
Unveiling the Membrane and Cell Wall Action of Antimicrobial Cyclic Lipopeptides: Modulation of the Spectrum of Activity

 , and
, and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Peptide Synthesis and Purification
2.3. Antibacterial Susceptibility Testing
2.4. Hemolysis Assays
2.5. Transmission Electron Microscopy (TEM) Observation
2.6. Large Unilamellar Vesicles Preparation
2.7. Kinetics of Insertion into Monolayers
2.8. Light Scattering
2.9. Fluorescence Assays for Lipid Mixing
2.10. Vesicle Leakage Experiment
3. Results and Discussion
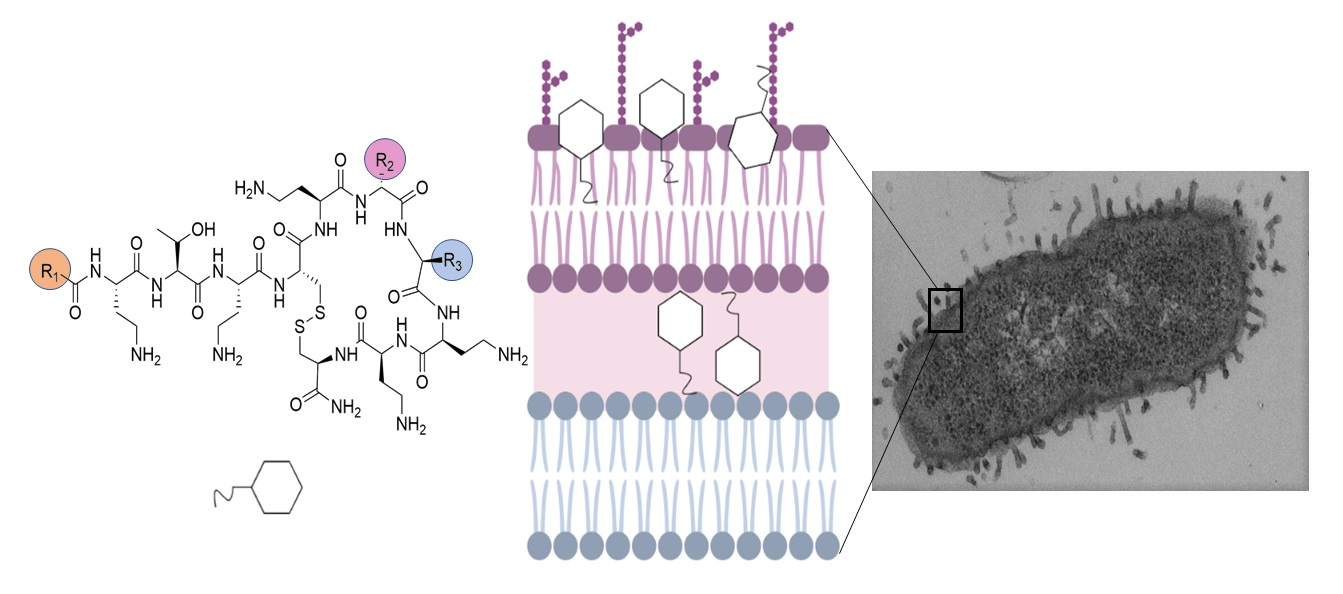
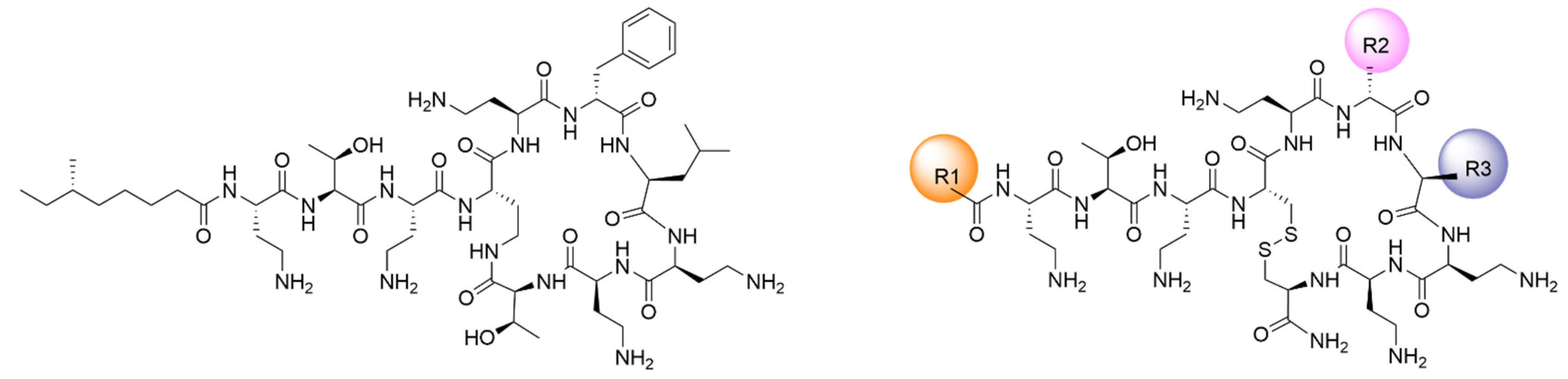
3.1. New Lipopeptides Inspired in Polymyxins with High Antibiotic Activity
3.2. Hemolytic Activity Is Low at the MIC
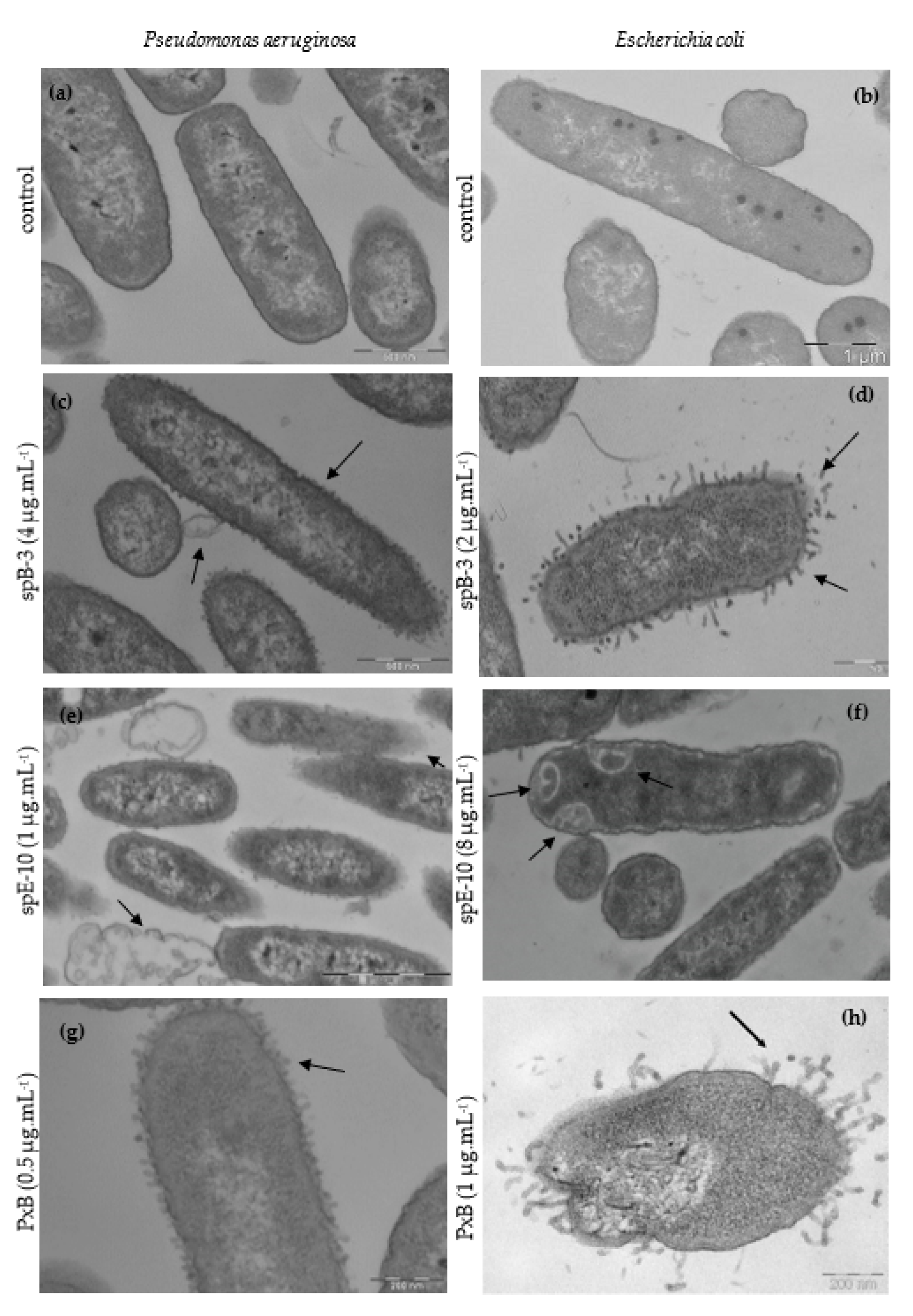
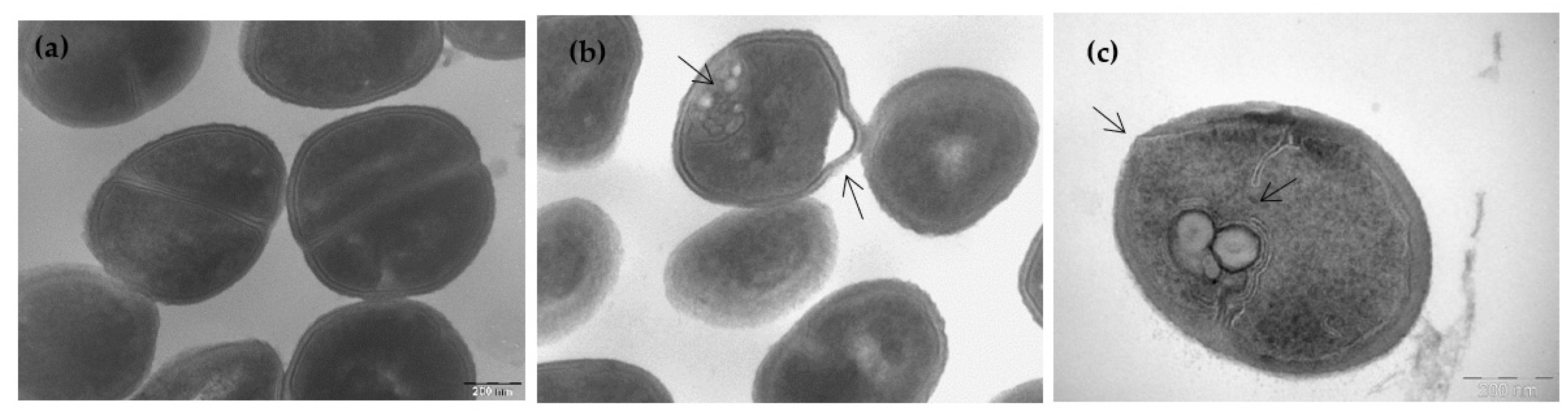
3.3. Ultrastructural Effects of Lipopeptide Treatment on Bacteria
3.4. Binding Affinity of the Lipopeptides Is Increased toward Bacterial-Like Model Membranes
3.5. Lipopeptide-Induced Aggregation and Lipid Mixing Is Selective for Anionic Vesicles
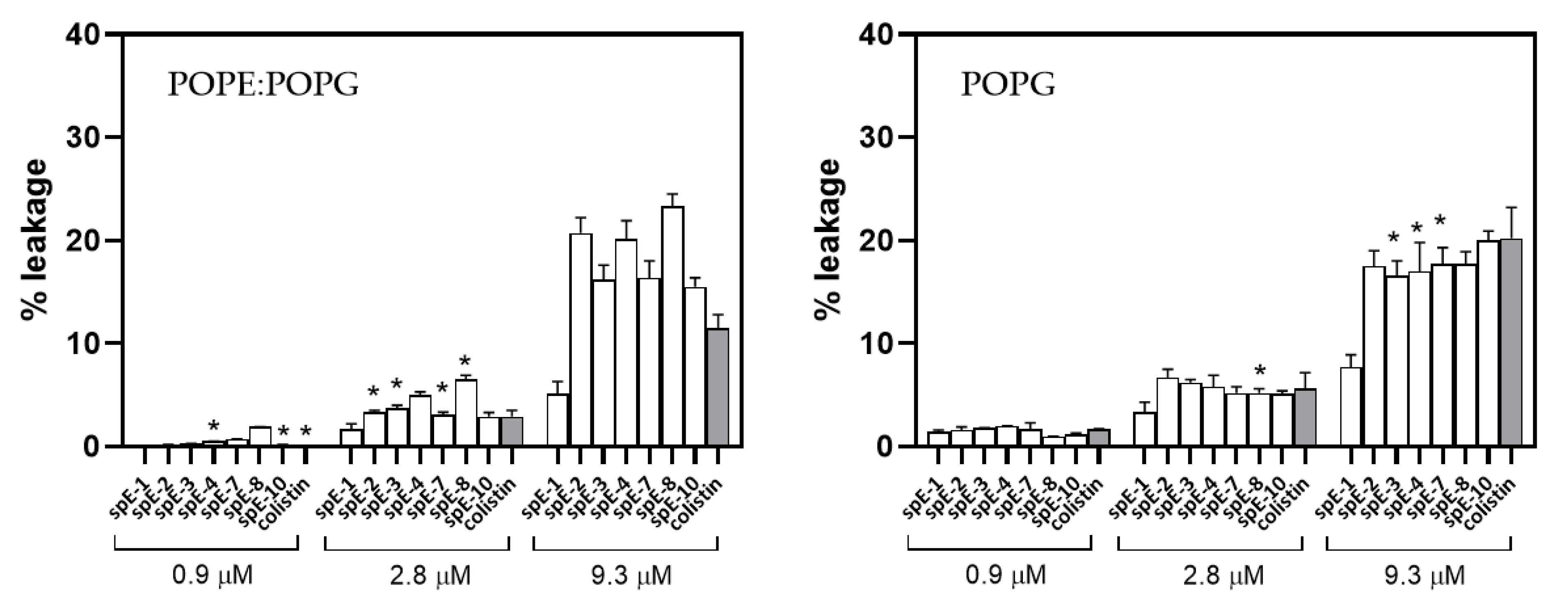
3.6. Leakage of Aqueous Contents Depends on Lipid Composition
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Interagency Coordination Group on Antimicrobial Resistance. No Time to Wait: Securing the Future from Drug-Resistant Infections. Report to the Secretary-General of the United Nations. 2019. Available online: https://www.who.int/antimicrobial-resistance/interagency-coordination-group/IACG_final_report_EN.pdf (accessed on 16 December 2021).
- 2020 Antibacterial Agents in Clinical and Preclinical Development: An Overview and Analysis; World Health Organization: Geneva, Switzerland, 2021; Licence: CC BY-NC-SA 3.0 IGO.
- World Health Organization (WHO). Antimicrobial Resistance: Global Report on Surveillance. 2014. Available online: https://www.who.int/drugresistance/documents/surveillancereport/en/ (accessed on 6 May 2021).
- Plackett, B. No money for new drugs. Nature 2020, 586, S50–S52. [Google Scholar] [CrossRef]
- Rabanal, F.; Cajal, Y. Therapeutic Potential of Antimicrobial Peptides. In New Weapons to Control Bacterial Growth; Villa, T., Vinas, M., Eds.; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Sierra, J.M.; Fusté, E.; Rabanal, F.; Vinuesa, T.; Viñas, M. An overview of antimicrobial peptides and the latest advances in their development. Expert Opin. Biol. Ther. 2017, 17, 663–676. [Google Scholar] [CrossRef]
- Patel, A.; Emerick, M.; Cabunoc, M.K.; Williams, M.H.; Preas, M.A.; Schrank, G.; Rabinowitz, R.; Luethy, P.; Johnson, J.K.; Leekha, S. Rapid Spread and Control of Multidrug-Resistant Gram-Negative Bacteria in COVID-19 Patient Care Units. Emerg. Infect. Dis. 2021, 27, 1234–1237. [Google Scholar] [CrossRef] [PubMed]
- Langford, B.J.; So, M.; Raybardhan, S.; Leung, V.; Westwood, D.; MacFadden, D.R.; Soucy, J.-P.R.; Daneman, N. Bacterial co-infection and secondary infection in patients with COVID-19: A living rapid review and meta-analysis. Clin. Microbiol. Infect. 2020, 26, 1622–1629. [Google Scholar] [CrossRef]
- Vaillancourt, M.; Jorth, P. The unrecognized threat of secondary bacterial infections with COVID-19. mBio 2020, 11, e01806-20. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liao, B.; Cheng, L.; Peng, X.; Xu, X.; Li, Y.; Hu, T.; Li, J.; Zhou, X.; Ren, B. The microbial coinfection in COVID-19. Appl. Microbiol. Biotechnol. 2020, 104, 7777–7785. [Google Scholar] [CrossRef] [PubMed]
- Rex, J.H. ND4BB: Addressing the antimicrobial resistance crisis. Nat. Rev. Microbiol. 2014, 12, 231–232. [Google Scholar] [CrossRef]
- Olliver, M.; Griestop, L.; Hughes, D.; Belfrage, A.K.; Gising, J.; Baranczewski, P.; Vingsbo Lundberg, C.; Karlén, A. ENABLE: An engine for European antibacterial drug discovery and development. Nat. Rev. Drug Discov. 2021, 20, 407–408. [Google Scholar] [CrossRef]
- van Hengel, A.J.; Marin, L. Research, Innovation, and Policy: An Alliance Combating Antimicrobial Resistance. Trends Microbiol. 2019, 27, 287–289. [Google Scholar] [CrossRef]
- Vaara, M. Polymyxins and Their Potential Next Generation as Therapeutic Antibiotics. Front. Microbiol. 2019, 10, 1689. [Google Scholar] [CrossRef]
- Biswas, S.; Brunel, J.M.; Dubus, J.C.; Reynaud-Gaubert, M.; Rolain, J.M. Colistin: An update on the antibiotic of the 21st century. Expert Rev. Anti-Infect. Ther. 2012, 10, 917–934. [Google Scholar] [CrossRef]
- Rabanal, F.; Cajal, Y. Recent advances and perspectives in the design and development of polymyxins. Nat. Prod. Rep. 2017, 34, 886–908. [Google Scholar] [CrossRef]
- Dubashynskaya, N.V.; Skorik, Y.A. Polymyxin Delivery Systems: Recent Advances and Challenges. Pharmaceuticals 2020, 13, 83. [Google Scholar] [CrossRef]
- Gallardo-Godoy, A.; Hansford, K.A.; Muldoon, G.; Becker, B.; Elliott, A.G.; Huang, J.X.; Pelingon, R.; Butler, M.S.; Blaskovich, M.A.T.; Cooper, M.A. Structure-function studies of polymyxin B lipononapeptides. Molecules 2019, 24, 553. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Patil, N.A.; Azad, M.A.K.; Wickremasinghe, H.; Yu, H.; Zhao, J.; Zhang, X.; Li, M.; Gong, B.; Wan, L.; et al. A novel chemical biology and computational approach to expedite the discovery of new-generation polymyxins against life-threatening Acinetobacter baumannii. Chem. Sci. 2021, 12, 12211. [Google Scholar] [CrossRef]
- Cajal, Y.; Rogers, J.; Berg, O.; Jain, M.K. Intermembrane molecular contacts by polymyxin B mediate exchange of phospholipids. Biochemistry 1996, 35, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Cajal, Y.; Ghanta, J.; Easwaran, K.; Surolia, A.; Jain, M.K. Specificity for the exchange of phospholipids through polymyxin B mediated intermembrane molecular contacts. Biochemistry 1996, 35, 5684–5695. [Google Scholar] [CrossRef]
- Oh, J.T.; Van Dyk, T.K.; Cajal, Y.; Dhurjati, P.S.; Sasser, M.; Jain, M.K. Osmotic stress in viable Escherichia coli as the basis for the antibiotic response to polymyxin B. Biochem. Biophys. Res. Commun. 1998, 246, 619–623. [Google Scholar] [CrossRef]
- Oh, J.T.; Cajal, Y.; Skowronska, E.M.; Belkin, S.; Chen, J.; Van Dyk, T.K.; Sasser, M.; Jain, M.K. Cationic peptide antimicrobials induce selective transcription of micF and osmY in Escherichia coli. Biochim. Biophys. Acta 2000, 1463, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Daugelavicius, R.; Bakiene, E.; Bamford, D.H. Stages of polymyxin B interaction with the Escherichia coli cell envelope. Antimicrob. Agents Chemother. 2000, 44, 2969–2978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd El-Gawad, M.; Zhong, L.L.; Shen, C.; Yang, Y.; Doi, Y.; Tian, G.B. Colistin and its role in the era of antibiotic resistance: An extended review (2000–2019). Emerg. Microbes Infect. 2020, 9, 868–885. [Google Scholar] [CrossRef] [Green Version]
- Rabanal, F.; Grau-Campistany, A.; Vila-Farrés, X.; Gonzalez-Linares, J.; Borràs, M.; Vila, J.; Manresa, A.; Cajal, Y. A bioinspired peptide scaffold with high antibiotic activity and low in vivo toxicity. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Grau-Campistany, A.; Manresa, A.; Pujol, M.; Rabanal, F.; Cajal, Y. Tryptophan-containing lipopeptide antibiotics derived from polymyxin B with activity against Gram positive and Gram negative bacteria. Biochem. Biophys. Acta-Biomembr. 2016, 1858, 333–343. [Google Scholar] [CrossRef]
- Grau-Campistany, A.; Pujol, M.; Marqués, A.M.; Manresa, A.; Rabanal, F.; Cajal, Y. Membrane interaction of a new synthetic antimicrobial lipopeptide sp-85 with broad spectrum activity. Colloids Surf. A 2015, 480, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Clausell, A.; Rabanal, F.; Garcia-Subirats, M.; Alsina, M.A.; Cajal, Y. Membrane association and contact formation by a synthetic analog of polymyxin B and its fluorescent derivatives. J. Phys. Chem. B 2006, 110, 4465–4471. [Google Scholar] [CrossRef]
- Velkov, T.; Thompson, P.E.; Nation, R.L.; Li, J. Structure-activity relationships of polymyxin antibiotics. J. Med. Chem. 2010, 53, 1898–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, M.; Iida, K.; Matsuo, T. Electron microscopic studies on mode of action of polymyxin. J. Bacteriol. 1969, 97, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, M.; Berditsch, M.; Hawecker, J.; Fotouhi-Ardakani, M.; Gerthsen, D.; Ulrich, A.S. Damage of the bacterial cell envelope by antimicrobial peptides Gramicidin S and PGLa as revealed by transmission and scanning electron microscopy. Antimicrob. Agents Chemother. 2010, 54, 3132–3142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velkov, T.; Roberts, K.D.; Nation, R.L.; Wang, J.; Thompson, P.E.; Li, J. Teaching ‘Old’ Polymyxins New Tricks: New-Generation Lipopeptides Targeting Gram-Negative ‘Superbugs’. ACS Chem. Biol. 2014, 9, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Santhana, L.R.; Hing, H.L.; Baharudin, O.; Hamidah, Z.-T.; Suhana, R.A.; Nor Asiha, C.P.; Vimala, B.; Paramsarvaran, S.; Sumarni, G.; Hanjeet, K. Mesosomes are a definite event in antibiotic-treated Staphylococcus aureus ATCC 25923. Trop. Biomed. 2007, 24, 105–109. [Google Scholar]
- Andrä, J.; Goldmann, T.; Ernst, C.M.; Peschel, A.; Gutsmann, T. Multiple peptide resistance factor (MprF)-mediated resistance of Staphylococcus aureus against antimicrobial peptides coincides with a modulated peptide interaction with artificial membranes comprising Lysyl-phosphatidylglycerol. JBC 2011, 286, 18692–18700. [Google Scholar] [CrossRef] [Green Version]
- Shimoda, M.; Ohki, K.; Shimamoto, Y.; Kohashi, O. Morphology of defensin-treated Staphylococcus aureus. Infect. Immun. 1995, 63, 2886–2891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigoreva, A.; Bardasheva, A.; Tupitsyna, A.; Amirkhanov, N.; Tikunova, N.; Pyshnyi, D.; Ryabchikova, E. Changes in the ultrastructure of Staphylococcus aureus treated with cationic peptides and chlorhexidine. Microorganisms 2020, 8, 1991. [Google Scholar] [CrossRef] [PubMed]
- Marsh, D. Lateral pressure in membranes. Biochim. Biophys. Acta Biomembr. 1996, 1286, 163–223. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Yu, Z.; Qin, W.; Lin, J.; Fang, S.; Qiu, J. Antibacterial mechanisms of polymyxin and bacterial resistance. BioMed Res. Int. 2015, 679109. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Rotem, S.; Mor, A.; Berno, B.; Epand, R.F. Bacterial membranes as predictors of antimicrobial potency. J. Am. Chem. Soc. 2008, 130, 14346–14352. [Google Scholar] [CrossRef] [PubMed]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Wölk, C.; Youssef, H.; Guttenberg, T.; Marbach, H.; Vizcay-Barrena, G.; Shen, C.; Brezesinski, G.; Harvey, R.D. Phase diagram for a lysyl-phosphatidylglycerol analog in biomimetic mixed monolayers with phosphatidylglycerol: Insights into the tunable properties of bacterial membranes. ChemPhysChem 2020, 21, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Faust, J.E.; Yang, P.-Y.; Huang, H.W. Action of Antimicrobial Peptides on Bacterial and Lipid Membranes: A Direct Comparison. Biophys. J. 2017, 112, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- CDC’s Antibiotic Resistance Threats in the United States. 2019 (2019 AR Threats Report). Available online: https://www.cdc.gov/drugresistance/biggest-threats.html#pse (accessed on 3 August 2021).
- Outbreak of VIM-Producing Carbapenem-Resistant Pseudomonas aeruginosa Linked to Medical Tourism to Mexico. 2019. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/27-02-2019-RRA-Pseudomonas%20aeruginosa%2C%20Carbapenems%2C%20Surgical%20site%20infection-Mexico-final%20for%20web.pdf. (accessed on 3 August 2021).
- María Díez-Aguilar, M.; Hernández-García, M.; Morosini, M.I.; Fluit, A.; Tunney, M.M.; Huertas, N.; del Campo, R.; Obrecht, D.; Bernardini, F.; Ekkelenkamp, M.; et al. Murepavadin antimicrobial activity against and resistance development in cystic fibrosis Pseudomonas aeruginosa isolates. J. Antimicrob. Chemother. 2021, 76, 984–992. [Google Scholar] [CrossRef] [PubMed]





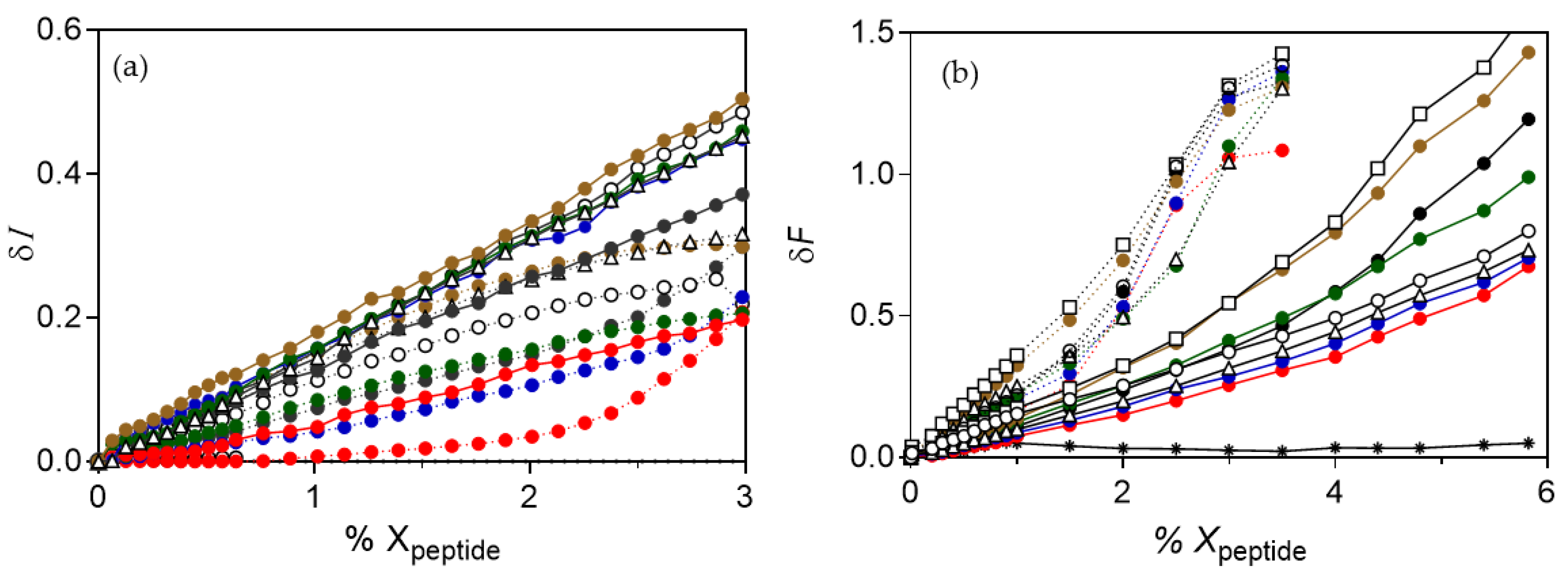
 ), spE-2 (
), spE-2 (  ), spE-3 (
), spE-3 (  ), spE-4 (
), spE-4 (  ), spE-7 (△), spE-8 (◻), spE-10 (◯), or PxE (
), spE-7 (△), spE-8 (◻), spE-10 (◯), or PxE (  ). In all cases, no representative changes were detected with POPC vesicles, as shown in (b) for PxE (∗). Each data is the mean of three independent experiments, with error being always below 10%.
), spE-2 ( ), spE-3 ( ), spE-4 ( ), spE-7 (△), spE-8 (◻), spE-10 (◯), or PxE ( ). In all cases, no representative changes were detected with POPC vesicles, as shown in (b) for PxE (∗). Each data is the mean of three independent experiments, with error being always below 10%.
). In all cases, no representative changes were detected with POPC vesicles, as shown in (b) for PxE (∗). Each data is the mean of three independent experiments, with error being always below 10%.
), spE-2 ( ), spE-3 ( ), spE-4 ( ), spE-7 (△), spE-8 (◻), spE-10 (◯), or PxE ( ). In all cases, no representative changes were detected with POPC vesicles, as shown in (b) for PxE (∗). Each data is the mean of three independent experiments, with error being always below 10%.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | MIC/µg mL−1 (µM) | ||||
|---|---|---|---|---|---|
| EC 1 | PA | SA | |||
| Series I-PxB | spB-1 | hexanoyl-Dab-Thr-Dab-Cys-Dab-Phe-Leu-Dab-Dab-Cys | >32 (>27) | >32 (>27) | >32 (>27) |
| spB-2 | octanoyl-Dab-Thr-Dab-Cys-Dab-Phe-Leu-Dab-Dab-Cys | 16 (13) | 8 (6.6) | >32 (26) | |
| spB-3 | decanoyl-Dab-Thr-Dab-Cys-Dab-Phe-Leu-Dab-Dab-Cys | 2 (1.6) | 4 (3.2) | 8 (6.5) | |
| spB-4 | dodecanoyl-Dab-Thr-Dab-Cys-Dab-Phe-Leu-Dab-Dab-Cys | 16 (13) | 8 (6.3) | 8 (6.3) | |
| Series II-PxB | spB-5 | octanoyl-Dab-Thr-Dab-Cys-Dab-Phe-Nle-Dab-Dab-Cys | 2 (1.7) | 4 (3.3) | >32 (>26) |
| spB-6 | nonanoyl-Dab-Thr-Dab-Cys-Dab-Phe-Nle-Dab-Dab-Cys | 2 (1.6) | 1 (0.82) | 16 (13) | |
| spB-7 | decanoyl-Dab-Thr-Dab-Cys-Dab-Phe-Nle-Dab-Dab-Cys | 2 (1.6) | 1 (0.81) | 4 (3.2) | |
| spB-8 | dodecanoyl-Dab-Thr-Dab-Cys-Dab-Phe-Nle-Dab-Dab-Cys | 4 (3.2) | 1 (0.79) | 2 (1.58) | |
| spB-9 | dodecanoyl-Dab-Thr-Dab-Cys-Dab-Phe-Nle-Arg-Dab-Cys | 4 (3.0) | 4 (3.0) | 2 (1.5) | |
| Series I-PxE | spE-1 | hexanoyl-Dab-Thr-Dab-Cys-Dab-Leu-Leu-Dab-Dab-Cys | >32 (>28) | >32 (>28) | >32 (>28) |
| spE-2 | octanoyl-Dab-Thr-Dab-Cys-Dab-Leu-Leu-Dab-Dab-Cys | 16 (14) | 4 (3.4) | >32 (>27) | |
| spE-3 | decanoyl-Dab-Thr-Dab-Cys-Dab-Leu-Leu-Dab-Dab-Cys | 8 (6.7) | 2 (1.7) | 8 (6.7) | |
| spE-4 | dodecanoyl-Dab-Thr-Dab-Cys-Dab-Leu-Leu-Dab-Dab-Cys | 8 (6.5) | 1 (0.81) | 8 (6.5) | |
| Series II-PxE | spE-7 | decanoyl-Dab-Thr-Dab-Cys-Dab-Nle-Nle-Dab-Dab-Cys | 2 (1.7) | 2 (1.7) | 8 (6.7) |
| spE-8 | dodecanoyl-Dab-Thr-Dab-Cys-Dab-Nle-Nle-Dab-Dab-Cys | 4 (3.3) | 4 (3.3) | 4 (3.3) | |
| spE-10 | heptanoyl-Dab-Thr-Dab-Cys-Dab-Aoc-Nle-Dab-Dab-Cys | 8 (6.7) | 1 (0.84) | >32 (>27) | |
| Controls | PxB | R1-Dab-Thr-Dab-Dab-Dab-Phe-Leu-Dab-Dab-Thr | 1 (0.83) | 0.5 (0.42) | >32 (>27) |
| PxE | R1-Dab-Thr-Dab-Dab-Dab-Leu-Leu-Dab-Dab-Thr | 1 (0.86) | 0.5 (0.43) | >32 (>27) | |
| Series I-PxB | Series II-PxB | Series I-PxE | Series II-PxE | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Composition * | spB-1 | spB-2 | spB-3 | spB-4 | spB-5 | spB-6 | spB-7 | spB-8 | spB-9 | spE-1 | spE-2 | spE-3 | spE-4 | spE-7 | spE-8 | spE-10 | PxB /PxE |
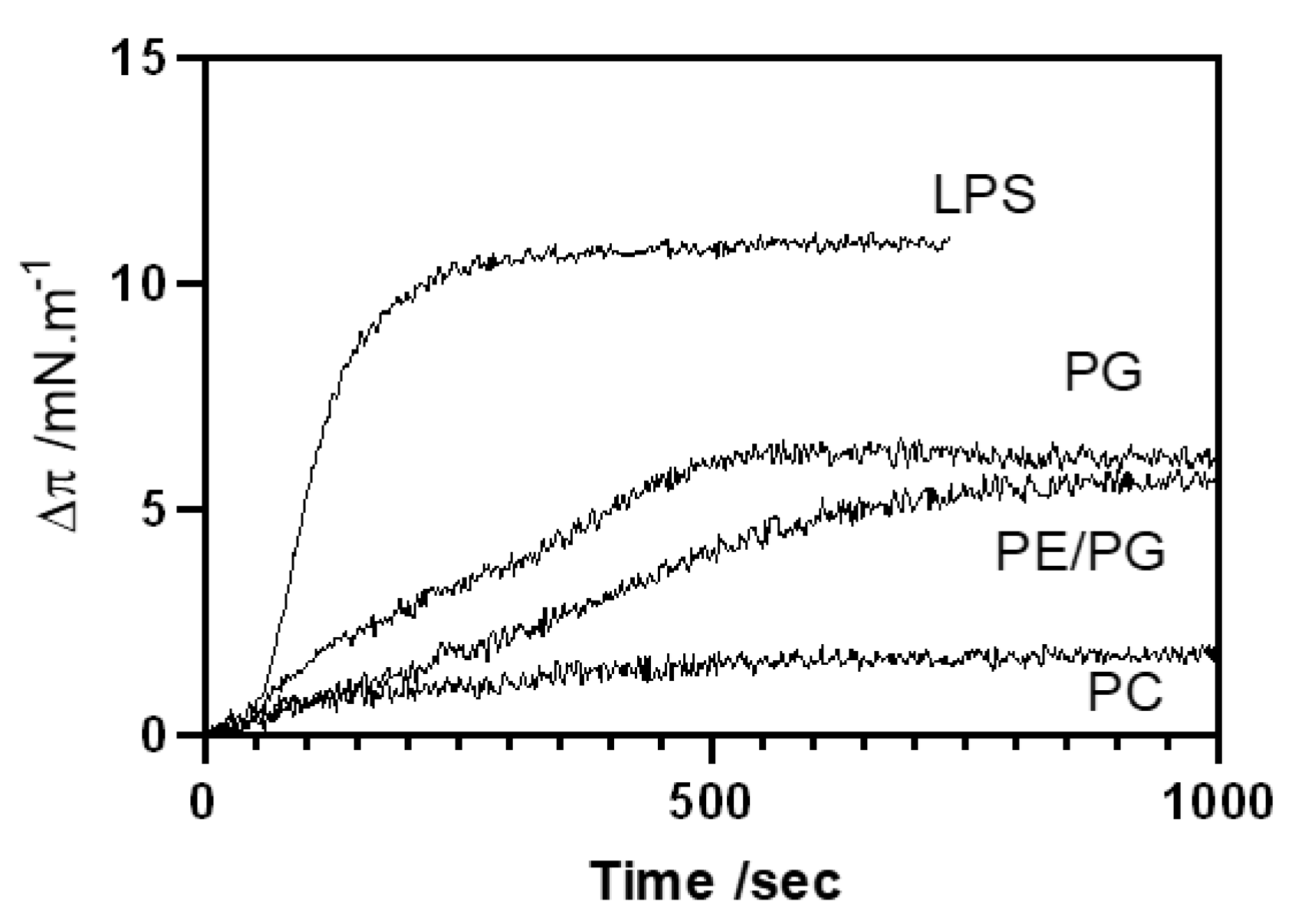
| LPS | 12.1 | 12.6 | 11.0 | 12.2 | 10.6 | 10.8 | 9.3 | 11.2 | 11.1 | 10.6 | 9.5 | 10.0 | 11.4 | 14.2 | 12.5 | 14.8 | 11.4/11.1 |
| POPE/POPG | 4.6 | 5.5 | 6.0 | 5.0 | 6.5 | 5.0 | 6.5 | 6.2 | 6.0 | 4.1 | 6.4 | 7.9 | 9.1 | 6.8 | 9.8 | 8.2 | 7.2/7.9 |
| POPG | 5.7 | 7.0 | 6.2 | 6.7 | 7.3 | 6.0 | 4.2 | 3.8 | 3.4 | 2.8 | 4.3 | 5.7 | 5.8 | 8.2 | 7.4 | 0.8 | 5.9/5.3 |
| POPC | 0.9 | 1.8 | 2.0 | 2.5 | 0.8 | 0.4 | 2.1 | 1.7 | 2.2 | 0.3 | 0.6 | 1.3 | 2.1 | 1.4 | 1.3 | 1.8 | 1.4/0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segovia, R.; Solé, J.; Marqués, A.M.; Cajal, Y.; Rabanal, F. Unveiling the Membrane and Cell Wall Action of Antimicrobial Cyclic Lipopeptides: Modulation of the Spectrum of Activity. Pharmaceutics 2021, 13, 2180. https://doi.org/10.3390/pharmaceutics13122180
Segovia R, Solé J, Marqués AM, Cajal Y, Rabanal F. Unveiling the Membrane and Cell Wall Action of Antimicrobial Cyclic Lipopeptides: Modulation of the Spectrum of Activity. Pharmaceutics. 2021; 13(12):2180. https://doi.org/10.3390/pharmaceutics13122180
Chicago/Turabian StyleSegovia, Roser, Judith Solé, Ana Maria Marqués, Yolanda Cajal, and Francesc Rabanal. 2021. "Unveiling the Membrane and Cell Wall Action of Antimicrobial Cyclic Lipopeptides: Modulation of the Spectrum of Activity" Pharmaceutics 13, no. 12: 2180. https://doi.org/10.3390/pharmaceutics13122180
APA StyleSegovia, R., Solé, J., Marqués, A. M., Cajal, Y., & Rabanal, F. (2021). Unveiling the Membrane and Cell Wall Action of Antimicrobial Cyclic Lipopeptides: Modulation of the Spectrum of Activity. Pharmaceutics, 13(12), 2180. https://doi.org/10.3390/pharmaceutics13122180