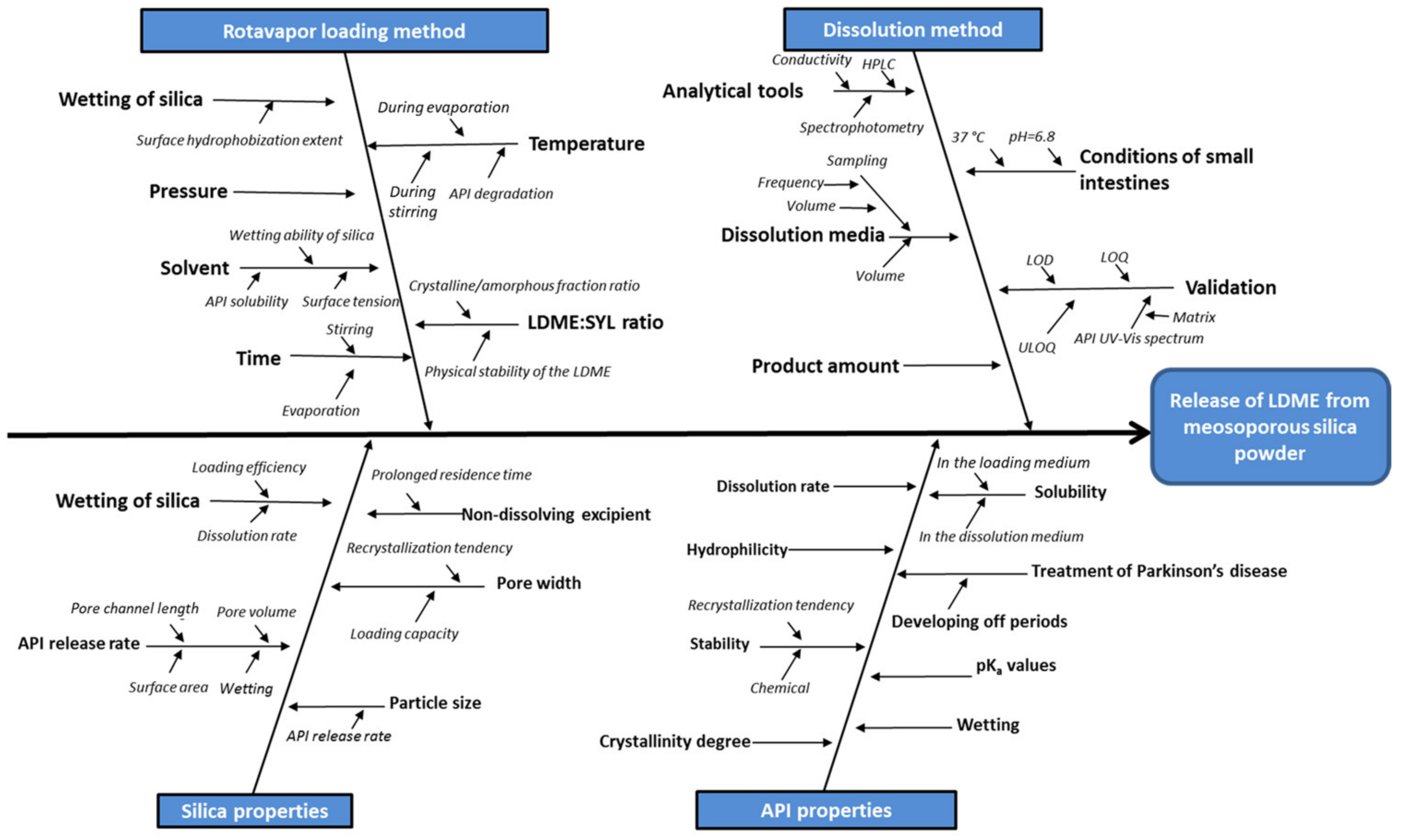
3.1. Evaluation of the Hydrophobization Reaction
To quantify the efficacy of carrier hydrophobization, we examined the surface of the hydrophobized silica particles applying contact angle measurements, particle charge titrations and infrared spectroscopy.
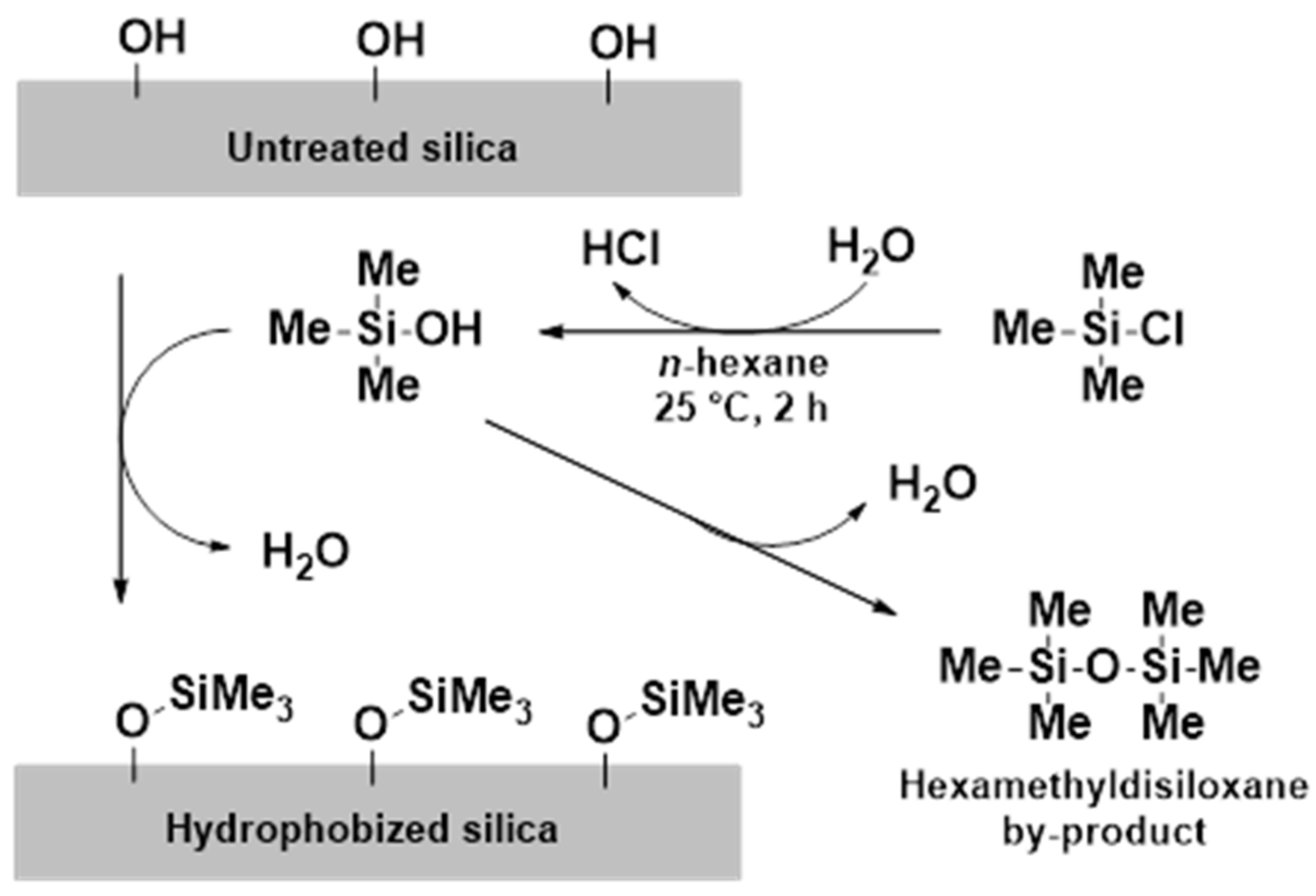
At first, the optimization of the hydrophobization reaction was carried out. The adsorbed water amount of SYL-0 proved to be a key factor because it might affect the hydrolysis of the functionalizing agent; therefore, the storage circumstances were standardized (SYL-0 was kept under 40 °C and 75% RH for 3 days). Besides, the containers have to be hydrophobized prior to the reaction to ensure reproducibility via preventing the unwanted reactant consumption by the -OH content of the glass. As a result of these preliminary measures, the standard deviations of the contact angles of SYL-1 and SYL-2 showed a significant decrease (p < 0.05). The evaporation of water content at 110 °C resulted in the formation of the so-called SYL-DRY product. The contact angle of SYL-DRY was 0°, as well as in the case of raw SYL-0 (kept under 40 °C and 75% RH for 3 days), which also confirmed the role of water in the reaction.
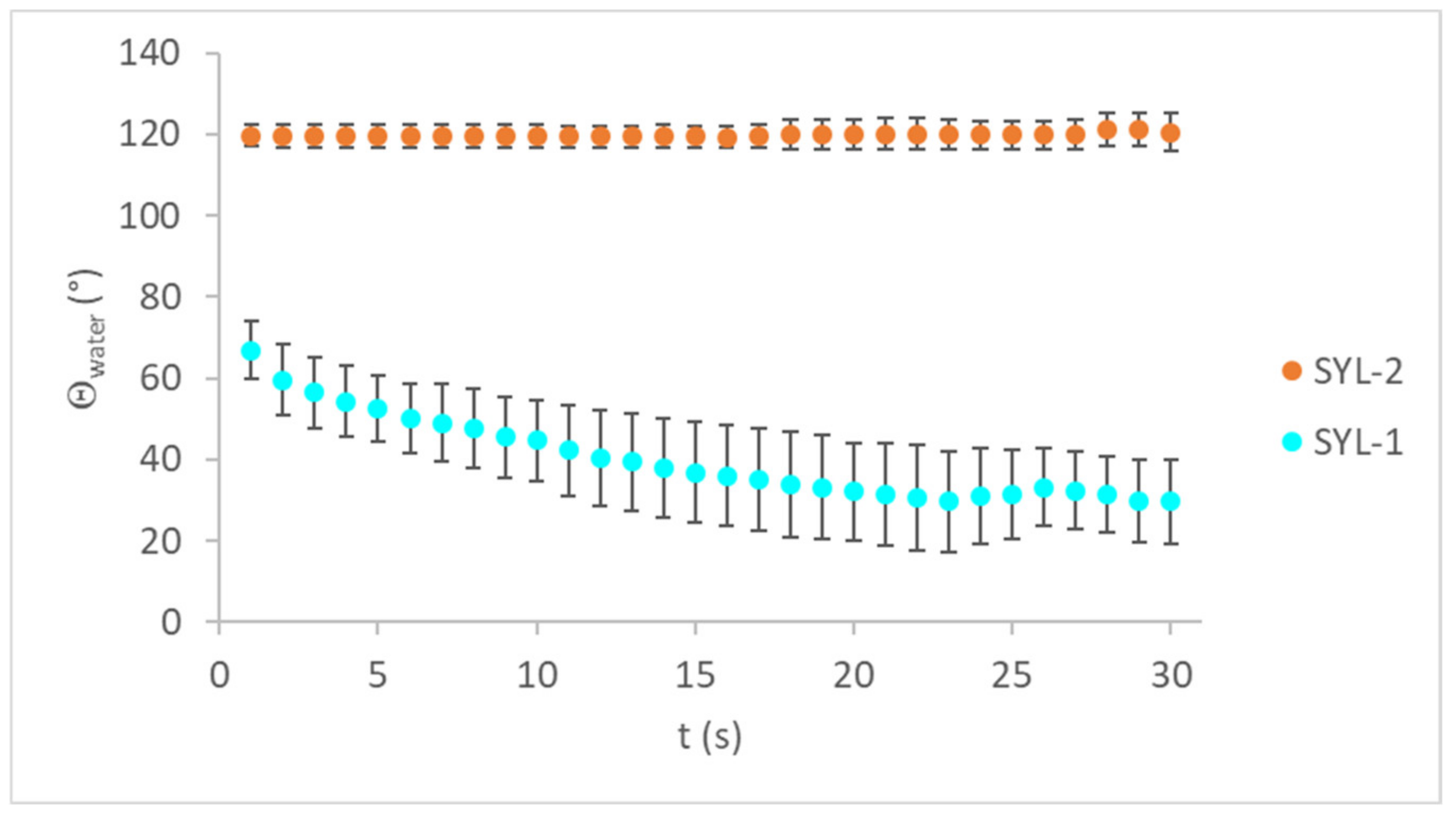
During surface functionalization, the water contact angles increased with increasing TMCS concentration (
Table 3), which can be translated to a decrease in wettability.
As the contact angles of SYLs were reduced, the water droplets had a lower tendency to spread on the pastille surface. The time-dependency of the water contact angles can be seen in
Figure 3.
The CTAB concentrations belonging to 0 streaming potential could give an estimation for the surface density of silanol groups. The concentration of TMCS in the reaction mixture reduced d
Si-OH, indicating that the Si-OH groups were functionalized during the reaction (
Table 4).
On the other hand, it was necessary to prove that -Si(CH
3)
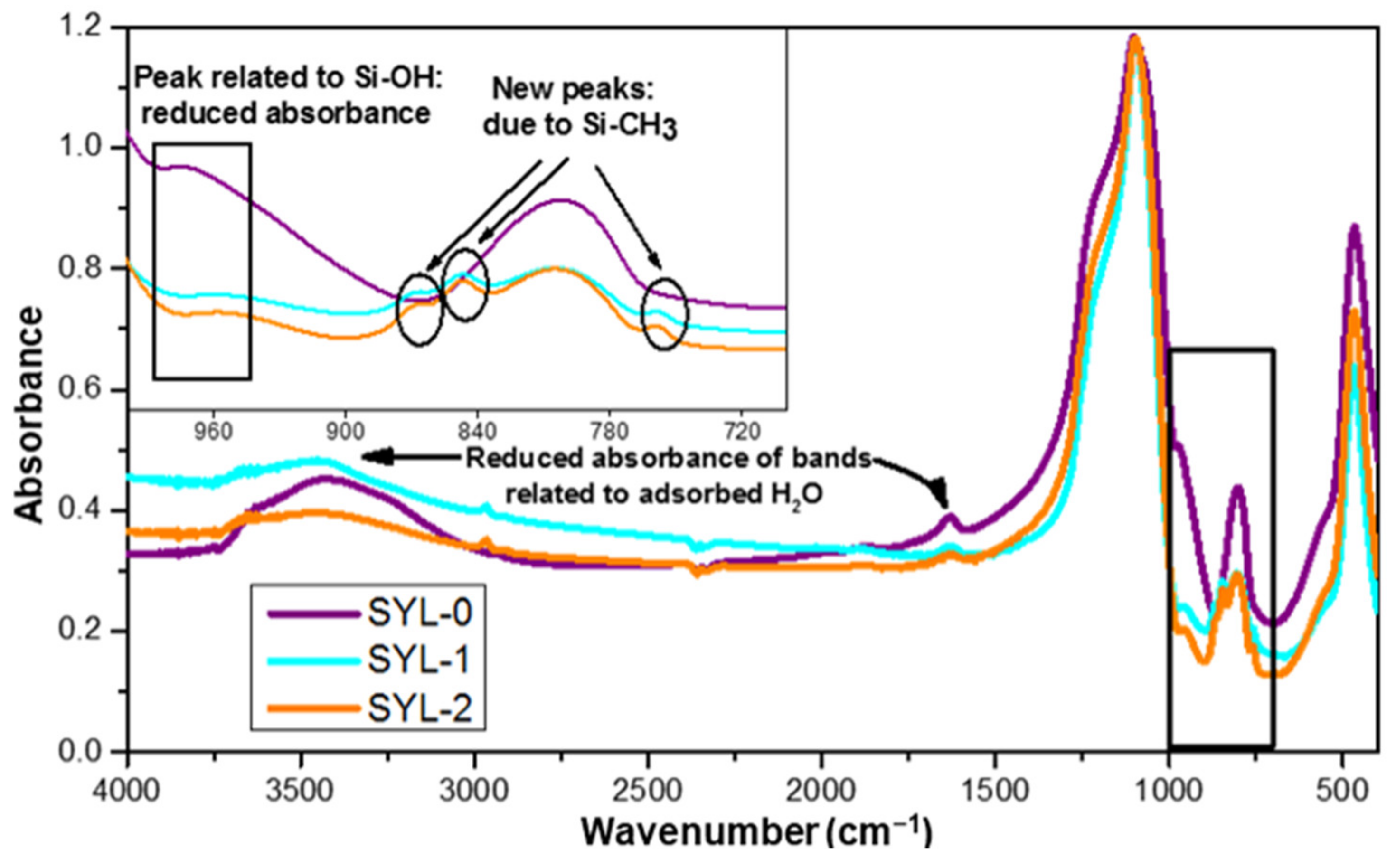
3 groups were covalently attached to the surface as a result of the reaction, and the surface density of silanol groups did not decrease because of the storage temperature of 110 °C after the reaction. The functionalization of SYL was proved by IR spectroscopy. The absorption of the samples in the mid-IR range could provide qualitative information on the changes in functional group composition.
Figure 4 shows a comparison of the spectra of SYL-1, SYL-2 and SYL-0. According to the analysis, the spectra of both functionalized silicas (SYL-1 and SYL-2) showed the same types of changes.
The band at 474 cm
−1 could be assigned to Si-O-Si bending vibrations, while at 804 cm
−1 it indicates the symmetric stretching vibrations of Si-O-Si. The very strong and broad IR band located at 1105 cm
−1 with a shoulder-type peak at 1188 cm
−1 can be assigned to the transversal optic and longitudinal optic modes of the Si-O-Si asymmetric stretching vibrations [
43]. As the reaction left the siloxane groups intact, the relative intensity of the bands did not differ. The band at 972 cm
−1 corresponded to the stretching vibrations of the Si–OH groups [
44]. The intensity of this band decreased drastically in the case of SYL-1 and SYL-2, compared to SYL-0, indicating a significant decline in the density of surface silanol groups. The presence of the formed Si-CH
3 bonds on the surface was confirmed by the new peaks in the range of 865–750 cm
−1 (peaks: 759 cm
−1, 850 cm
−1 and 865 cm
−1) [
45]. Meanwhile, the band at 1633 cm
−1 belongs to the bending vibration, and the peak at 3435 cm
−1 belongs to the stretching vibration of adsorbed H
2O molecules [
43]. The intensity of both bands decreased in the SYL derivative spectra despite the fact that both samples were stored at 70 °C, at the same RH, which means that the hydrophobized SYL could adsorb a smaller amount of water. This can be attributed to the lower surface -OH content, which otherwise could render the surfaces able to make strong H-bonds. Besides, new peaks appear at 2906 cm
−1 and 2964 cm
−1 which are corresponding to the C-H vibration of the trimethylsilyl groups.
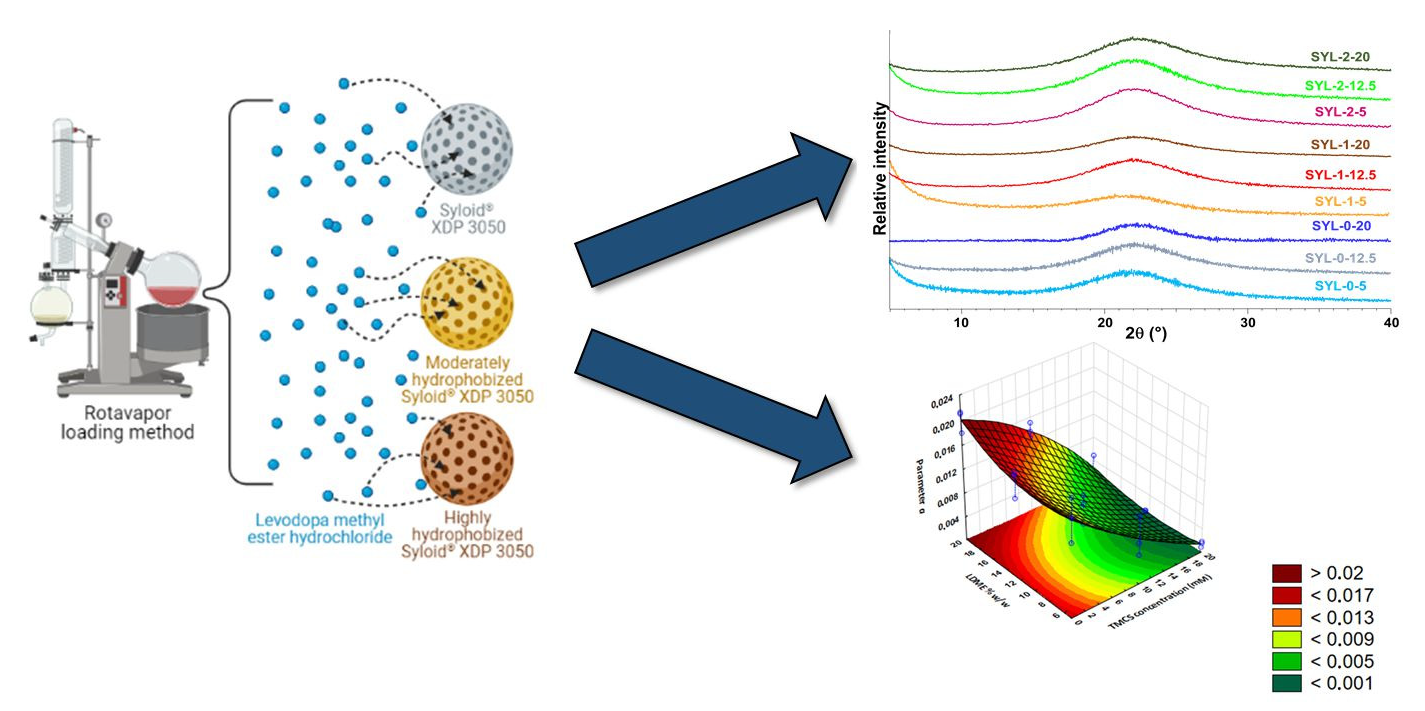
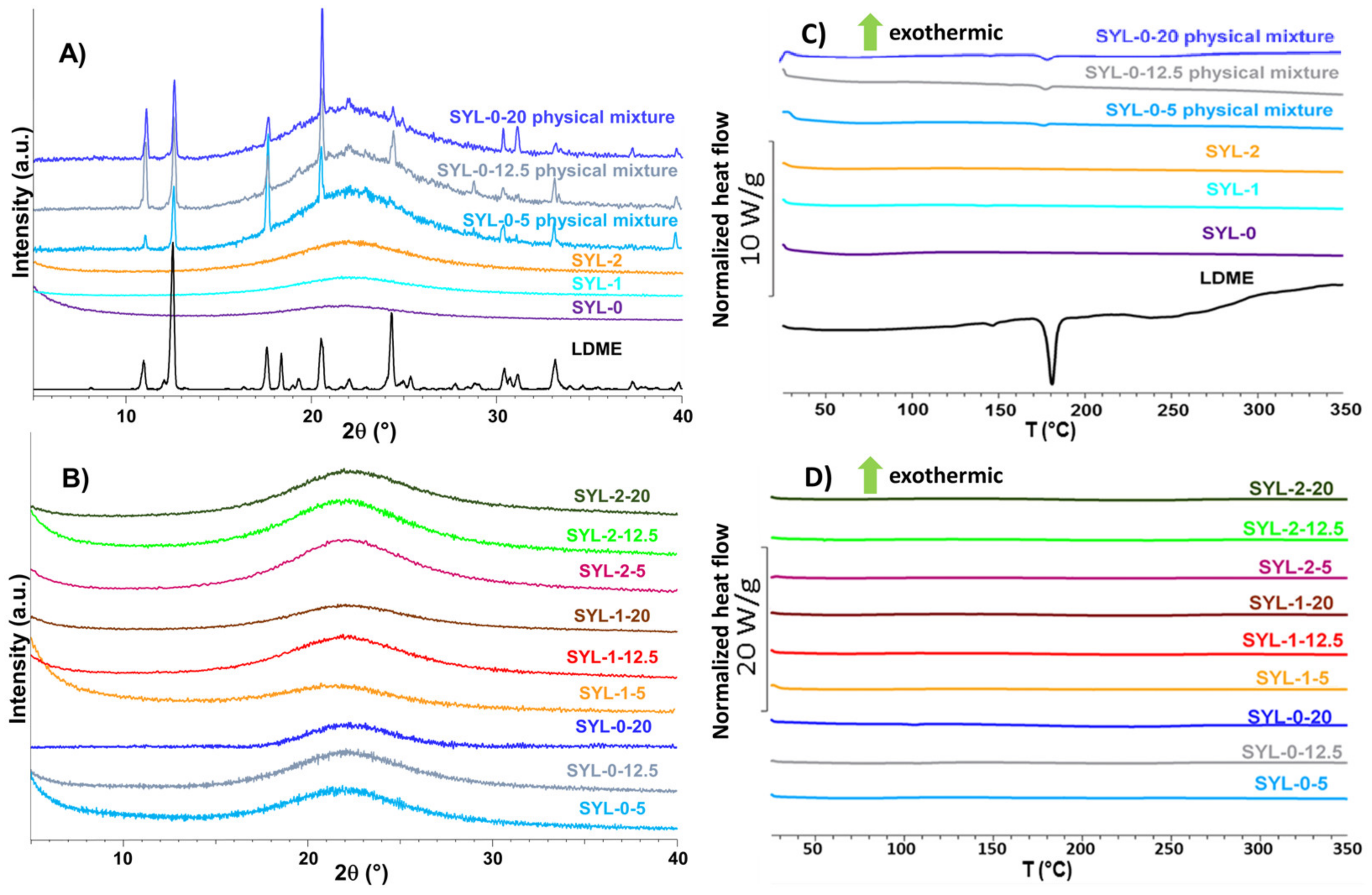
Based on the diffractograms, it was proven that amorphous mesoporous silica particles were used as raw material, and during the hydrophobization reaction, this amorphous nature remained unchanged (see SYL-0, SYL-1 and SYL-2 in
Figure 5C). The storage at 70 °C did not affect the surface area, which is in good accordance with the literature (sintering occurs only above 550 °C) [
46].
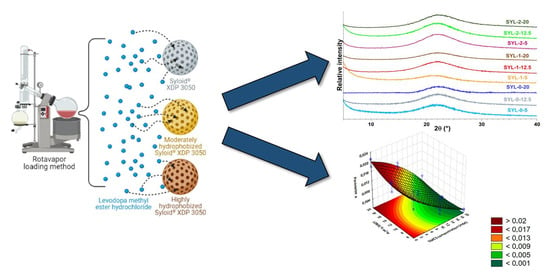
3.4. Distribution of LDME in the Products, Its Effect on Particle Size and BET Specific Surface Area
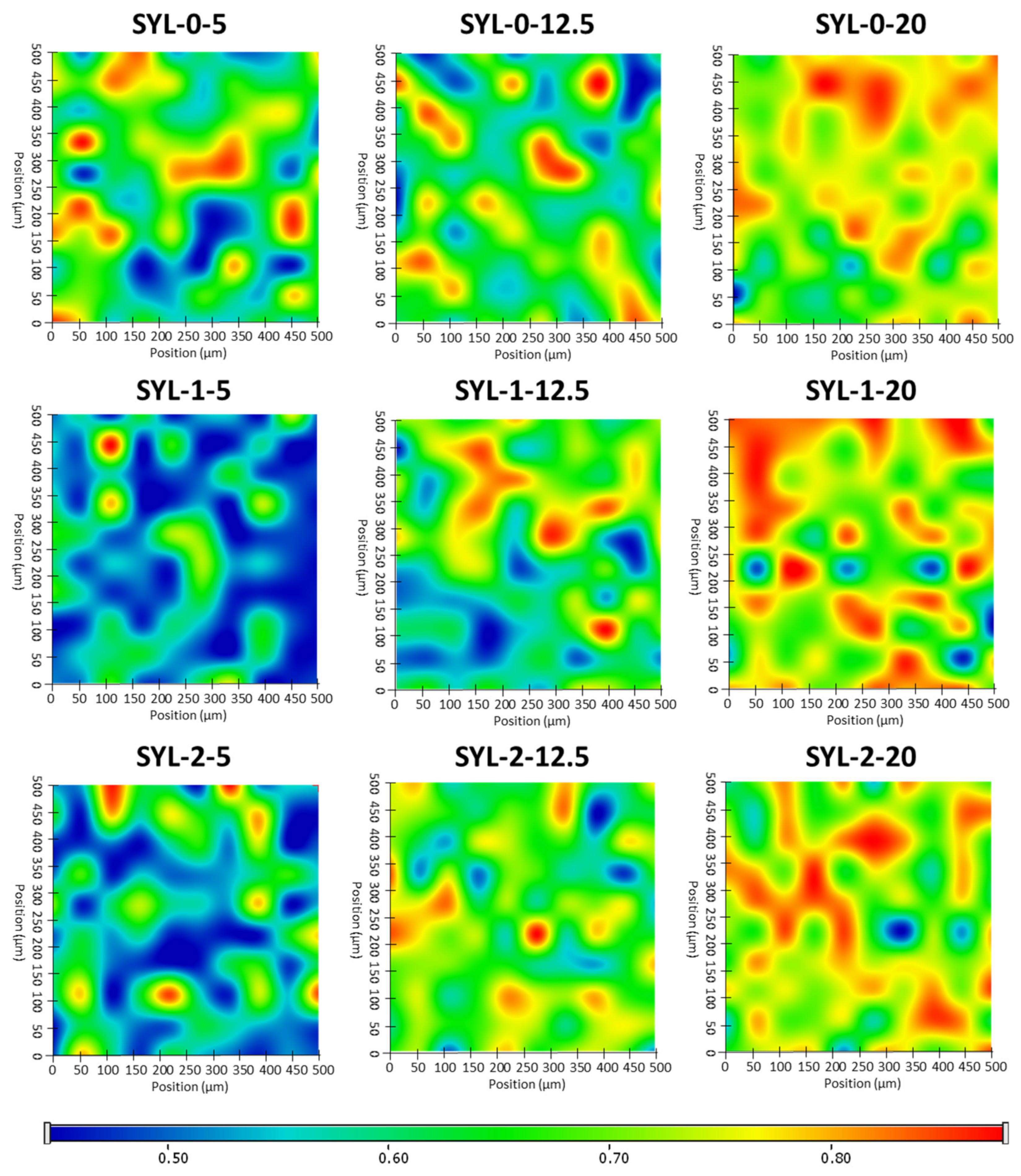
Raman mapping was carried out to determine the distribution of LDME in different SYL products (
Figure 6). For this purpose, the Raman spectrum of pure LDME was used as a reference to visualize the chemical map of the products showing the amount of LDME. Not surprisingly, increasing the amount of LDME during the loading process resulted in a higher final drug content, as evidenced by the greater extent of regions on the chemical map, marked by red colour. The effect of hydrophobization was remarkable only in the case of low drug content: the Raman map of SYL 1-5 and SYL 2-5 indicates that LDME can be found only sporadically in comparison to SYL 0-5, which can be attributed to the higher extent of interactions between the polar API and the polar functional groups of the surface. In the hydrophobic products, the API might develop a stronger interaction between the API molecules than between the surface APIs.
The study of the products and the SYL carriers proved that LDME was likely to be loaded into the pores and did not recrystallize on the SYL surface, as no significant change was detected among the samples compared to the given SYL (
Table 5). Every product was compared to the SYL, which had the same hydrophobicity as the silica used in the product (for example, SYL-2-12.5 was not significantly different from SYL-2). The silylation process resulted in a decreased nitrogen adsorption capacity, i.e., decreased specific areas (
Table 5). Thus, the BET-specific surface area (A
s) of the original SYL-0—283 m
2/g—decreased to approximately 244 m
2/g in SYL-1, 235 m
2/g in SYL-2. This phenomenon had been observed in the literature earlier after the hydrophobization of mesoporous silica with TMCS [
47]. Besides, the BET surface area also decreased after the incorporation of the API into the pores. These results also confirm the success of the loading process.
3.5. Stability of the Products
The chemical stability of the powders was appropriate upon storage in the freezer; however, the powders at 40 °C, 75% RH became light brown. According to the additional HPLC measurement, drug content decreased in these samples, indicating that the LDME-containing samples should be stored in the freezer just as the API itself. However, the residual API remained amorphous in the products within the 5–20% w/w range of the initial API content.
It makes sense to interpret the results of the physical stability investigation only in such cases where the API did not suffer chemical decomposition. Thus, only the results of samples stored at −20 °C are shown. A weak sign of crystalline LDME appeared in the X-ray diffractogram in the case of the SYL-0-20 product after 3 months at −20 °C, which probably means that the 20% w/w LDME ratio could be too high for all API molecules to be incorporated into the pores. Thus, the amorphous API remained stable in the SYLs at lower than 20% w/w LDME content.
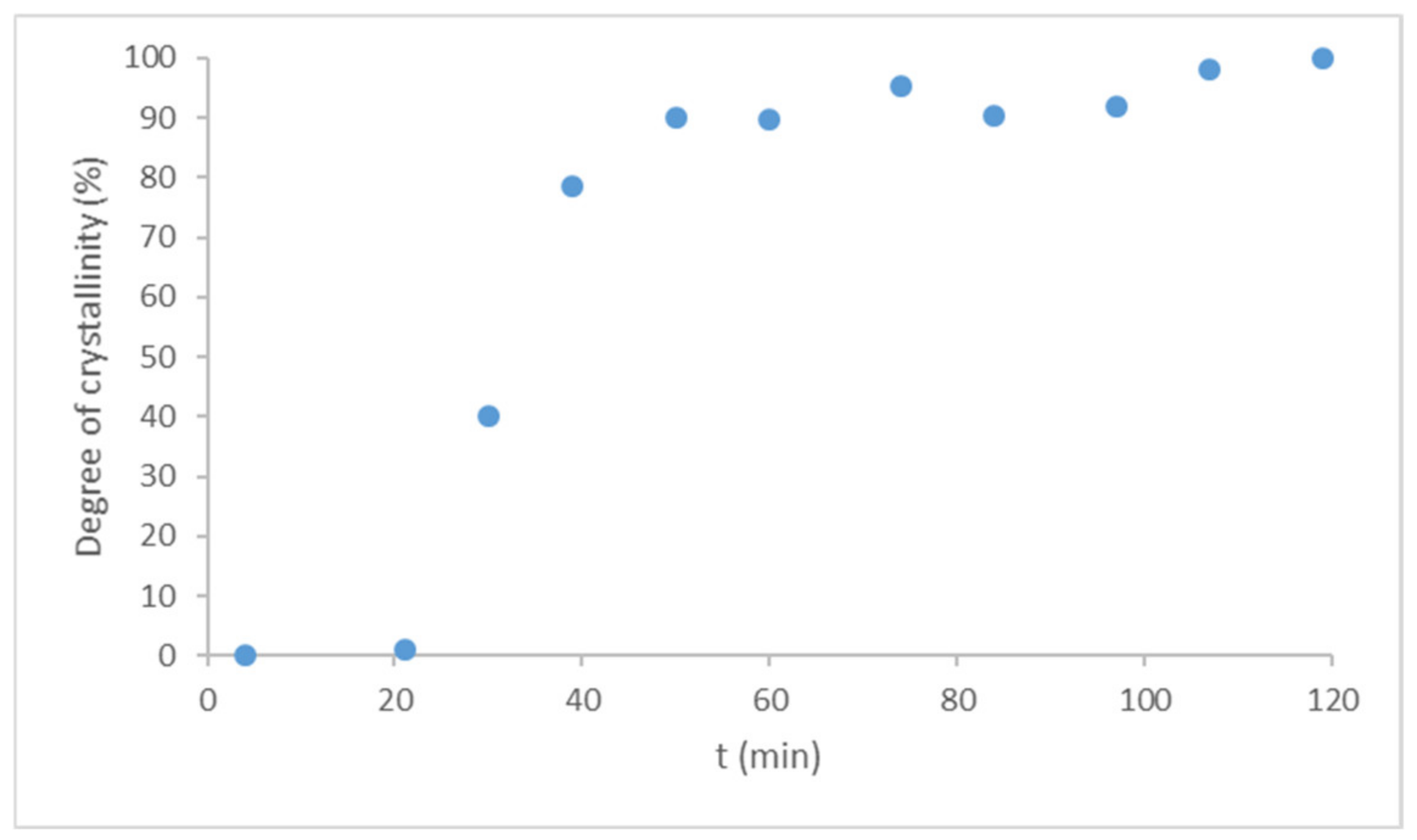
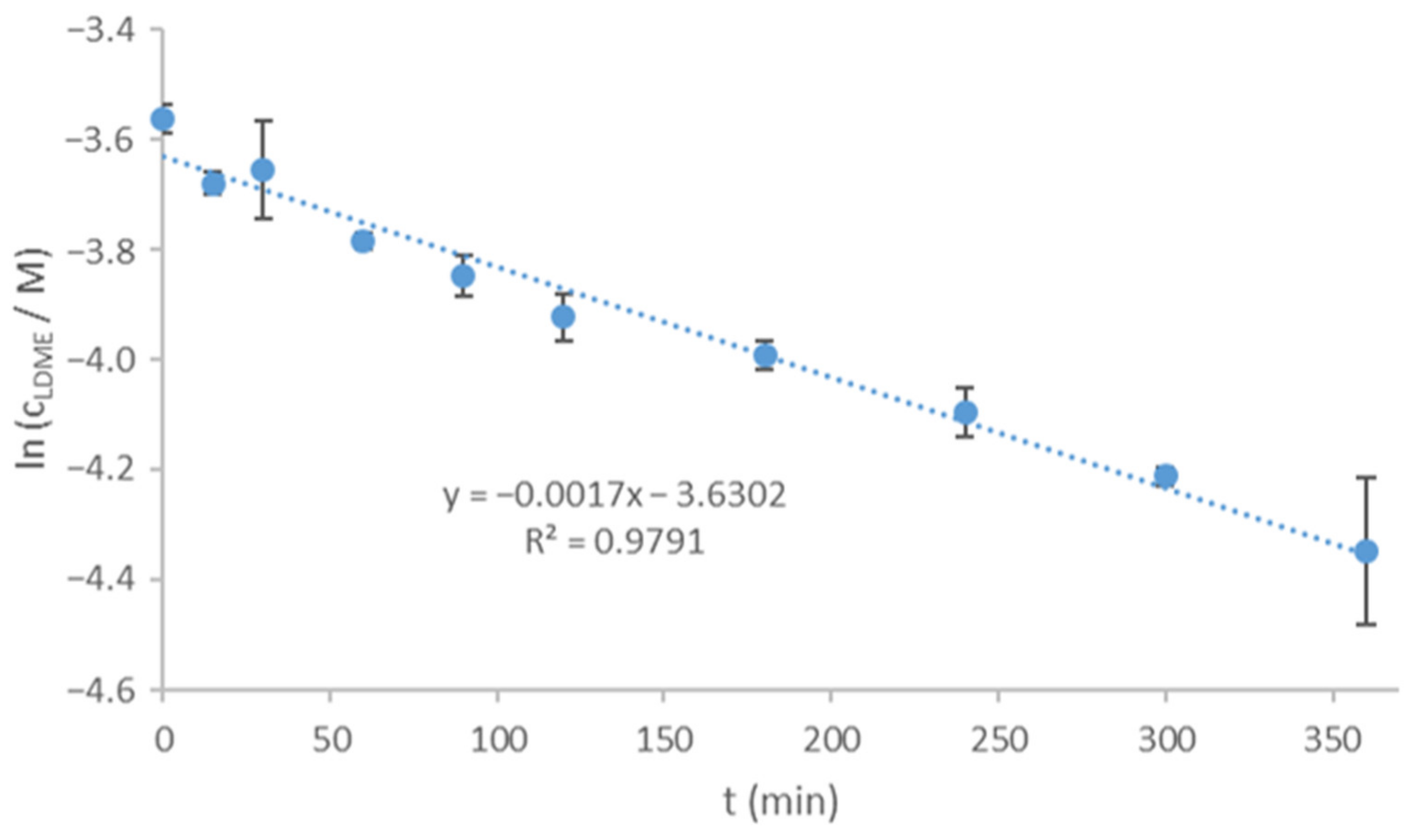
In comparison, LDME was directly amorphized in the sample holder of XRPD under the same conditions as used in the rotavapor. The amorphized raw LDME recrystallized due to weak scraping and after leaving it untouched for one day under the conditions of the stability tests. The amorphization kinetics of the pure LDME was investigated at 40 °C, 75% RH. The recrystallization process took place in about 100 min under these circumstances (
Figure 7).
Compared to the reference, the recrystallization rate of the products turned out to be slower by orders of magnitude. The loading of LDME into the pores of MPSs seems to be a quite reliable tool to prevent recrystallization if the% w/w of the API is under a certain ratio. Thus, it is important to determine the storage conditions and the related maximum drug loading below which the API remains in the mesopores and in an amorphous state, resulting in the desired stability of the products.
3.7. Release Studies
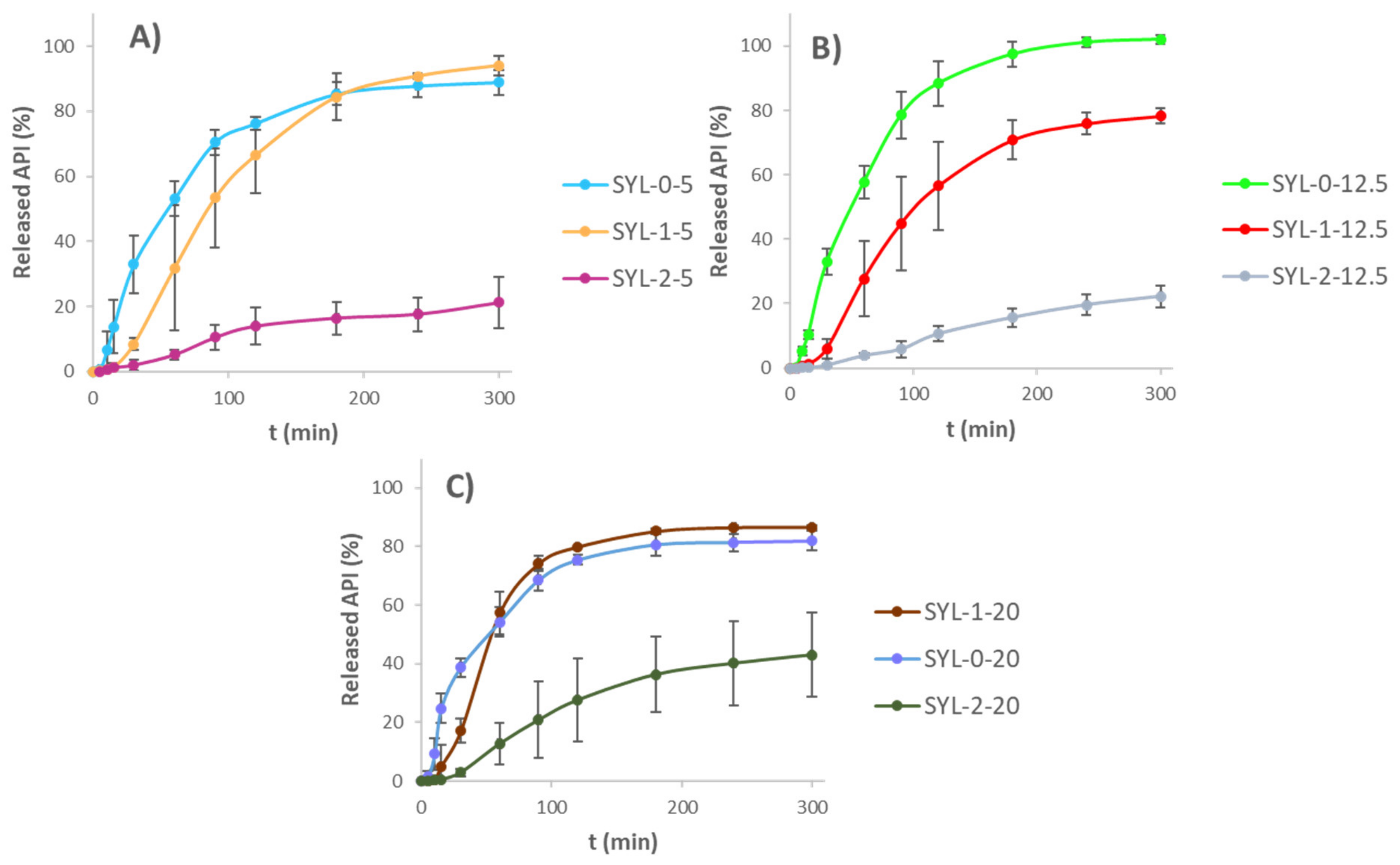
Dissolution kinetics were investigated for 5 h. The resulting curves are shown in
Figure 9. As a general tendency, the mean release rate of LDME could be distinguished according to the hydrophobicity of SYL. As hydrophobicity increased, the release rate showed an unambiguous decrease. There was a spectacular difference detected between the SYL-2-containing samples and the other two samples; however, the difference was noticeably smaller in the case of 20%
w/
w LDME-containing samples. A probable explanation for this is the much poorer aqueous wetting of SYL-2 compared to the other two SYLs.
The kinetics of the release curves were determined. The first-order kinetics of the reference LDME was not changed in the case of dissolution from SYL-0-20, SYL-1-20 and SYL-2-20 products. It was probably due to the high loading content. Thanks to this, the diffusion of water molecules was not really hindered in the pores at the beginning, and they could contact the API molecules easily, as polar API molecules covered the pore surface presumably thickly. Thus, the relative increase in SYL and the decrease in LDME resulted in the change of release kinetics. The release of SYL-0-5, SYL-0-12.5, SYL-1-5 and SYL-1-12.5 products fitted to the Hixson–Crowell model the most closely. It could be due to the decrease in the surface area as a consequence of the release of the API. Compared to these, the release of SYL-2-5 and SYL-2-12.5 products was zero order; therefore, SYL-2 with API content in the range of 5-12.5 w/w% may be appropriate for maintaining a constant blood level, but if the product contains 20% w/w LDME, it will not be proper for this purpose.
To interpret the results statistically,
parameters a and
b were determined based on the procedure detailed in
Section 2.15. In the following part, we would like to focus on
parameter a, which characterizes IRR, i.e., as this parameter increases, so does the drug release rate. The parameters of the release curves can be seen in
Table 6. After the determination of
parameters a and
b, which can model the release kinetics, the similarity factor (f
2) was defined between the model and the actual release results. According to this value, the models fitted close to the actual release results; therefore, we suggest that this model be used in the case of products with such release kinetics during the evaluation of drug release.
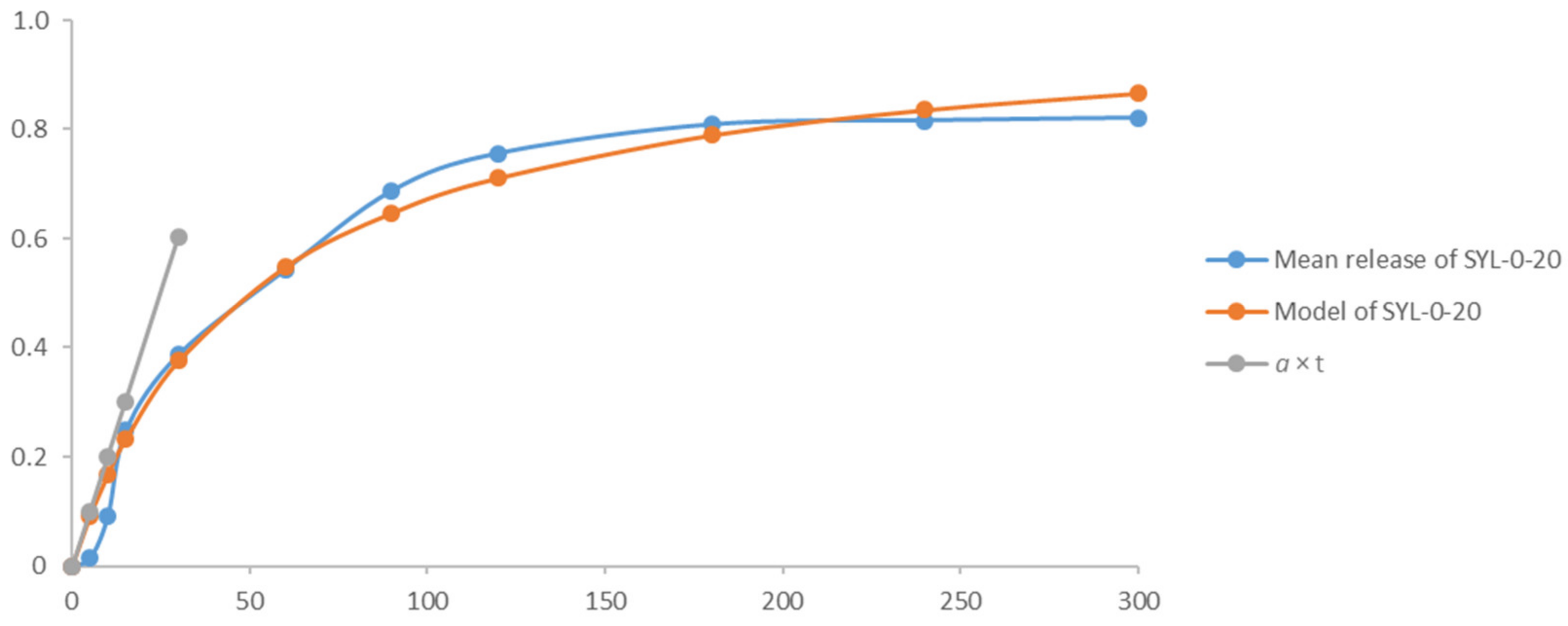
Figure A1 presents the methodology of the
parameters a and
b showing the model curve (based on Equation (3)) and the actual release results of SYL-0-20. Besides, the curve based on the
a × t (Equation (4)) is also shown which fits the initial stage of the model curve.
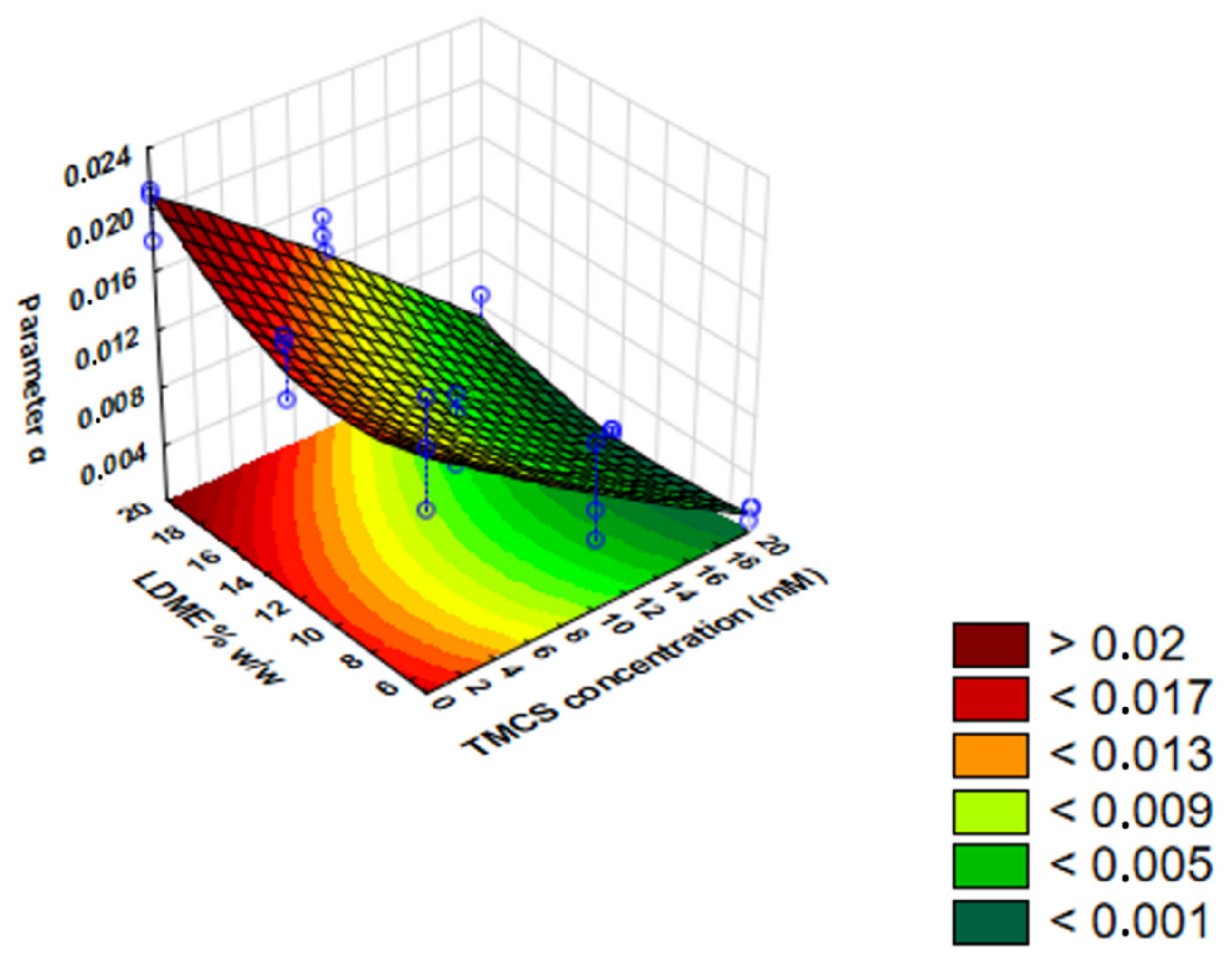
Based on the results, a polynomial equation was constructed to give the influence of the independent variables (cTMCS and LDME% w/w) on the dependent variable (IRR).
The IRR of LDME can be described by Equation (7):
where
x belongs to the TMCS concentration;
y belongs to the LDME mass percent prior to the preparation of the product. The members of the equation highlighted in bold have a significant influence on IRR. The statistical parameters of this equation are: R
2 = 0.93462, adjusted R
2 = 0.90556, mean square residual = 0.0000048. By the removal of the
y2, the
x y2 member from the polynomial equation fits the most closely to the measured points. As a consequence, there is a negative linear relationship between the IRR and the hydrophobization extent. After the removal of these points, the result is Equation (8):
The statistical parameters of this equation are: R
2 = 0.93381, adjusted R
2 = 0.91395, mean square residual = 0.0000044. The members of the equation highlighted in bold have a significant influence on IRR. This equation defines the surface plot below (
Figure 10).
Based on the IRR results, we can see that both factors have a significant linear effect on the release rate. Hydrophobization had a significant retarding effect on release. The results also indicate that LDME% w/w had a positive significant effect and a negative quadratic effect on parameter a.
According to the Tukey HSD results (see
Table A2), the release rate decreased significantly in all products compared to the LDME reference except for SYL-0-20. In the case of samples with the same LDME content and different wettability, the increasing degree of hydrophobization had a significant retarding effect on LDME release.
Parameter a decreased significantly for SYL-2-containing products at a certain API ratio compared to the SYL-0-containing products; however, there was no significant difference between the SYL-0 and SYL-1 samples, which indicates that 10 mM of TMCS was not enough to decelerate the release rate, but 20 mM was enough.
In contrast to the comparison of individual values with the help of the Tukey HSD test, according to the result of the ANOVA evaluation of the 32 full-factorial design, the API% w/w also had a significant effect on parameter a, but it does not seem so obvious when the individual IRR values are compared. This finding emphasizes the importance of the methodology of the ANOVA evaluation, as it holistically compares all products and not only individual values. Summarizing the data, even if the product is stable, the ratio of the API and the hydrophobicity of the mesoporous carrier must be taken into consideration when the drug release rate is supposed to be regulated in mesoporous silica systems because it has a significant effect on the release properties.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










