
Effects of p-Cresol on Oxidative Stress, Glutathione Depletion, and Necrosis in HepaRG Cells: Comparisons to Other Uremic Toxins and the Role of p-Cresol Glucuronide Formation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. HepaRG Cell Maintenance and Differentiation
2.3. HepaRG Cell Treatment
2.4. DCF Assay for the Measurement of Cellular Oxidative Stress
2.5. GSH Assay for the Measurement of Total Cellular Glutathione Concentration
2.6. LDH Assay for the Measurement of Cellular Necrosis
2.7. Quantification of p-Cresol Sulfate and p-Cresol Glucuronide Concentrations in HepaRG Cell Culture
2.8. Quantification of p-Cresol Concentrations in HepaRG Cell Culture
2.9. Statistical Analysis
3. Results
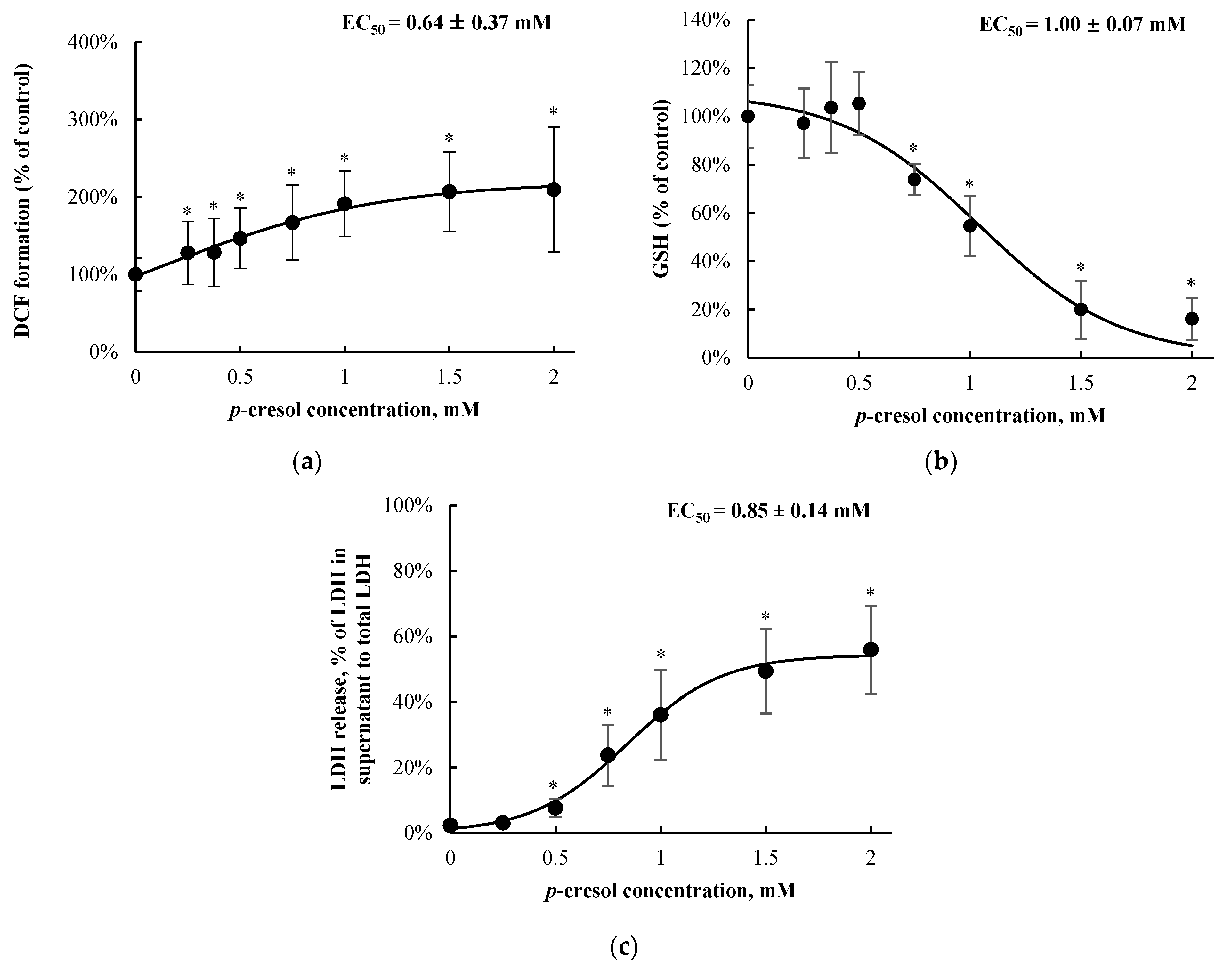
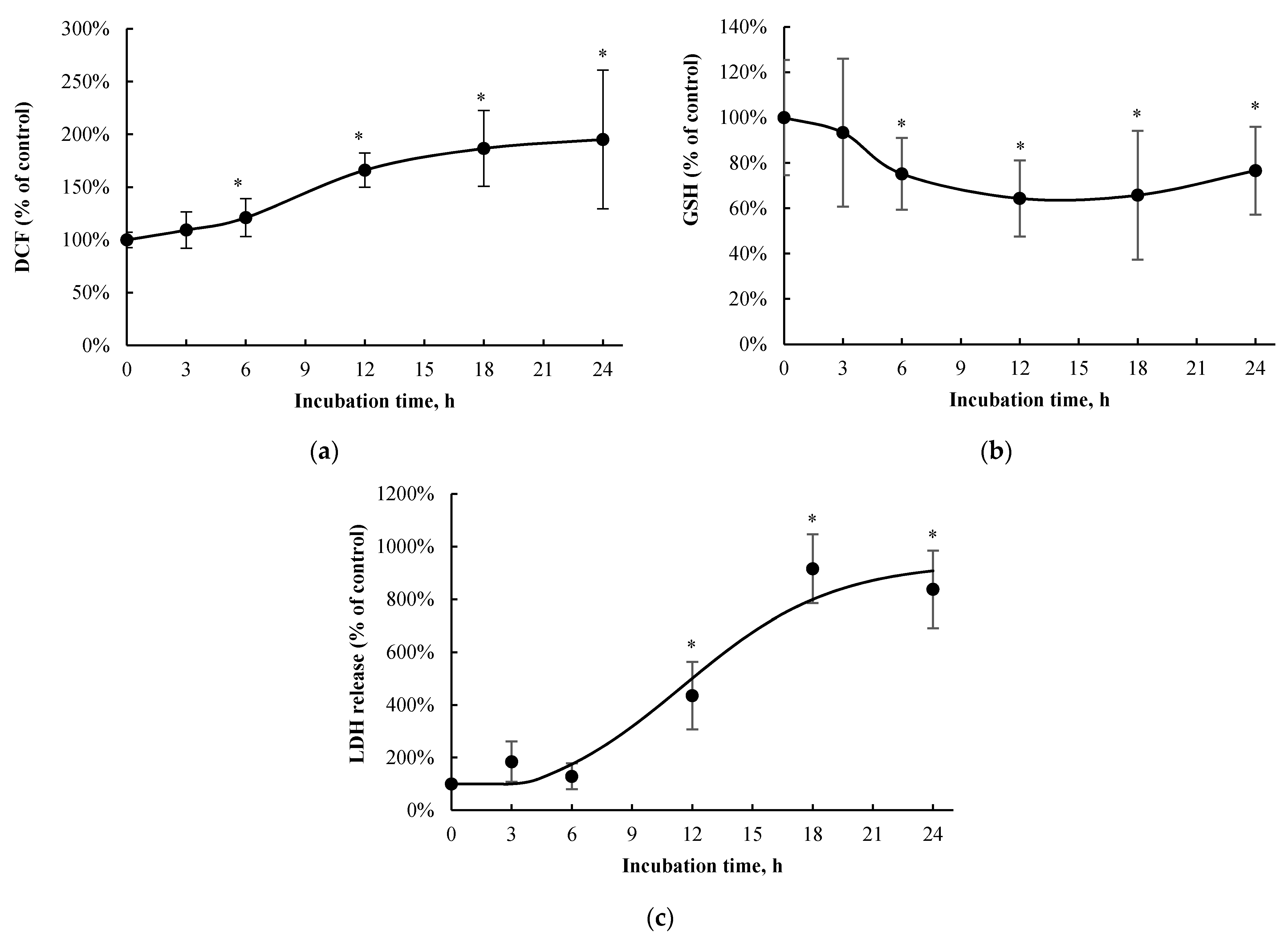
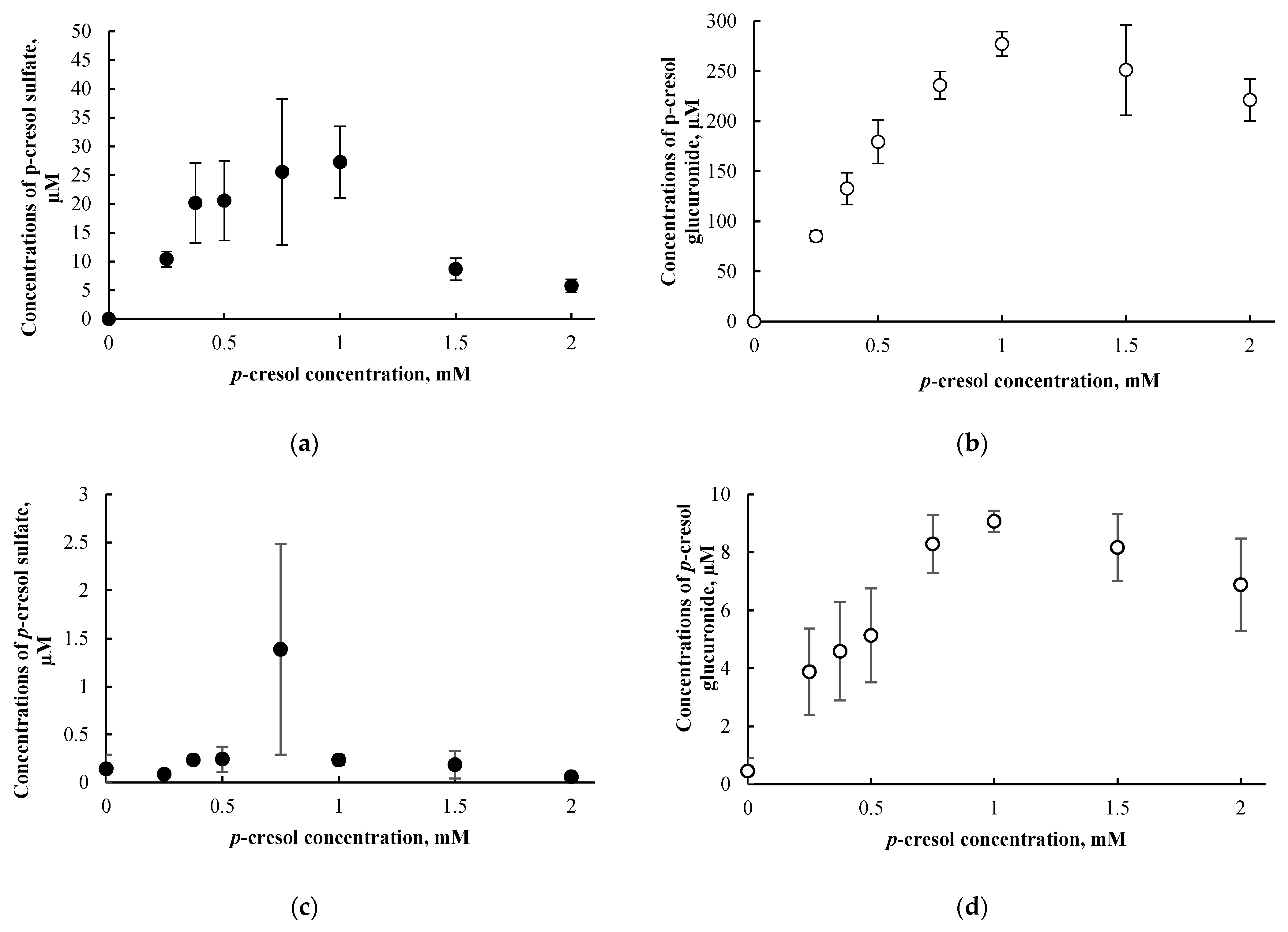
3.1. Positive Control and Concentration/Time-Course Responses of p-Cresol in HepaRG Cells
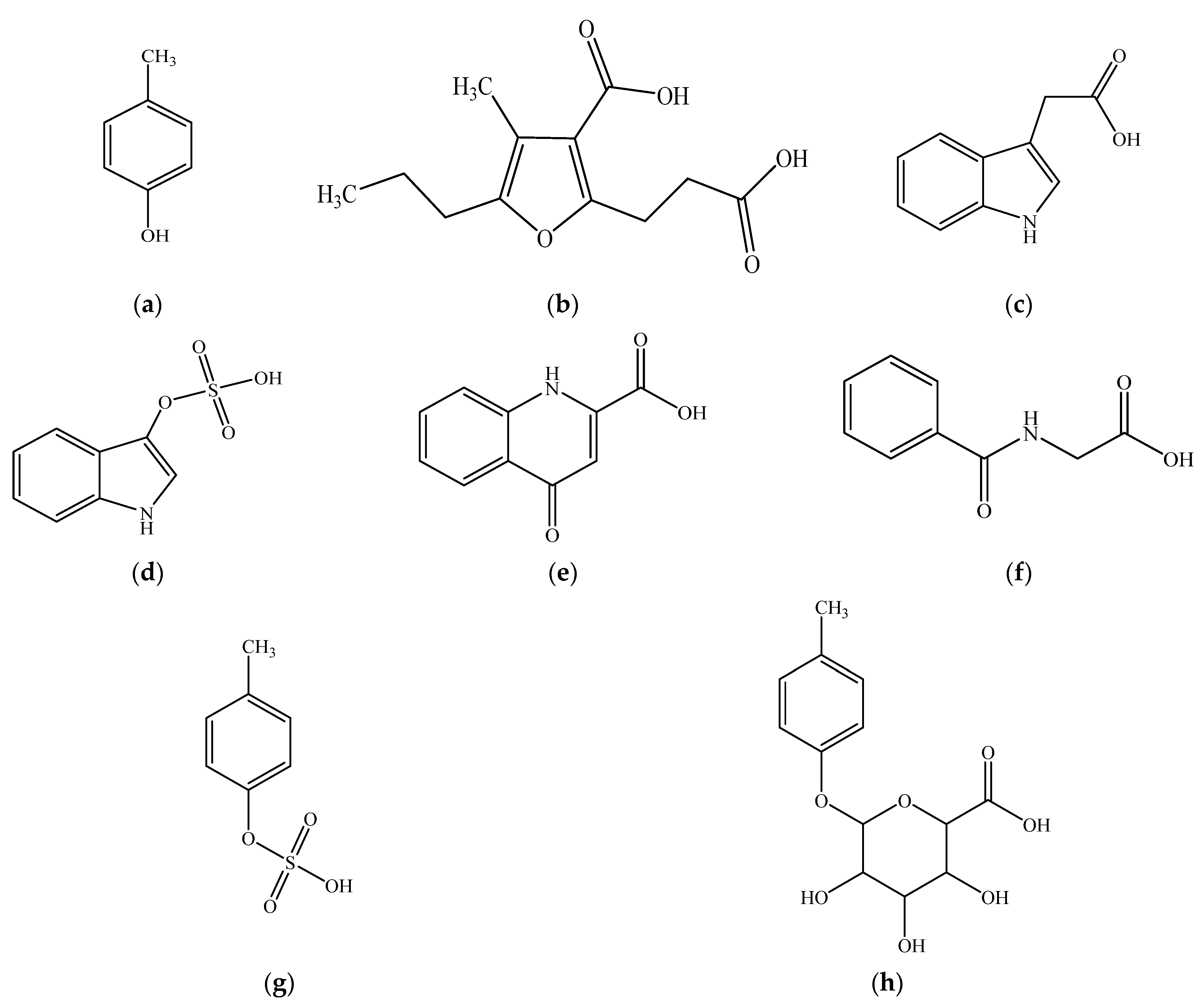
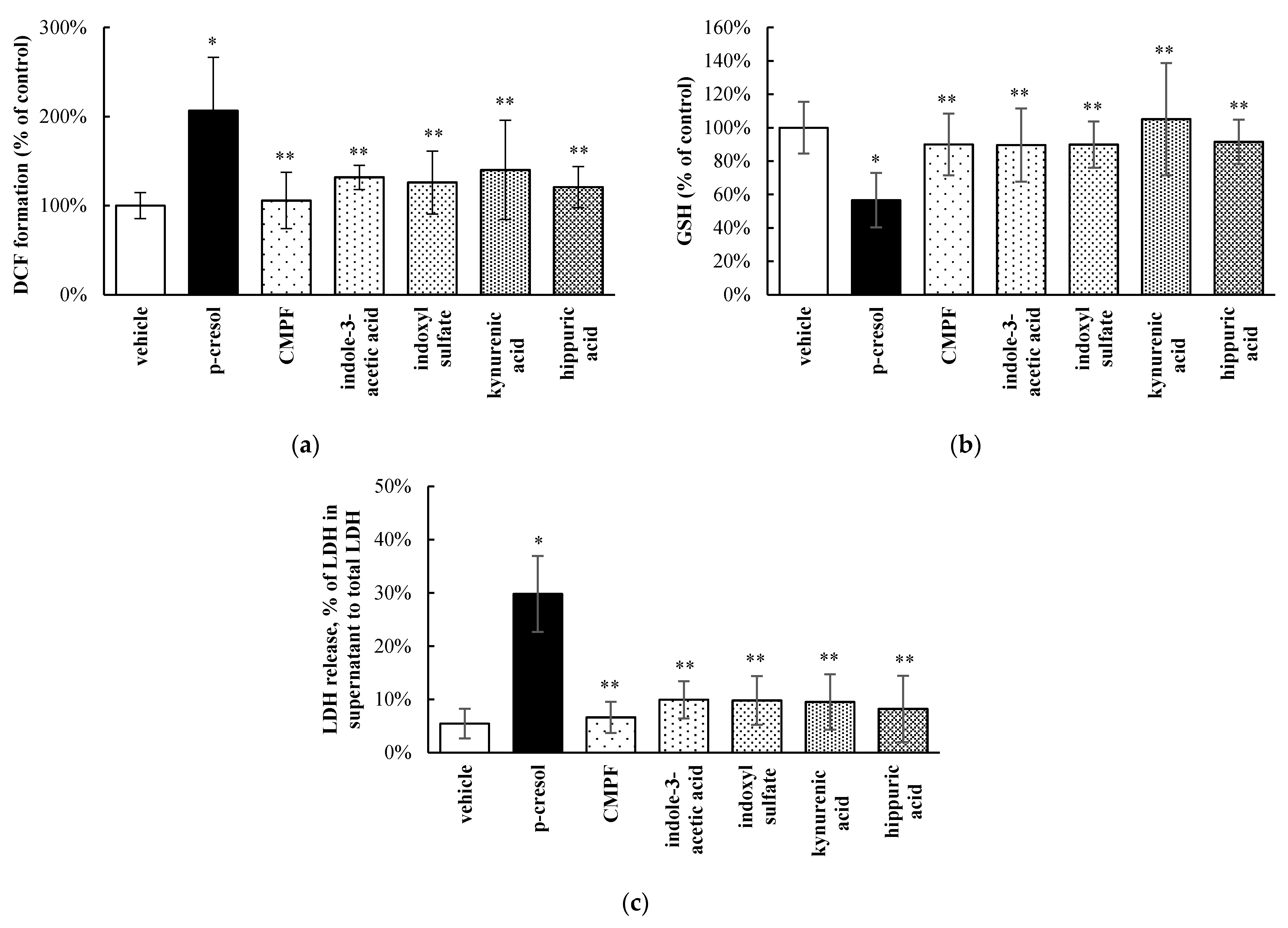
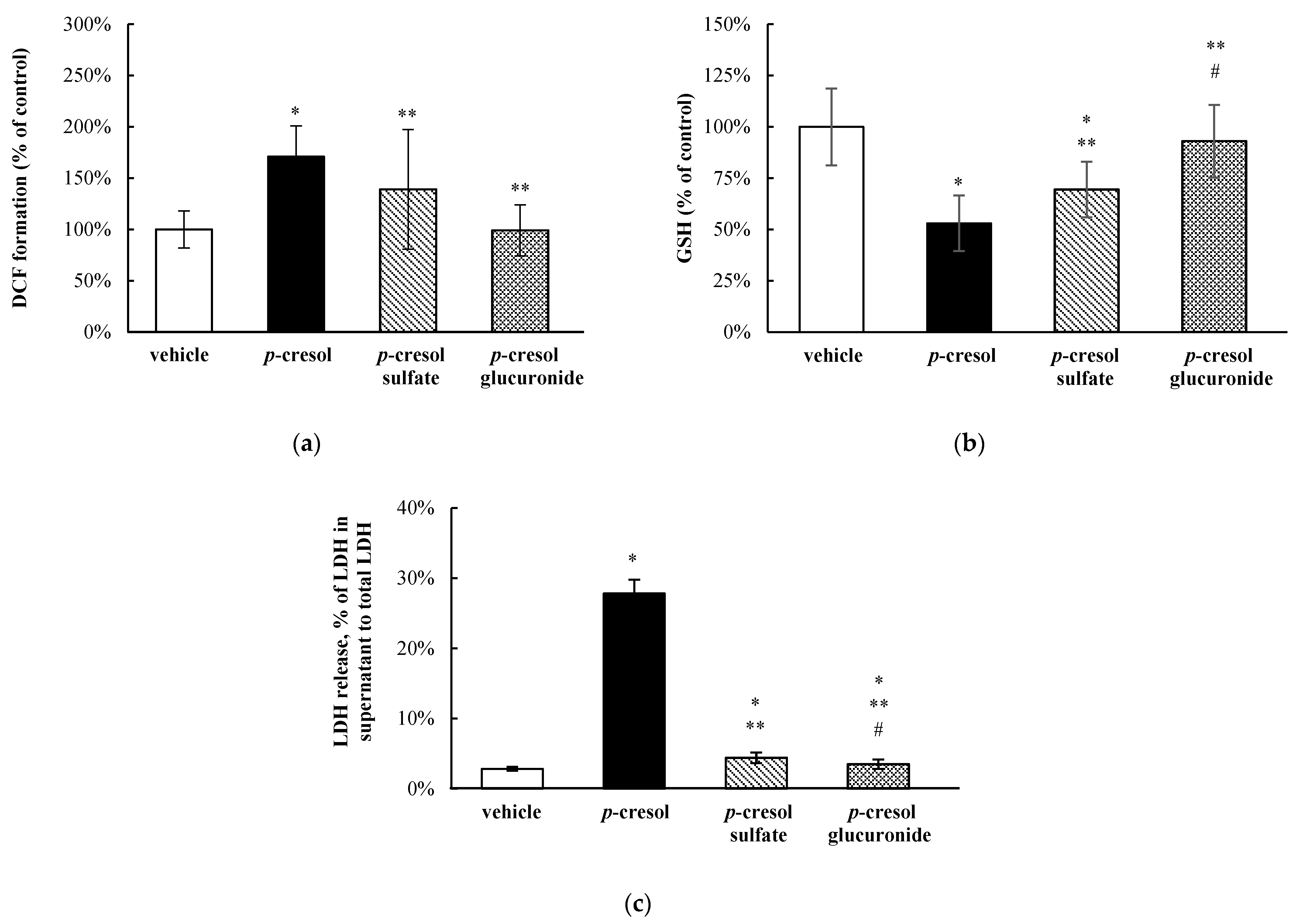
3.2. Relative Toxic Effects of p-Cresol in Comparison to Other Uremic Toxins in HepaRG Cells
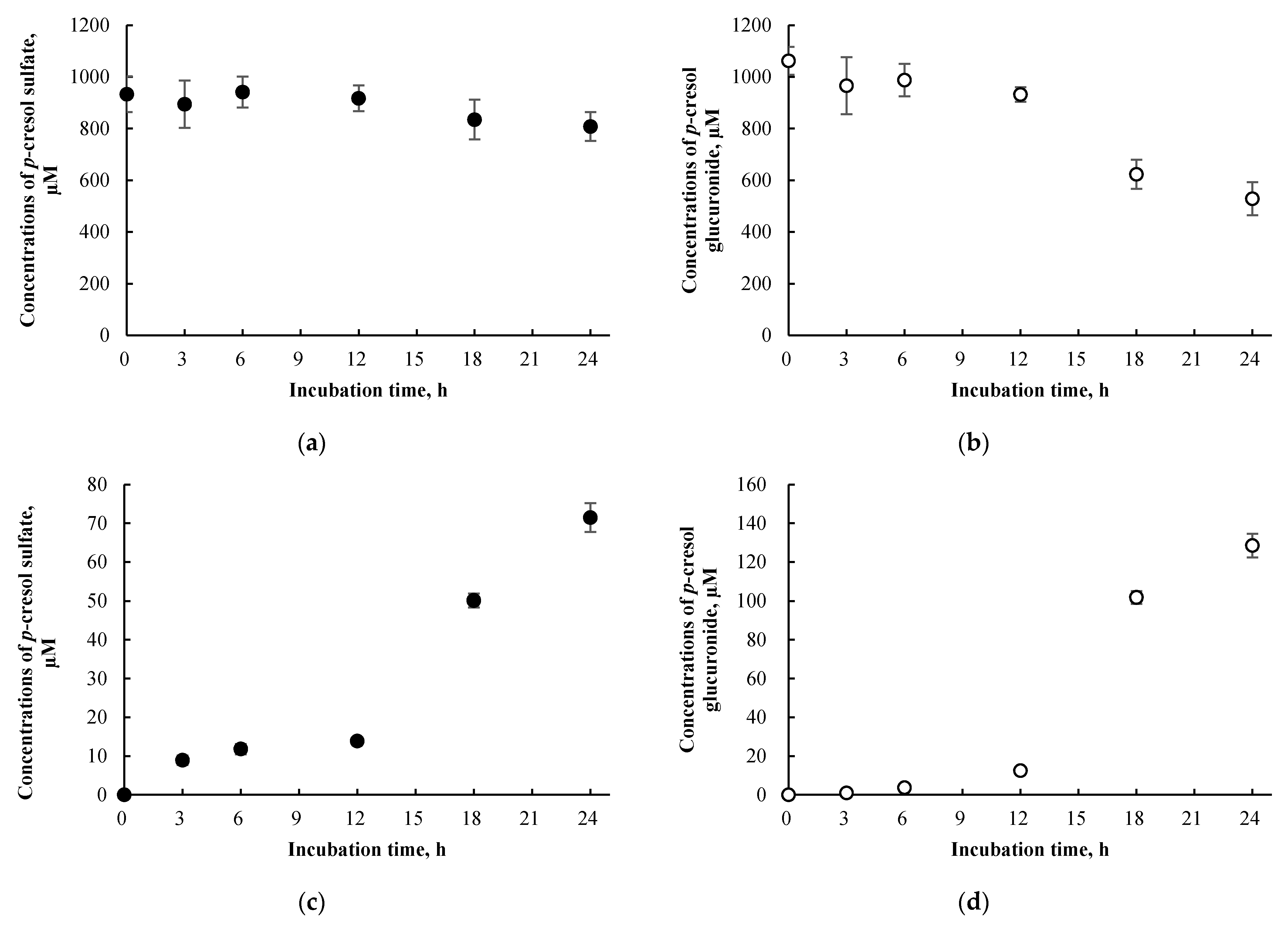
3.3. p-Cresol Sulfate and Glucuronide Concentrations in HepaRG Cells Treated with p-Cresol
3.4. Relative Toxic Effects of p-Cresol and p-Cresol Conjugated Metabolites
3.5. p-Cresol Sulfate and Glucuronide Concentrations in HepaRG cells Treated Exogenously with p-Cresol Sulfate and p-Cresol Glucuronide
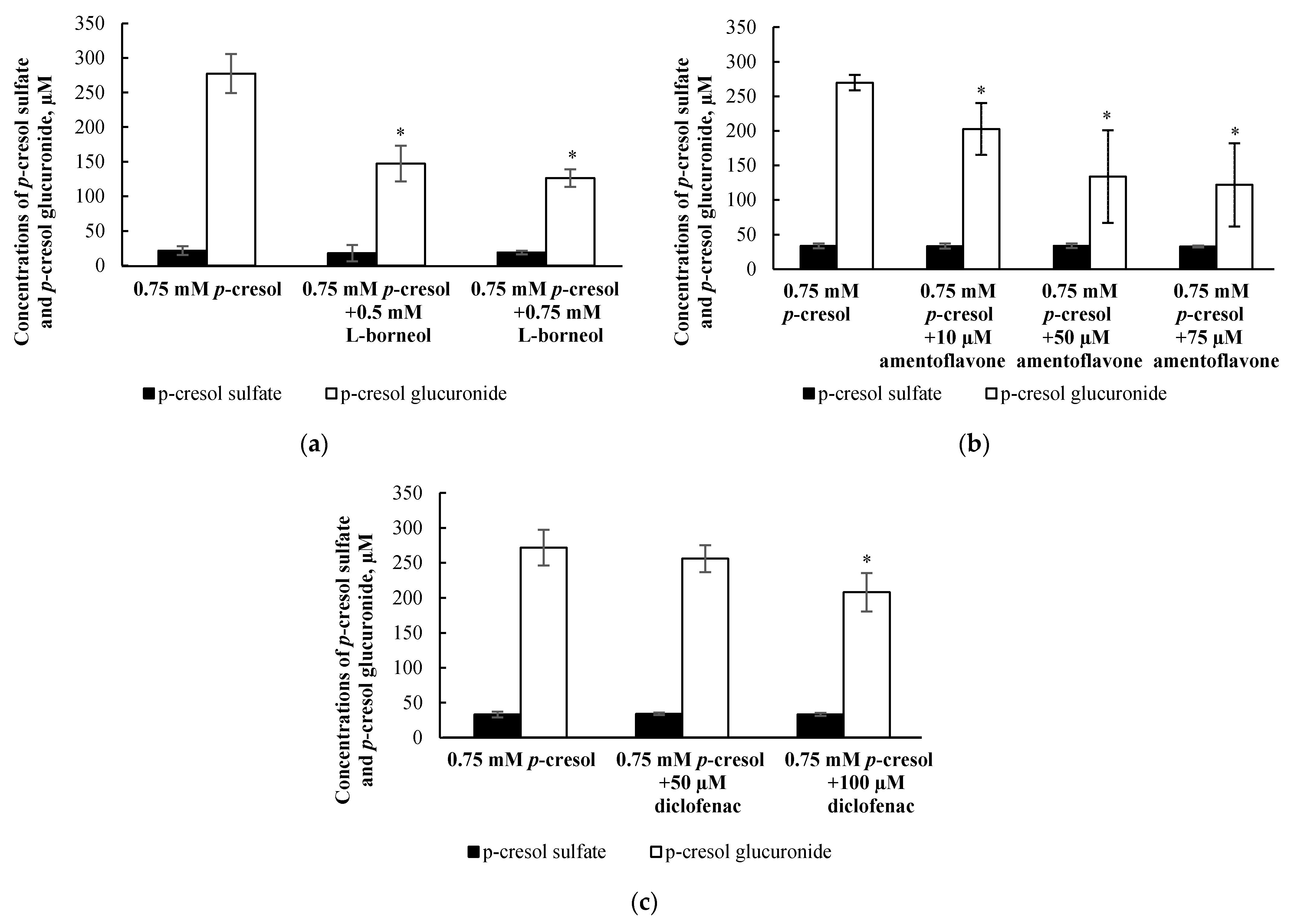
3.6. Determination of Conditions for the Selective Attenuation of p-Cresol Glucuronide Formation in HepaRG Cells Using Chemical Inhibitors
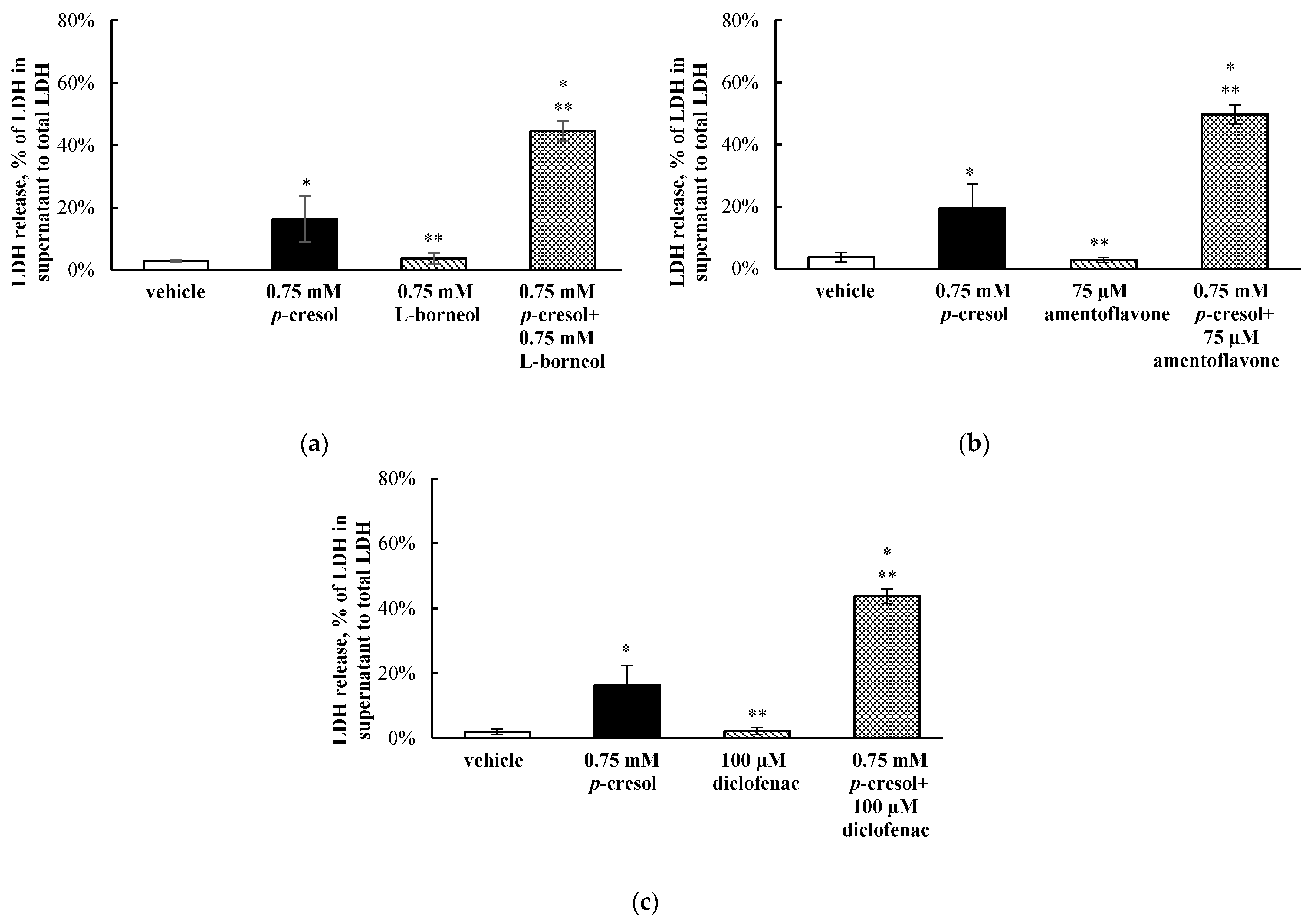
3.7. Effects of l-Borneol, Amentoflavone, or Diclofenac on p-Cresol Generated Cellular Necrosis
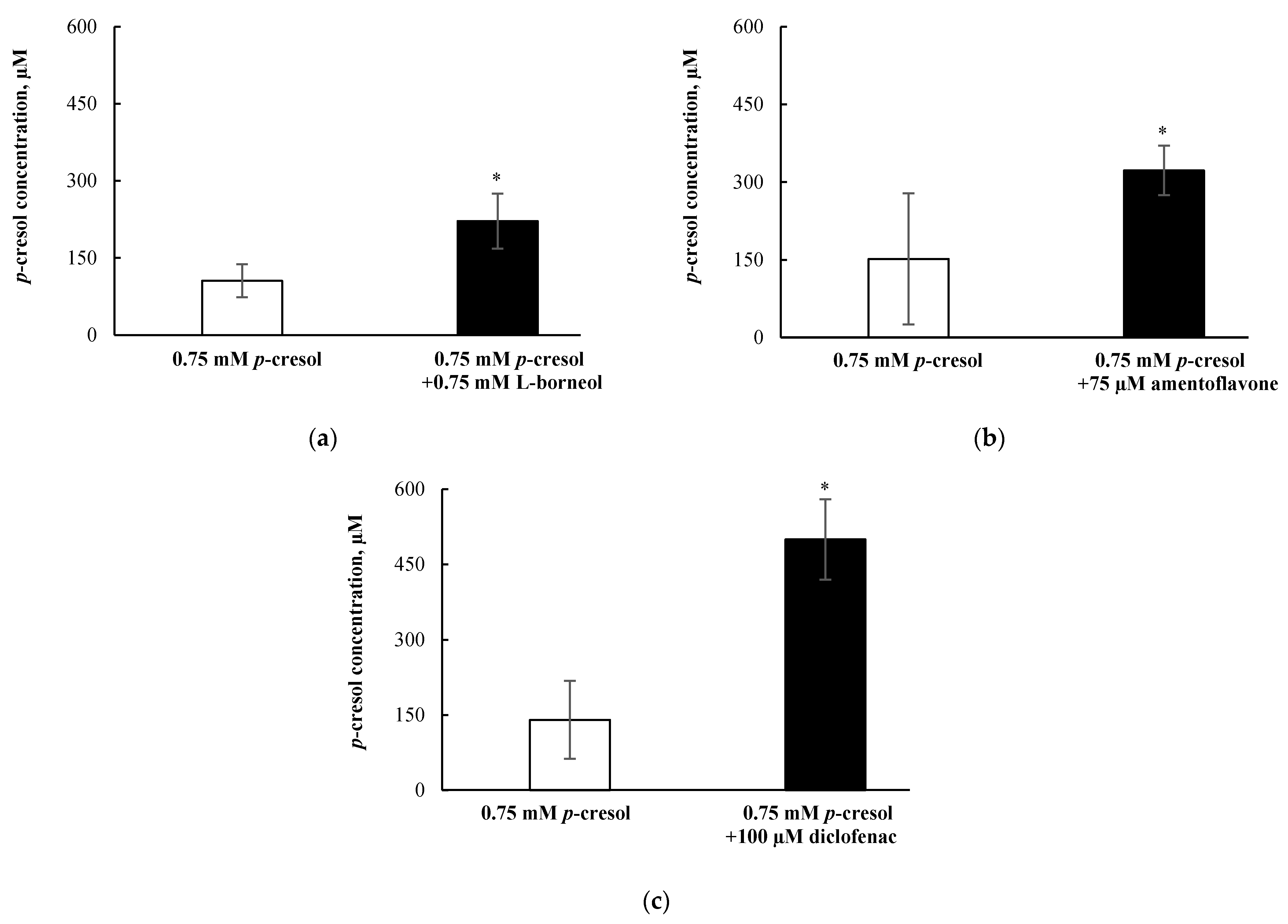
3.8. Effects of l-Borneol-borneol, Amentoflavone, or Diclofenac on Cellular p-Cresol Concentrations
3.9. Validation of LC/MS/MS and UPLC Assays for the Quantification of p-Cresol Sulfate, p-Cresol Glucuronide, and p-Cresol
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vanholder, R.; De Smet, R.; Lesaffer, G. p-Cresol: A Toxin Revealing Many Neglected but Relevant Aspects of Uraemic Toxicity. Nephrol. Dial. Transplant. 1999, 14, 2813–2815. [Google Scholar] [CrossRef] [Green Version]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and Clinical Impact of Organic Uremic Retention Solutes: A Comprehensive Update. Toxins 2018, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Glorieux, G.; Vanholder, R.; Van Biesen, W.; Pletinck, A.; Schepers, E.; Neirynck, N.; Speeckaert, M.; De Bacquer, D.; Verbeke, F. Free p-Cresyl Sulfate shows the Highest Association with Cardiovascular Outcome in Chronic Kidney Disease. Nephrol. Dial. Transplant. 2021, 36, 998–1005. [Google Scholar] [CrossRef]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Poesen, R.; Evenepoel, P.; de Loor, H.; Kuypers, D.; Augustijns, P.; Meijers, B. Metabolism, Protein Binding, and Renal Clearance of Microbiota-Derived p-Cresol in Patients with CKD. Clin. J. Am. Soc. Nephrol. 2016, 11, 1136–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liabeuf, S.; Glorieux, G.; Lenglet, A.; Diouf, M.; Schepers, E.; Desjardins, L.; Choukroun, G.; Vanholder, R.; Massy, Z.A. European Uremic Toxin (EUTox) Work Group. Does p-Cresyl Glucuronide have the Same Impact on Mortality as Other Protein-Bound Uremic Toxins? PLoS ONE 2013, 8, e67168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutsaers, H.A.; Caetano-Pinto, P.; Seegers, A.E.; Dankers, A.C.; van den Broek, P.H.; Wetzels, J.F.; van den Brand, J.A.; van den Heuvel, L.P.; Hoenderop, J.G.; Wilmer, M.J.; et al. Proximal Tubular Efflux Transporters Involved in Renal Excretion of p-cresyl Sulfate and p-Cresyl Glucuronide: Implications for Chronic Kidney Disease Pathophysiology. Toxicol. In Vitro 2015, 29, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Ezawa, A.; Kikuchi, K.; Tsuruta, Y.; Niwa, T. Protein-Bound Uremic Toxins in Hemodialysis Patients Measured by Liquid Chromatography/Tandem Mass Spectrometry and their Effects on Endothelial ROS Production. Anal. Bioanal Chem. 2012, 403, 1841–1850. [Google Scholar] [CrossRef] [PubMed]
- Chinnappa, S.; Tu, Y.K.; Yeh, Y.C.; Glorieux, G.; Vanholder, R.; Mooney, A. Association between Protein-Bound Uremic Toxins and Asymptomatic Cardiac Dysfunction in Patients with Chronic Kidney Disease. Toxins 2018, 10, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meert, N.; Schepers, E.; Glorieux, G.; Van Landschoot, M.; Goeman, J.L.; Waterloos, M.A.; Dhondt, A.; Van der Eycken, J.; Vanholder, R. Novel Method for Simultaneous Determination of p-Cresyl Sulphate and p-Cresyl Glucuronide: Clinical Data and Pathophysiological Implications. Nephrol. Dial. Transplant. 2012, 27, 2388–2396. [Google Scholar] [CrossRef] [Green Version]
- Mutsaers, H.A.; Wilmer, M.J.; Reijnders, D.; Jansen, J.; van den Broek, P.H.; Forkink, M.; Schepers, E.; Glorieux, G.; Vanholder, R.; van den Heuvel, L.P.; et al. Uremic Toxins Inhibit Renal Metabolic Capacity through Interference with Glucuronidation and Mitochondrial Respiration. Biochim. Biophys. Acta 2013, 1832, 142–150. [Google Scholar] [CrossRef]
- London, J.A.; Wang, E.C.S.; Barsukov, I.L.; Yates, E.A.; Stachulski, A.V. Synthesis and Toxicity Profile in 293 Human Embryonic Kidney Cells of the Β D-Glucuronide Derivatives of Ortho-, Meta- and Para-Cresol. Carbohydr. Res. 2020, 499, 108225. [Google Scholar] [CrossRef]
- Weigand, K.M.; Schirris, T.J.J.; Houweling, M.; van den Heuvel, J.J.M.W.; Koenderink, J.B.; Dankers, A.C.A.; Russel, F.G.M.; Greupink, R. Uremic Solutes Modulate Hepatic Bile Acid Handling and Induce Mitochondrial Toxicity. Toxicol. In Vitro 2019, 56, 52–61. [Google Scholar] [CrossRef]
- Kitagawa, A. Effects of Cresols (O-, M-, and P-Isomers) on the Bioenergetic System in Isolated Rat Liver Mitochondria. Drug Chem. Toxicol. 2001, 24, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Abreo, K.; Sella, M.; Gautreaux, S.; De Smet, R.; Vogeleere, P.; Ringoir, S.; Vanholder, R. P-Cresol, a Uremic Compound, Enhances the Uptake of Aluminum in Hepatocytes. J. Am. Soc. Nephrol. 1997, 8, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.C.; Perera, K.; Fisher, R.; Brendel, K. Cresol Isomers: Comparison of Toxic Potency in Rat Liver Slices. Toxicol. Appl. Pharmacol. 1994, 125, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.C.; Perera, K.; London, R. Studies on the Mechanism of Hepatotoxicity of 4-Methylphenol (p-Cresol): Effects of Deuterium Labeling and Ring Substitution. Chem. Biol. Interact. 1996, 101, 1–11. [Google Scholar] [CrossRef]
- Ikematsu, N.; Kashiwagi, M.; Hara, K.; Waters, B.; Matsusue, A.; Takayama, M.; Kubo, S.I. Organ Distribution of Endogenous p-Cresol in Hemodialysis Patients. J. Med. Investig. 2019, 66, 81–85. [Google Scholar] [CrossRef]
- Yan, Z.; Zhong, H.M.; Maher, N.; Torres, R.; Leo, G.C.; Caldwell, G.W.; Huebert, N. Bioactivation of 4-Methylphenol (p-Cresol) Via Cytochrome P450-Mediated Aromatic Oxidation in Human Liver Microsomes. Drug Metab. Dispos. 2005, 33, 1867–1876. [Google Scholar]
- Ashraf, M.N.; Asghar, M.W.; Rong, Y.; Doschak, M.R.; Kiang, T.K.L. Advanced in Vitro HepaRG Culture Systems for Xenobiotic Metabolism and Toxicity Characterization. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 437–458. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a Human Hepatoma Cell Line by Hepatitis B Virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [Green Version]
- Guillouzo, A.; Corlu, A.; Aninat, C.; Glaise, D.; Morel, F.; Guguen-Guillouzo, C. The Human Hepatoma HepaRG Cells: A Highly Differentiated Model for Studies of Liver Metabolism and Toxicity of Xenobiotics. Chem. Biol. Interact. 2007, 168, 66–73. [Google Scholar] [CrossRef]
- Rong, Y.; Kiang, T.K.L. Development and Validation of a Sensitive Liquid-Chromatography Tandem Mass Spectrometry Assay for Mycophenolic Acid and Metabolites in HepaRG Cell Culture: Characterization of Metabolism Interactions between p-Cresol and Mycophenolic Acid. Biomed. Chromatogr. 2019, 33, e4549. [Google Scholar] [CrossRef]
- Biopredic International. HepaRG Cell Line Specifications. 2016. Available online: https://www.biopredic.com/ (accessed on 3 February 2021).
- Liang, F.; Fang, Y.; Cao, W.; Zhang, Z.; Pan, S.; Xu, X. Attenuation of Tert-Butyl Hydroperoxide (T-BHP)-Induced Oxidative Damage in HepG2 Cells by Tangeretin: Relevance of the Nrf2-ARE and MAPK Signaling Pathways. J. Agric. Food Chem. 2018, 66, 6317–6325. [Google Scholar] [CrossRef] [PubMed]
- Kiang, T.K.; Teng, X.W.; Karagiozov, S.; Surendradoss, J.; Chang, T.K.; Abbott, F.S. Role of Oxidative Metabolism in the Effect of Valproic Acid on Markers of Cell Viability, Necrosis, and Oxidative Stress in Sandwich-Cultured Rat Hepatocytes. Toxicol. Sci. 2010, 118, 501–509. [Google Scholar] [CrossRef] [Green Version]
- Kiang, T.K.; Teng, X.W.; Surendradoss, J.; Karagiozov, S.; Abbott, F.S.; Chang, T.K. Glutathione Depletion by Valproic Acid in Sandwich-Cultured Rat Hepatocytes: Role of Biotransformation and Temporal Relationship with Onset of Toxicity. Toxicol. Appl. Pharmacol. 2011, 252, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Prokopienko, A.J.; Nolin, T.D. Microbiota-Derived Uremic Retention Solutes: Perpetrators of Altered Nonrenal Drug Clearance in Kidney Disease. Expert Rev. Clin. Pharmacol. 2018, 11, 71–82. [Google Scholar] [CrossRef]
- Watkins, J.B.; Klaassen, C.D. Chemically-Induced Alteration of UDP-Glucuronic Acid Concentration in Rat Liver. Drug Metab. Dispos. 1983, 11, 37–40. [Google Scholar]
- Lv, X.; Zhang, J.B.; Wang, X.X.; Hu, W.Z.; Shi, Y.S.; Liu, S.W.; Hao, D.C.; Zhang, W.D.; Ge, G.B.; Hou, J.; et al. Amentoflavone is a Potent Broad-Spectrum Inhibitor of Human UDP-Glucuronosyltransferases. Chem. Biol. Interact. 2018, 284, 48–55. [Google Scholar] [CrossRef]
- Uchaipichat, V.; Mackenzie, P.I.; Guo, X.H.; Gardner-Stephen, D.; Galetin, A.; Houston, J.B.; Miners, J.O. Human Udp-Glucuronosyltransferases: Isoform Selectivity and Kinetics of 4-Methylumbelliferone and 1-Naphthol Glucuronidation, Effects of Organic Solvents, and Inhibition by Diclofenac and Probenecid. Drug Metab. Dispos. 2004, 32, 413–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rong, Y.; Kiang, T.K.L. Characterizations of Human UDP-Glucuronosyltransferase Enzymes in the Conjugation of p-Cresol. Toxicol. Sci. 2020, 176, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhong, Z.; Xu, Z.; Chen, L.; Wang, Y. 2′,7′-Dichlorodihydrofluorescein as a Fluorescent Probe for Reactive Oxygen Species Measurement: Forty Years of Application and Controversy. Free Radic. Res. 2010, 44, 587–604. [Google Scholar] [CrossRef]
- Cayman Chemical. Glutathione Assay Kit Booklet. 2016. Available online: https://www.caymanchem.com/product/703002/glutathione-assay-kit (accessed on 3 February 2021).
- Roche. Cytotoxicity Detection Kit (LDH) Protocol. 2016. Available online: https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/roche/bulletin/1/11644793001bul.pdf (accessed on 3 February 2021).
- Cuoghi, A.; Caiazzo, M.; Bellei, E.; Monari, E.; Bergamini, S.; Palladino, G.; Ozben, T.; Tomasi, A. Quantification of p-Cresol Sulphate in Human Plasma by Selected Reaction Monitoring. Anal. Bioanal Chem. 2012, 404, 2097–2104. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Bioanalytical Method Validation Guidance for Industry. 2018. Available online: https://www.fda.gov/files/drugs/published/bioanalytical-method-validation-guidance-for-industry.pdf (accessed on 1 February 2021).
- Surendradoss, J.; Chang, T.K.; Abbott, F.S. Evaluation of in Situ Generated Valproyl 1-O-Beta-Acyl Glucuronide in Valproic Acid Toxicity in Sandwich-Cultured Rat Hepatocytes. Drug Metab. Dispos. 2014, 42, 1834–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, G.; Glorieux, G.; Thornalley, P.; Schepers, E.; Meert, N.; Jankowski, J.; Jankowski, V.; Argiles, A.; Anderstam, B.; Brunet, P.; et al. Review on Uraemic Toxins III: Recommendations for Handling Uraemic Retention Solutes in Vitro—Towards a Standardized Approach for Research on Uraemia. Nephrol. Dial. Transplant. 2007, 22, 3381–3390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U.S. Environmental Protection Agency. Provisional Peer-Reviewed Toxicity Values for 4-Methylphenol (p-Cresol). 2010. Available online: https://Cfpub.epa.gov/ncea/pprtv/documents/Methylphenol4.pdf (accessed on 3 February 2021).
- Sheu, J.J.; Yang, C.C.; Wallace, C.G.; Chen, K.H.; Shao, P.L.; Sung, P.H.; Li, Y.C.; Chu, Y.C.; Guo, J.; Yip, H.K. Uremic Toxic Substances are Essential Elements for Enhancing Carotid Artery Stenosis After Balloon-Induced Endothelial Denudation: Worsening Role of the Adventitial Layer. Am. J. Transl. Res. 2020, 12, 7144–7159. [Google Scholar]
- Chang, M.C.; Chang, H.H.; Chan, C.P.; Yeung, S.Y.; Hsien, H.C.; Lin, B.R.; Yeh, C.Y.; Tseng, W.Y.; Tseng, S.K.; Jeng, J.H. p-Cresol Affects Reactive Oxygen Species Generation, Cell Cycle Arrest, Cytotoxicity and Inflammation/Atherosclerosis-Related Modulators Production in Endothelial Cells and Mononuclear Cells. PLoS ONE 2014, 9, e114446. [Google Scholar] [CrossRef] [Green Version]
- Wong, X.; Carrasco-Pozo, C.; Escobar, E.; Navarrete, P.; Blachier, F.; Andriamihaja, M.; Lan, A.; Tomé, D.; Cires, M.J.; Pastene, E.; et al. Deleterious Effect of P-Cresol on Human Colonic Epithelial Cells Prevented by Proanthocyanidin-Containing Polyphenol Extracts from Fruits and Proanthocyanidin Bacterial Metabolites. J. Agric. Food Chem. 2016, 64, 3574–3583. [Google Scholar] [CrossRef]
- Idziak, M.; Pędzisz, P.; Burdzińska, A.; Gala, K.; Pączek, L. Uremic Toxins Impair Human Bone Marrow-Derived Mesenchymal Stem Cells Functionality in Vitro. Exp. Toxicol. Pathol. 2014, 66, 187–194. [Google Scholar] [CrossRef]
- Rong, Y.; Kiang, T. Characterization of Human Sulfotransferases Catalyzing the Formation of p-Cresol Sulfate and Identification of Mefenamic Acid as a Potent Metabolism Inhibitor and Potential Therapeutic Agent for Detoxification. Toxicol. Appl. Pharmacol. 2021, 425, 115553. [Google Scholar] [CrossRef]
- James, M.O.; Ambadapadi, S. Interactions of Cytosolic Sulfotransferases with Xenobiotics. Drug Metab. Rev. 2013, 45, 401–414. [Google Scholar] [CrossRef]
- James, M.O. Enzyme Kinetics of Conjugating Enzymes: PAPS Sulfotransferase. Methods Mol. Biol. 2014, 1113, 187–201. [Google Scholar]
- Hart, S.N.; Li, Y.; Nakamoto, K.; Subileau, E.A.; Steen, D.; Zhong, X.B. A Comparison of Whole Genome Gene Expression Profiles of HepaRG Cells and HepG2 Cells to Primary Human Hepatocytes and Human Liver Tissues. Drug Metab. Dispos. 2010, 38, 988–994. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, P.R.; Athersuch, T.J.; Stachulski, A.V.; Wilson, I.D. Acyl Glucuronide Reactivity in Perspective. Drug Discov. Today 2020, 25, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Van Vleet, T.R.; Liu, H.; Lee, A.; Blomme, E.A.G. Acyl Glucuronide Metabolites: Implications for Drug Safety Assessment. Toxicol. Lett. 2017, 272, 1–7. [Google Scholar] [CrossRef]
- Tassaneeyakul, W.; Birkett, D.J.; Miners, J.O. Inhibition of Human Hepatic Cytochrome P4502E1 by Azole Antifungals, CNS-Active Drugs and Non-Steroidal Anti-Inflammatory Agents. Xenobiotica 1998, 28, 293–301. [Google Scholar] [CrossRef]
- Karjalainen, M.J.; Neuvonen, P.J.; Backman, J.T. In Vitro Inhibition of CYP1A2 by Model Inhibitors, Anti-Inflammatory Analgesics and Female Sex Steroids: Predictability of in Vivo Interactions. Basic Clin. Pharmacol. Toxicol. 2008, 103, 157–165. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, S.; Rong, Y.; Kiang, T.K.L. Effects of p-Cresol on Oxidative Stress, Glutathione Depletion, and Necrosis in HepaRG Cells: Comparisons to Other Uremic Toxins and the Role of p-Cresol Glucuronide Formation. Pharmaceutics 2021, 13, 857. https://doi.org/10.3390/pharmaceutics13060857
Zhu S, Rong Y, Kiang TKL. Effects of p-Cresol on Oxidative Stress, Glutathione Depletion, and Necrosis in HepaRG Cells: Comparisons to Other Uremic Toxins and the Role of p-Cresol Glucuronide Formation. Pharmaceutics. 2021; 13(6):857. https://doi.org/10.3390/pharmaceutics13060857
Chicago/Turabian StyleZhu, Sang, Yan Rong, and Tony K. L. Kiang. 2021. "Effects of p-Cresol on Oxidative Stress, Glutathione Depletion, and Necrosis in HepaRG Cells: Comparisons to Other Uremic Toxins and the Role of p-Cresol Glucuronide Formation" Pharmaceutics 13, no. 6: 857. https://doi.org/10.3390/pharmaceutics13060857
APA StyleZhu, S., Rong, Y., & Kiang, T. K. L. (2021). Effects of p-Cresol on Oxidative Stress, Glutathione Depletion, and Necrosis in HepaRG Cells: Comparisons to Other Uremic Toxins and the Role of p-Cresol Glucuronide Formation. Pharmaceutics, 13(6), 857. https://doi.org/10.3390/pharmaceutics13060857