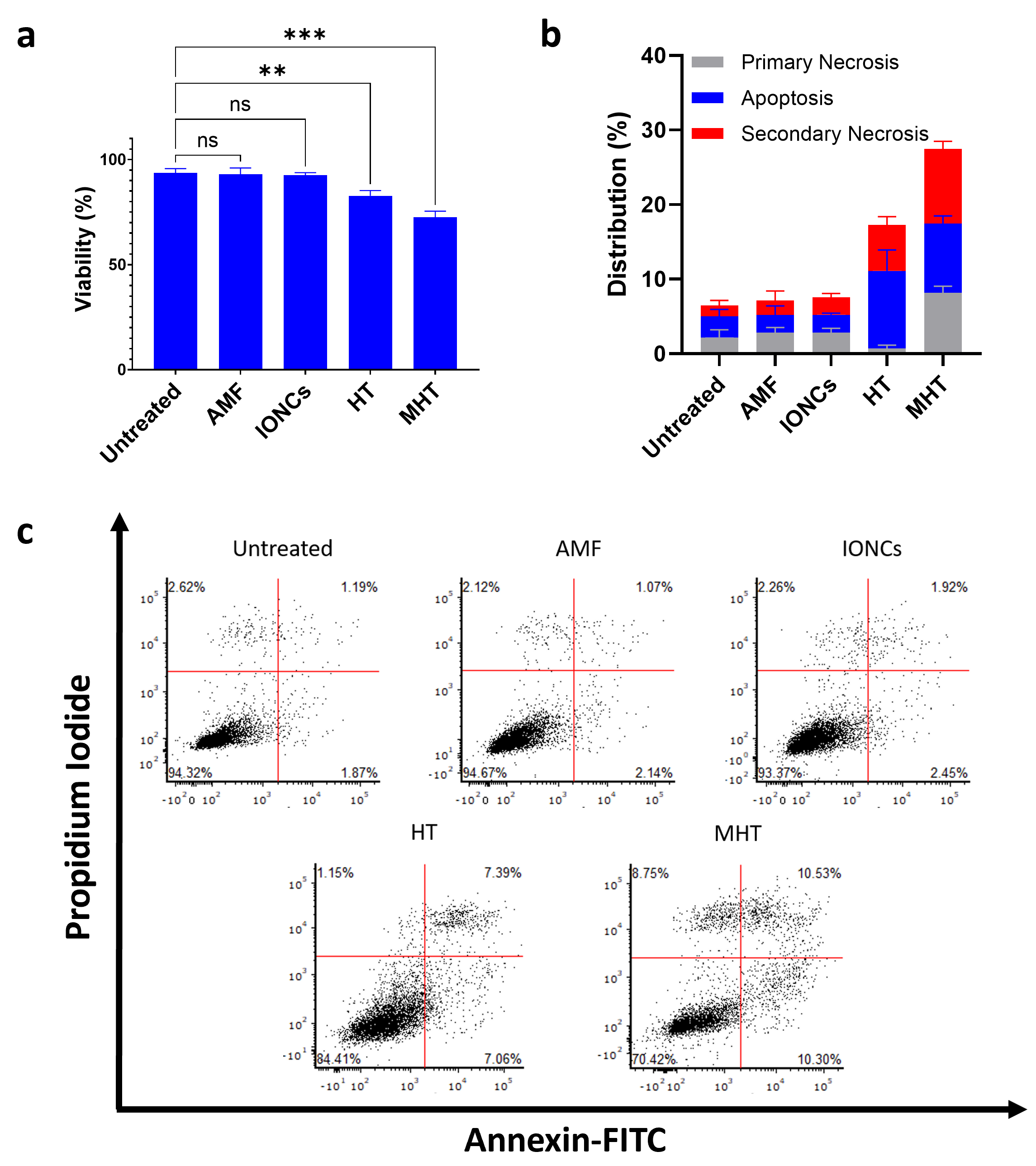
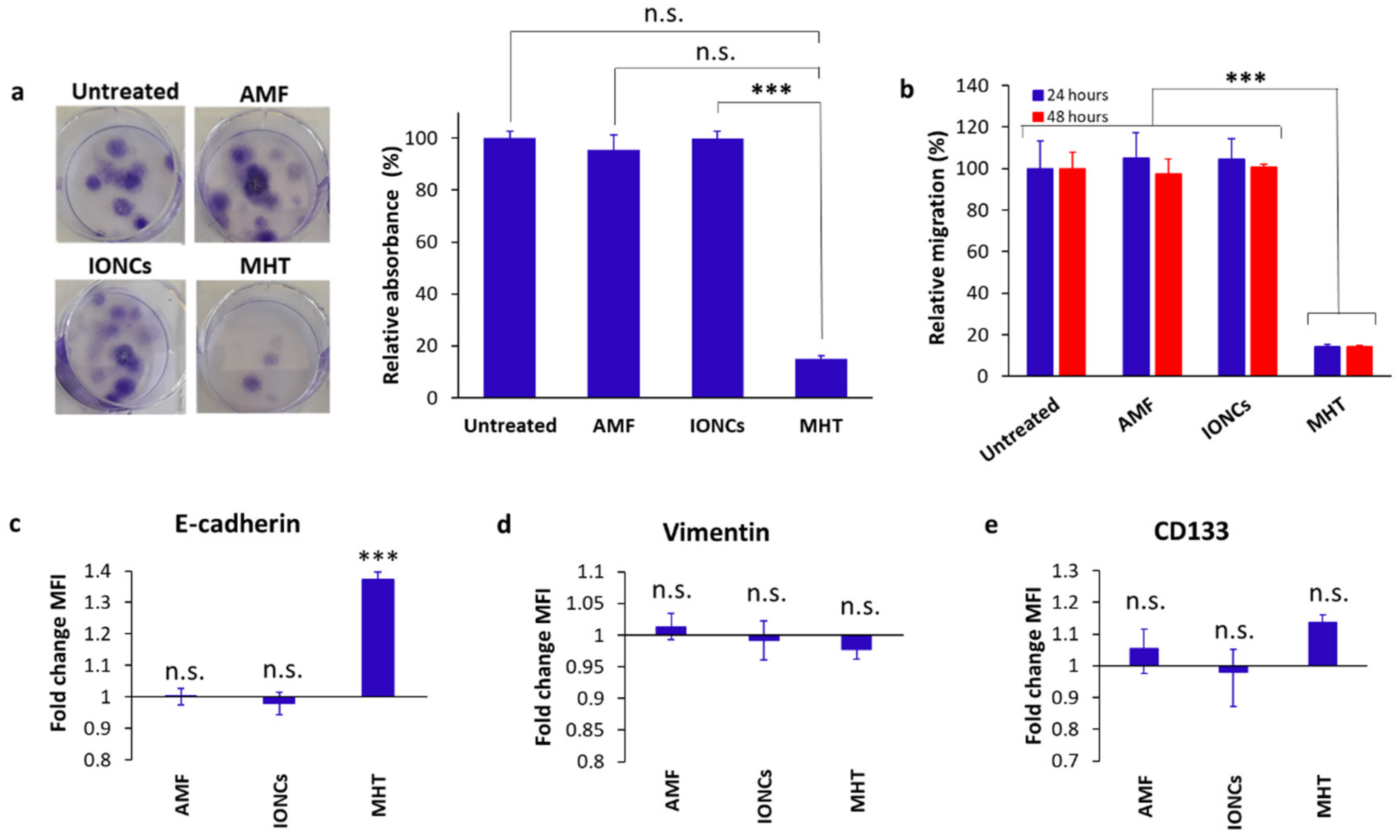
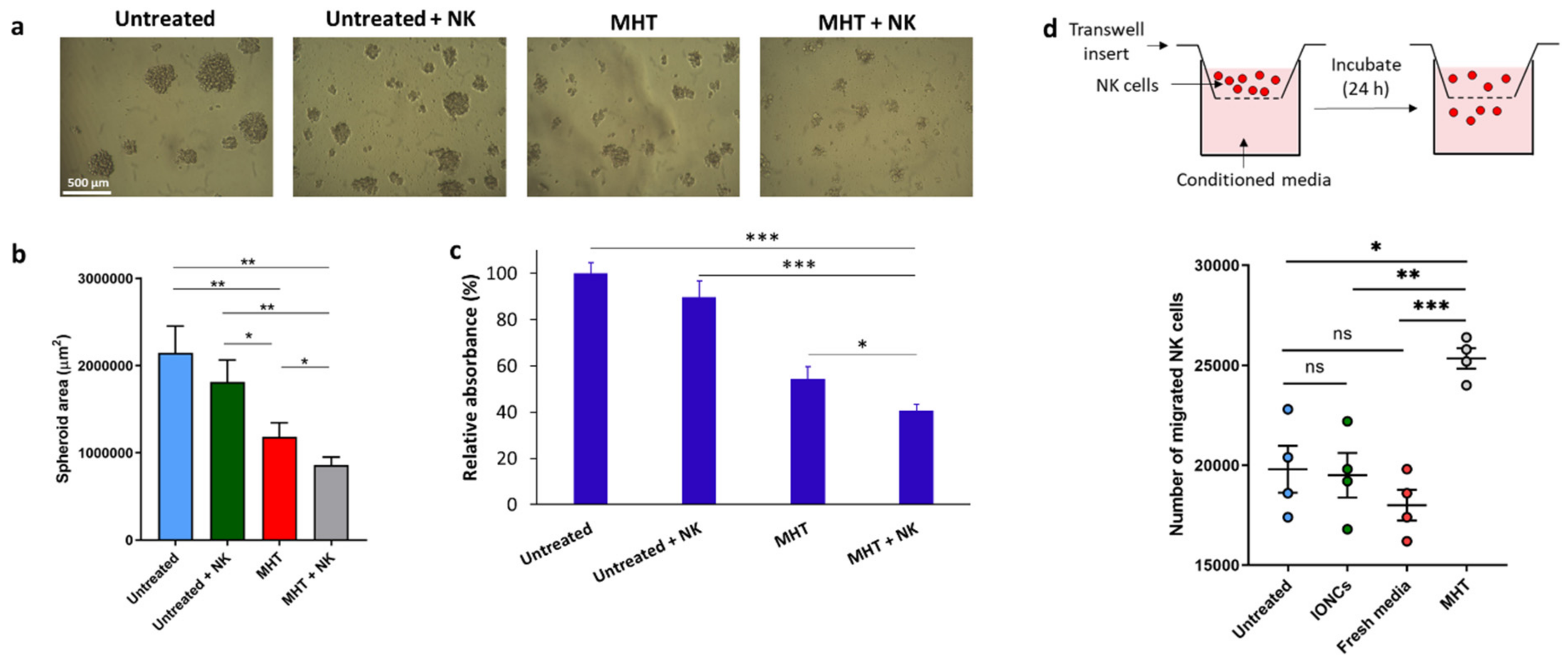
2.2. Mild MHT Induces Apoptosis and Impacts Tumor-Initiating and Migratory Abilities of U87 Cells
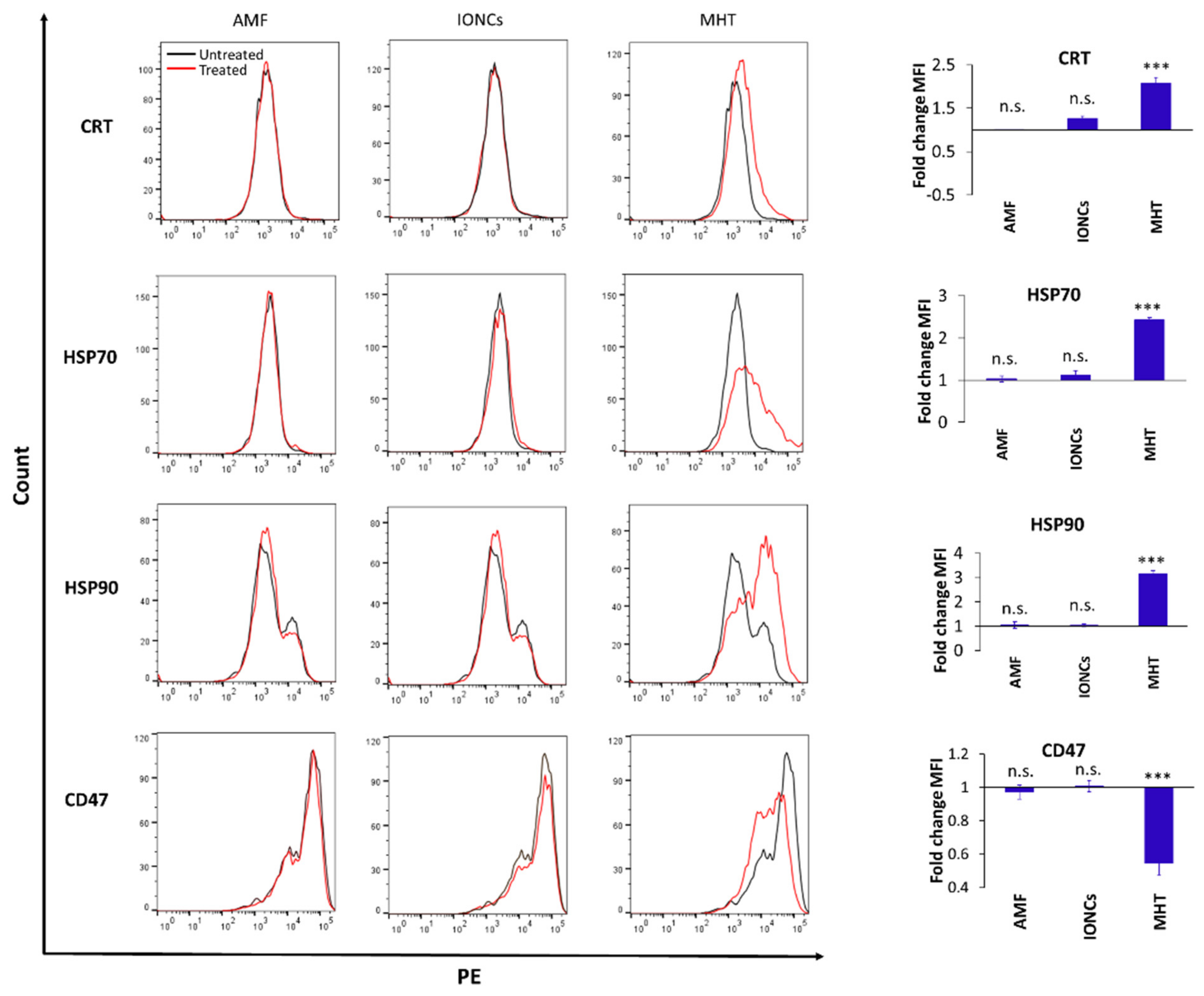
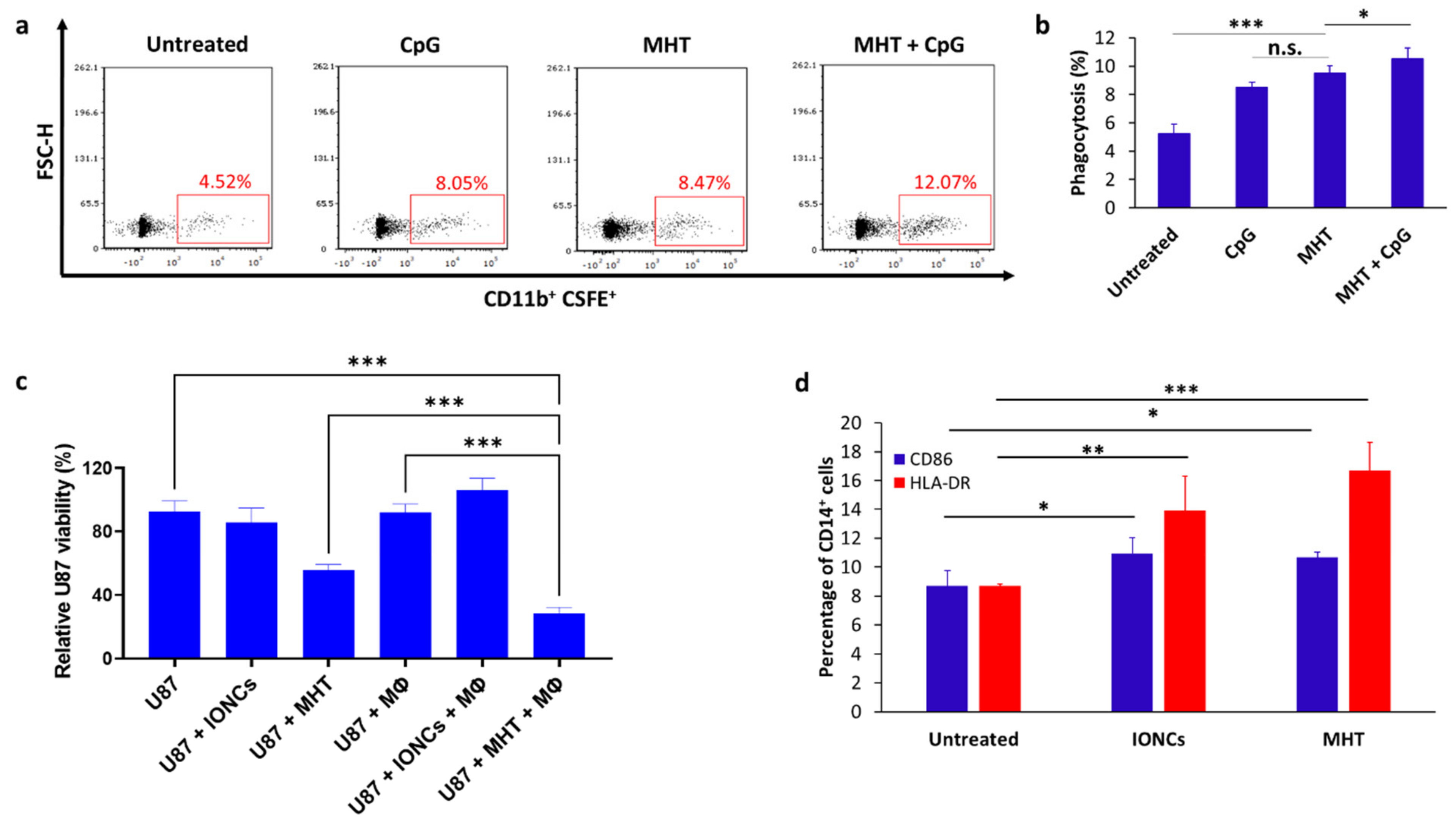
2.3. Mild MHT Induces ICD in U87 Cells and Enhances Macrophages’ Antitumoral Functions
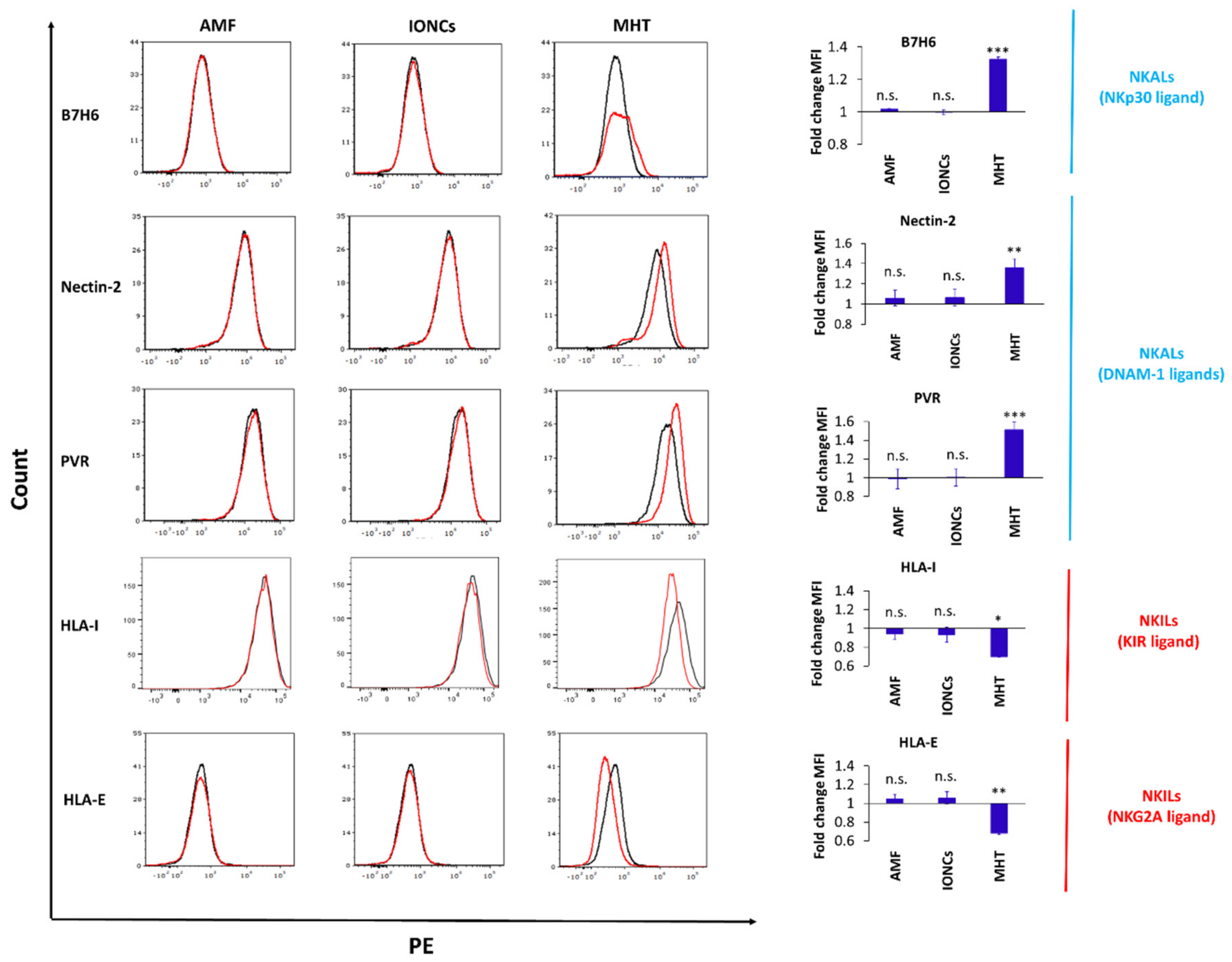
2.4. Mild MHT Alters the Expression of NK Cell-Activating and Inhibitory Ligands on U87 Cells
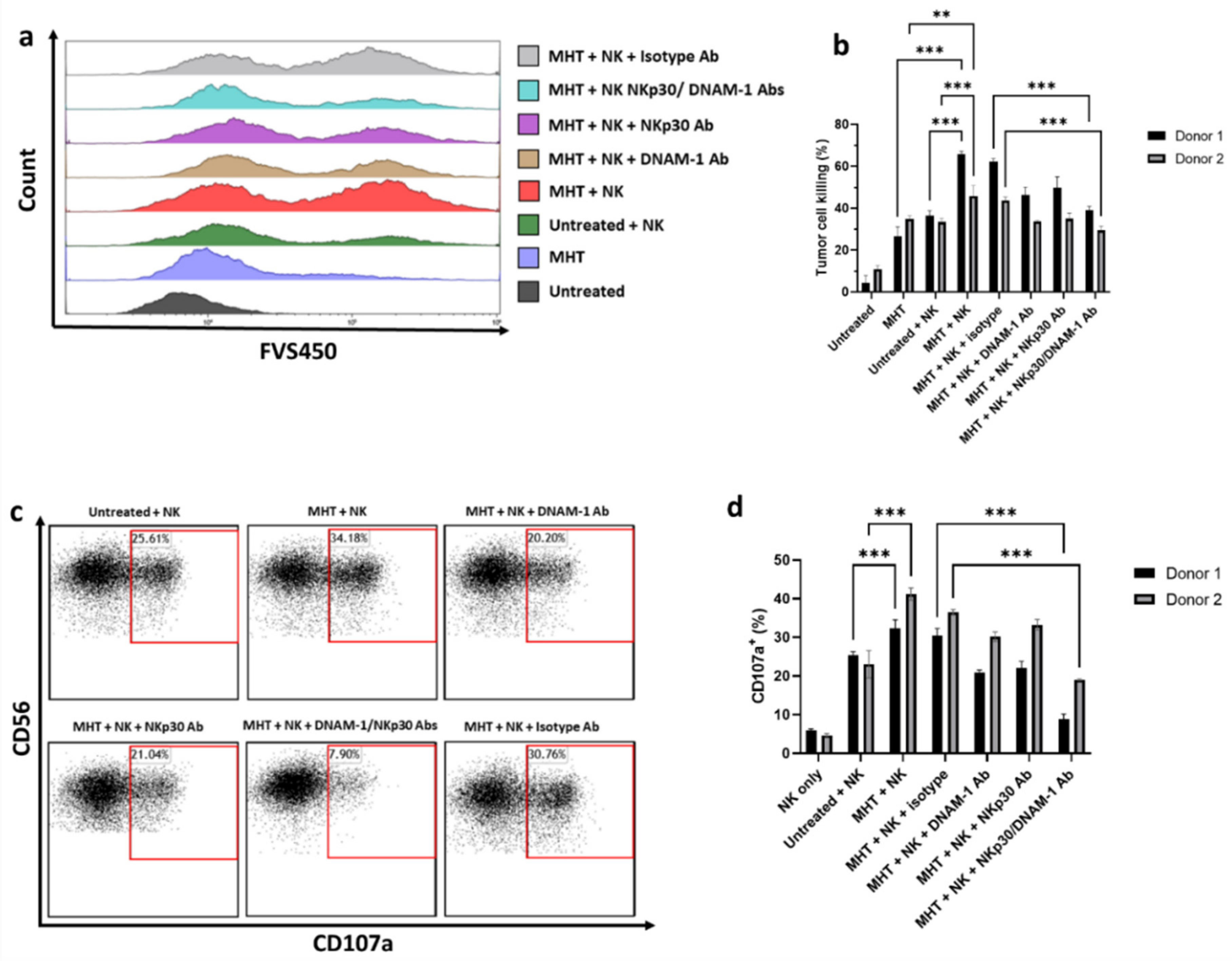
2.5. Mild MHT Sensitizes U87 Cells to NK Cell-Mediated Functions
3. Discussion
4. Conclusions
5. Experimental Section
5.1. Reagents
5.2. Synthesis of the IONCs
5.3. Synthesis of Gallic Acid PEG (GA-PEG) Ligand
5.4. Ligand Exchange and Water Transfer of Cubic Iron Oxide Nanoparticles (IONCs-GA-PEG)
5.5. SAR Measurements
5.6. Cell Lines and Cell Cultures
5.7. Cell Treatment with MHT
5.8. Cell Viability Assay
5.9. Annexin V-FITC/Propidium Iodide Assay
5.10. Flow Cytometry Analysis of Surface Markers on U87 Cells
5.11. Cell Cycle Analysis
5.12. Transwell Migration Assay
5.13. Colony Formation Assay
5.14. In Vitro Phagocytosis, Killing and Activation of THP-1-Derived Macrophages
5.15. HMGB-1 ELISA
5.16. NK Cell Preparation
5.17. NK Cell Functional Assays
5.18. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008-2012. Neuro-Oncology 2015, 17, iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Costa, A.; Osório, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy. Glioblastoma 2017, 11, 197–241. [Google Scholar] [CrossRef] [Green Version]
- Dobosz, P.; Dzieciątkowski, T. The Intriguing History of Cancer Immunotherapy. Front. Immunol. 2019, 10, 2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Barone, A.; Hazarika, M.; Theoret, M.R.; Mishra-Kalyani, P.; Chen, H.; He, K.; Sridhara, R.; Subramaniam, S.; Pfuma, E.; Wang, Y.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Patients with Unresectable or Metastatic Melanoma. Clin. Cancer Res. 2017, 23, 5661–5665. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Su, W.; Lu, T.; Wang, Y.; Dong, Y.; Qin, Y.; Liu, D.; Sun, L.; Jiao, W. First-Line Immune-Checkpoint Inhibitors in Non-Small Cell Lung Cancer: Current Landscape and Future Progress. Front. Pharmacol. 2020, 11, 578091. [Google Scholar] [CrossRef]
- Pusztai, L.; Karn, T.; Safonov, A.; Abu-Khalaf, M.M.; Bianchini, G. New Strategies in Breast Cancer: Immunotherapy. Clin. Cancer Res. 2016, 22, 2105–2110. [Google Scholar] [CrossRef] [Green Version]
- Johdi, N.A.; Sukor, N.F. Colorectal Cancer Immunotherapy: Options and Strategies. Front. Immunol. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Deleuze, A.; Saout, J.; Dugay, F.; Peyronnet, B.; Mathieu, R.; Verhoest, G.; Bensalah, K.; Crouzet, L.; Laguerre, B.; Belaud-Rotureau, M.-A.; et al. Immunotherapy in Renal Cell Carcinoma: The Future Is Now. Int. J. Mol. Sci. 2020, 21, 2532. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, T.; Therkelsen, K.E.; Ahmad, S.; Nagpal, S. Current State of Immunotherapy for Treatment of Glioblastoma. Curr. Treat. Options Oncol. 2019, 20, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Decordova, S.; Shastri, A.; Tsolaki, A.G.; Yasmin, H.; Klein, L.; Singh, S.K.; Kishore, U. Molecular Heterogeneity and Immunosuppressive Microenvironment in Glioblastoma. Front. Immunol. 2020, 11, 1402. [Google Scholar] [CrossRef]
- Antunes, A.R.P.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. eLife 2020, 9, e52176. [Google Scholar] [CrossRef]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef] [Green Version]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef] [Green Version]
- Angell, H.K.; Bruni, D.; Barrett, J.C.; Herbst, R.; Galon, J. The Immunoscore: Colon Cancer and Beyond. Clin. Cancer Res. 2020, 26, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Ghosh, S. Lifting the innate immune barriers to antitumor immunity. J. Immunother. Cancer 2019, 8, e000695. [Google Scholar] [CrossRef]
- Corrales, L.; Matson, V.; Flood, B.; Spranger, S.; Gajewski, T.F. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 2017, 27, 96–108. [Google Scholar] [CrossRef] [Green Version]
- Moynihan, K.D.; Irvine, D.J. Roles for Innate Immunity in Combination Immunotherapies. Cancer Res. 2017, 77, 5215–5221. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Fan, X.; Deng, H.; Brezski, R.J.; Rycyzyn, M.; Jordan, R.E.; Strohl, W.; Zou, Q.; Zhang, N.; An, Z. Trastuzumab Triggers Phagocytic Killing of High HER2 Cancer Cells In Vitro and In Vivo by Interaction with Fcγ Receptors on Macrophages. J. Immunol. 2015, 194, 4379–4386. [Google Scholar] [CrossRef] [Green Version]
- Woan, K.V.; Miller, J.S. Harnessing Natural Killer Cell Antitumor Immunity: From the Bench to Bedside. Cancer Immunol. Res. 2019, 7, 1742–1747. [Google Scholar] [CrossRef] [Green Version]
- Kmiecik, J.; Zimmer, J.; Chekenya, M. Natural killer cells in intracranial neoplasms: Presence and therapeutic efficacy against brain tumours. J. Neuro-Oncol. 2014, 116, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Golán, I.; De La Fuente, L.R.; Costoya, J.A. NK Cell-Based Glioblastoma Immunotherapy. Cancers 2018, 10, 522. [Google Scholar] [CrossRef] [Green Version]
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain Tumor Microenvironment and Host State: Implications for Immunotherapy. Clin. Cancer Res. 2019, 25, 4202–4210. [Google Scholar] [CrossRef] [Green Version]
- Castriconi, R.; Daga, A.; Dondero, A.; Zona, G.; Poliani, P.L.; Melotti, A.; Griffero, F.; Marubbi, D.; Spaziante, R.; Bellora, F.; et al. NK Cells Recognize and Kill Human Glioblastoma Cells with Stem Cell-Like Properties. J. Immunol. 2009, 182, 3530–3539. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.-J.; Yang, J.-I.; Kim, O.; Ahn, E.-J.; Kang, W.D.; Lee, J.-H.; Moon, K.-S.; Lee, K.-H.; Cho, D. Human U87 glioblastoma cells with stemness features display enhanced sensitivity to natural killer cell cytotoxicity through altered expression of NKG2D ligand. Cancer Cell Int. 2017, 17, 22. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Nakazawa, T.; Nakamura, M.; Nishimura, F.; Matsuda, R.; Omoto, K.; Shida, Y.; Murakami, T.; Nakagawa, I.; Motoyama, Y.; et al. Ex vivo-expanded highly purified natural killer cells in combination with temozolomide induce antitumor effects in human glioblastoma cells in vitro. PLoS ONE 2019, 14, e0212455. [Google Scholar] [CrossRef]
- Cooper, M.A. Natural killer cells might adapt their inhibitory receptors for memory. Proc. Natl. Acad. Sci. USA 2018, 115, 11357–11359. [Google Scholar] [CrossRef] [Green Version]
- Stary, V.; Stary, G. NK Cell-Mediated Recall Responses: Memory-Like, Adaptive, or Antigen-Specific? Front. Cell. Infect. Microbiol. 2020, 10, 208. [Google Scholar] [CrossRef]
- Nikzad, R.; Angelo, L.S.; Aviles-Padilla, K.; Le, D.T.; Singh, V.K.; Bimler, L.; Vukmanovic-Stejic, M.; Vendrame, E.; Ranganath, T.; Simpson, L.; et al. Human natural killer cells mediate adaptive immunity to viral antigens. Sci. Immunol. 2019, 4, eaat8116. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, A.T.; Glavey, S.V.; Bezman, N.A.; Jhatakia, A.; Guerriero, J.L.; Manier, S.; Moschetta, M.; Mishima, Y.; Roccaro, A.; Detappe, A.; et al. Antibody-Dependent Cellular Phagocytosis by Macrophages is a Novel Mechanism of Action of Elotuzumab. Mol. Cancer Ther. 2018, 17, 1454–1463. [Google Scholar] [CrossRef] [Green Version]
- Seidel, U.J.E.; Schlegel, P.; Lang, P. Natural Killer Cell Mediated Antibody-Dependent Cellular Cytotoxicity in Tumor Immunotherapy with Therapeutic Antibodies. Front. Immunol. 2013, 4, 76. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Gong, J.; Murshid, A. Extracellular HSPs: The Complicated Roles of Extracellular HSPs in Immunity. Front. Immunol. 2016, 7, 159. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Vandenberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Van Gool, S.W.; Agostinis, P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell–driven rejection of high-grade glioma. Sci. Transl. Med. 2016, 8, 328ra27. [Google Scholar] [CrossRef]
- Humeau, J.; Sauvat, A.; Cerrato, G.; Xie, W.; Loos, F.; Iannantuoni, F.; Bezu, L.; Lévesque, S.; Paillet, J.; Pol, J.; et al. Inhibition of transcription by dactinomycin reveals a new characteristic of immunogenic cell stress. EMBO Mol. Med. 2020, 12, e11622. [Google Scholar] [CrossRef]
- Hayashi, K.; Nikolos, F.; Lee, Y.C.; Jain, A.; Tsouko, E.; Gao, H.; Kasabyan, A.; Leung, H.E.; Osipov, A.; Jung, S.Y.; et al. Tipping the immunostimulatory and inhibitory DAMP balance to harness immunogenic cell death. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, K.; Zhao, R.; Ji, T.; Wang, X.; Yang, X.; Zhang, Y.; Cheng, K.; Liu, S.; Hao, J.; et al. Inducing enhanced immunogenic cell death with nanocarrier-based drug delivery systems for pancreatic cancer therapy. Biomaterials 2016, 102, 187–197. [Google Scholar] [CrossRef]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2009, 29, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Fucikova, J.; Kralikova, P.; Fialová, A.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Špíšek, R. Human Tumor Cells Killed by Anthracyclines Induce a Tumor-Specific Immune Response. Cancer Res. 2011, 71, 4821–4833. [Google Scholar] [CrossRef] [Green Version]
- Wan, C.; Sun, Y.; Tian, Y.; Lu, L.; Dai, X.; Meng, J.; Huang, J.; He, Q.; Wu, B.; Zhang, Z.; et al. Irradiated tumor cell–derived microparticles mediate tumor eradication via cell killing and immune reprogramming. Sci. Adv. 2020, 6, eaay9789. [Google Scholar] [CrossRef] [Green Version]
- Adkins, I.; Sadilkova, L.; Hradilova, N.; Tomala, J.; Kovar, M.; Spisek, R. Severe, but not mild heat-shock treatment induces immunogenic cell death in cancer cells. OncoImmunology 2017, 6, e1311433. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zheng, J.; Sun, W.; Zhao, X.; Li, Y.; Gong, N.; Wang, Y.; Ma, X.; Zhang, T.; Zhao, L.-Y.; et al. Ferrimagnetic Vortex Nanoring-Mediated Mild Magnetic Hyperthermia Imparts Potent Immunological Effect for Treating Cancer Metastasis. ACS Nano 2019, 13, 8811–8825. [Google Scholar] [CrossRef]
- Guevara, M.L.; Persano, F.; Persano, S. Nano-immunotherapy: Overcoming tumour immune evasion. Semin. Cancer Biol. 2021, 69, 238–248. [Google Scholar] [CrossRef]
- Persano, S.; Das, P.; Pellegrino, T. Magnetic Nanostructures as Emerging Therapeutic Tools to Boost Anti-Tumour Immunity. Cancers 2021, 13, 2735. [Google Scholar] [CrossRef]
- Mukherjee, S.; Liang, L.; Veiseh, O. Recent Advancements of Magnetic Nanomaterials in Cancer Therapy. Pharmaceutics 2020, 12, 147. [Google Scholar] [CrossRef] [Green Version]
- Mahmoudi, K.; Bouras, A.; Bozec, D.; Ivkov, R.; Hadjipanayis, C. Magnetic hyperthermia therapy for the treatment of glioblastoma: A review of the therapy’s history, efficacy and application in humans. Int. J. Hyperth. 2018, 34, 1316–1328. [Google Scholar] [CrossRef] [Green Version]
- Thiesen, B.; Jordan, A. Clinical applications of magnetic nanoparticles for hyperthermia. Int. J. Hyperth. 2008, 24, 467–474. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, F.; Shao, D.; Chang, Z.; Wang, L.; Hu, H.; Zheng, X.; Li, X.; Chen, F.; Tu, Z.; et al. Janus Nanobullets Combine Photodynamic Therapy and Magnetic Hyperthermia to Potentiate Synergetic Anti-Metastatic Immunotherapy. Adv. Sci. 2019, 6, 1901690. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.; Bin Chen, G.; Liang, C.; Xu, J.; Dong, Z.L.; Han, X.; Wang, C.; Liu, Z. Iron Nanoparticles for Low-Power Local Magnetic Hyperthermia in Combination with Immune Checkpoint Blockade for Systemic Antitumor Therapy. Nano Lett. 2019, 19, 4287–4296. [Google Scholar] [CrossRef]
- Toraya-Brown, S.; Sheen, M.R.; Zhang, P.; Chen, L.; Baird, J.R.; Demidenko, E.; Turk, M.J.; Hoopes, P.J.; Conejo-Garcia, J.R.; Fiering, S. Local Hyperthermia Treatment of Tumors Induces CD8+ T Cell-Mediated Resistance against Distal and Secondary Tumors. In Handbook of Immunological Properties of Engineered Nanomaterials, 2nd ed.; World Scientific: Singapore, 2016; Volume 3, pp. 309–334. [Google Scholar] [CrossRef]
- Dayanc, B.E.; Beachy, S.H.; Ostberg, J.R.; Repasky, E.A. Dissecting the role of hyperthermia in natural killer cell mediated anti-tumor responses. Int. J. Hyperth. 2008, 24, 41–56. [Google Scholar] [CrossRef]
- Muñoz, N.M.; Dupuis, C.; Williams, M.; Dixon, K.; McWatters, A.; Avritscher, R.; Bouchard, R.; Kaseb, A.; Schachtschneider, K.M.; Rao, A.; et al. Molecularly targeted photothermal ablation improves tumor specificity and immune modulation in a rat model of hepatocellular carcinoma. Commun. Biol. 2020, 3, 1–10. [Google Scholar] [CrossRef]
- Lee, S.; Son, B.; Park, G.; Kim, H.; Kang, H.; Jeon, J.; Youn, H.; Youn, B. Immunogenic Effect of Hyperthermia on Enhancing Radiotherapeutic Efficacy. Int. J. Mol. Sci. 2018, 19, 2795. [Google Scholar] [CrossRef] [Green Version]
- DeMaria, O.; Cornen, S.; Daëron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56. [Google Scholar] [CrossRef]
- Pellegrino, T. Process for the Gram-Scale Preparation of Cubic Ferrite Nanocrystals for Biomedical Applications. Patent WO/2020/222133, 5 November 2020. [Google Scholar]
- Guardia, P.; Riedinger, A.; Nitti, S.; Pugliese, G.; Marras, S.; Genovese, A.; Materia, M.E.; Lefevre, C.; Manna, L.; Pellegrino, T. One pot synthesis of monodisperse water soluble iron oxide nanocrystals with high values of the specific absorption rate. J. Mater. Chem. B 2014, 2, 4426–4434. [Google Scholar] [CrossRef]
- Grauer, O.; Jaber, M.; Hess, K.; Weckesser, M.; Schwindt, W.; Maring, S.; Wölfer, J.; Stummer, W. Combined intracavitary thermotherapy with iron oxide nanoparticles and radiotherapy as local treatment modality in recurrent glioblastoma patients. J. Neuro-Oncol. 2019, 141, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachet, M.; Liang, Y.Y.; Oehler, R. The immune response to secondary necrotic cells. Apoptosis 2017, 22, 1189–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, W.; Wu, X.; Ma, S.; Wang, Y.; Nalin, A.P.; Zhu, Z.; Zhang, J.; Benson, D.M.; He, K.; Caligiuri, M.A.; et al. The Mechanism of Anti–PD-L1 Antibody Efficacy against PD-L1–Negative Tumors Identifies NK Cells Expressing PD-L1 as a Cytolytic Effector. Cancer Discov. 2019, 9, 1422–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juneja, V.R.; McGuire, K.A.; Manguso, R.T.; LaFleur, M.W.; Collins, N.; Nicholas Haining, W.; Freeman, G.J.; Sharpe, A.H. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 2017, 214, 895–904. [Google Scholar] [CrossRef]
- Hottinger, A.F.; Pacheco, P.; Stupp, R. Tumor treating fields: A novel treatment modality and its use in brain tumors. Neuro-Oncology 2016, 18, 1338–1349. [Google Scholar] [CrossRef] [Green Version]
- Lok, E.; San, P.; Hua, V.; Phung, M.; Wong, E.T. Analysis of physical characteristics of Tumor Treating Fields for human glioblastoma. Cancer Med. 2017, 6, 1286–1300. [Google Scholar] [CrossRef]
- Maraka, S.; Asmaro, K.; Walbert, T.; Lee, I. Cerebral edema induced by laser interstitial thermal therapy and radiotherapy in close succession in patients with brain tumor. Lasers Surg. Med. 2018, 50, 917–923. [Google Scholar] [CrossRef]
- Aaes, T.L.; Verschuere, H.; Kaczmarek, A.; Heyndrickx, L.; Wiernicki, B.; Delrue, I.; De Craene, B.; Taminau, J.; Delvaeye, T.; Bertrand, M.J.M.; et al. Immunodominant AH1 Antigen-Deficient Necroptotic, but Not Apoptotic, Murine Cancer Cells Induce Antitumor Protection. J. Immunol. 2020, 204, 775–787. [Google Scholar] [CrossRef]
- Sadhukha, T.; Niu, L.; Wiedmann, T.S.; Panyam, J. Effective Elimination of Cancer Stem Cells by Magnetic Hyperthermia. Mol. Pharm. 2013, 10, 1432–1441. [Google Scholar] [CrossRef]
- Spaggiari, G.M.; Contini, P.; Dondero, A.; Carosio, R.; Puppo, F.; Indiveri, F.; Zocchi, M.R.; Poggi, A. Soluble HLA class I induces NK cell apoptosis upon the engagement of killer-activating HLA class I receptors through FasL-Fas interaction. Blood 2002, 100, 4098–4107. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Persano, S.; Vicini, F.; Poggi, A.; Fernandez, J.L.C.; Rizzo, G.M.R.; Gavilán, H.; Silvestri, N.; Pellegrino, T. Elucidating the Innate Immunological Effects of Mild Magnetic Hyperthermia on U87 Human Glioblastoma Cells: An In Vitro Study. Pharmaceutics 2021, 13, 1668. https://doi.org/10.3390/pharmaceutics13101668
Persano S, Vicini F, Poggi A, Fernandez JLC, Rizzo GMR, Gavilán H, Silvestri N, Pellegrino T. Elucidating the Innate Immunological Effects of Mild Magnetic Hyperthermia on U87 Human Glioblastoma Cells: An In Vitro Study. Pharmaceutics. 2021; 13(10):1668. https://doi.org/10.3390/pharmaceutics13101668
Chicago/Turabian StylePersano, Stefano, Francesco Vicini, Alessandro Poggi, Jordi Leonardo Castrillo Fernandez, Giusy Maria Rita Rizzo, Helena Gavilán, Niccolo Silvestri, and Teresa Pellegrino. 2021. "Elucidating the Innate Immunological Effects of Mild Magnetic Hyperthermia on U87 Human Glioblastoma Cells: An In Vitro Study" Pharmaceutics 13, no. 10: 1668. https://doi.org/10.3390/pharmaceutics13101668
APA StylePersano, S., Vicini, F., Poggi, A., Fernandez, J. L. C., Rizzo, G. M. R., Gavilán, H., Silvestri, N., & Pellegrino, T. (2021). Elucidating the Innate Immunological Effects of Mild Magnetic Hyperthermia on U87 Human Glioblastoma Cells: An In Vitro Study. Pharmaceutics, 13(10), 1668. https://doi.org/10.3390/pharmaceutics13101668