1. Introduction
The epithelial mucin 1 (MUC1) is a functional protein with important roles in the survival of cells. However, MUC1 is also distinctly associated with many malignancies, where several original characteristics of the protein are lost. As opposed to the typical expression, which is restricted to the apical surface of normal epithelial cells, MUC1 is found to be overexpressed in many adenocarcinomas, such as those of the breast, lung, ovary, pancreas, prostate, and numerous other epithelial organs. Other cancers also expressing MUC1 to a substantial degree include multiple myeloma, lymphoma and particular leukaemias [
1]. Greater expression is due to the loss of polarisation. However, MUC1 displays many different features on tumour cells compared with the native protein, such as significantly reduced glycosylation due to the lessening number of tandem repeats present in the extracellular domain, combined with the extra addition of sialic acids, which also distort the cellular adhesion properties on malignant cells [
2]. Interestingly, MUC1 can also operate as an adhesive molecule due to the attached carbohydrate structures, such as sialyl Lewis (a) and sialyl Lewis (x) in cancers of the pancreas and the colon [
3].
Several MUC1 isoforms are generated through alternative splicing, and these are often found expressed within the same type of cells or tissues. The different alternative spliced variants vary in structure from the native MUC1 protein by lacking either the transmembrane domain, cytoplasmic domain, or the tandem repeat regions. Hence, each of the isoforms also differs in their functional properties, and most of the isoforms are overexpressed in cancer cells compared with cells obtained from benign tumours or normal epithelial cells [
4].
The alternatively spliced variant MUC1/Y is identical to the MUC1 regarding the transmembrane domain and the cytoplasmic tail. However, this splice variant is lacking the tandem repeat region and the flanking region. This splice variant, like MUC1, possesses sequences that are also observed in cytokine receptors, suggesting that MUC1/Y can perform as a cytokine receptor and possibly be involved in similar ligand interactions [
5]. Although MUC1/Y shares similarities with MUC1, the splice variant does not have a heterodimeric component, whereupon cleavage of the protein can occur [
6]. Furthermore, this membrane-bound cell protein goes through transphosphorylation within the cell on the tyrosine and serine residues, which are part of the cytoplasmic domain; this is found to occur in breast malignancy upon binding to the MUC1/SEC protein [
7,
8]. The interaction between MUC1/Y and MUC1/SEC, which occurs at the extracellular domain, is not only thought to induce phosphorylation but is responsible for instigating many biological processes, such as changes to cell morphology and prompting of the signalling cascade. Triggers to initiate the signalling cascade are thought to occur via the interaction of the cytoplasmic phosphorylated residues to the GRB2 protein, a similar mechanism to that observed with MUC1. One of the many effects of the signal transduction produced by the MUC1/Y protein is the enhancement of tumour progression. Interestingly, in cancers of the breast, the expression of this splice variant is seemingly advantageous as MUC1/Y is apparently found to be present in malignant tissue yet undetectable in neighbouring normal tissue of the same organ, thus establishing MUC1/Y as a potential marker for identifying malignancy [
6,
9].
Obermair et al. [
10] demonstrated the extensive expression of MUC1/Y after screening eight different cervical cancer cell lines, whereby seven out of eight were found to express this splice variant. Similarly, when investigating primary ovarian tumour samples, it was observed that MUC1/Y is more frequently expressed in malignant than in benign tumours. From the 55 ovarian cancer samples that were analysed, 54 of those samples expressed MUC1/Y, proposing that MUC1/Y is a strong candidate for indicating the presence of malignancy. Whereas the MUC1/Y splice variant is coexpressed with the splice variant MUC1/SEC in benign tumours, this is not the case in malignant tumours [
4].
We previously selected aptamers against the tandem repeat peptide of the MUC1 glycoprotein [
11] and the glycosylated form of MUC1 [
12]. Additionally, we utilized such aptamers for diagnostic applications [
12] and radiolabelled them for diagnostic and therapeutic applications [
13,
14,
15]. Finally, we coupled these aptamers to nanoparticles to improve their biodistribution and tumour delivery properties [
16,
17,
18]. In this work, we demonstrate, for the first time, the development of a DNA aptamer against a MUC1/Y alternative splice variant aiming to obtain novel biopharmaceutics with improved specificity for MUC1/Y positive cancers. We characterized these aptamers with a series of biochemical and biophysical techniques and evaluated them in preclinical models for their therapeutic potential.
2. Materials and Methods
2.1. Aptamer Targets—MUC1/Y Peptides: 10mer and 20mer
Since the MUC1/Y splice variant is a transmembrane protein, the full protein cannot be used in the selection procedure due to the complexities involved in its isolation and purification. Thus, two synthetic peptides were designed composed of amino acids flanking the splice site present in the MUC1/Y protein. The MUC1/Y peptides used as targets for selection consist of 10 and 20 amino acids, referred to as 10mer peptide and 20mer peptide, respectively. The 10mer peptide sequence (TEKNAFNSSL) was selected based on five amino acids before the splice site, and five amino acids after the splice site, distinguishing the MUC1/Y from the MUC1 extracellular domain protein sequence. The 20mer peptide sequence (SVPSSTEKNAFNSSLEDPST) is an extension to the 10mer peptide of five additional amino acids on either side; hence, 10 amino acids before and 10 amino acids after the splice site [
19]. Both peptides were HPLC purified by the Oligonucleotide and Peptide synthesis unit of the Queen’s Medical Centre, University of Nottingham, UK.
2.2. Single Stranded Aptamer Library and Primers
The SELEX single-stranded oligonucleotide library consists of a central random region of 25 nt flanked by primer hybridization sequences comprising 23 nt on one side and 24 nt on the opposite end. The library sequence used was 72 nt in length: 5′-GGGAGACAAGAATAAACGCTCAA (random 25 nt region) TTCGACAGGAGGCTCACAACAGGC–3′. The amplification primers are as follows: 5′-end forward primer GGGAGACAAGAATAAACGCTCAA & 3′-end reverse primer GCCTGTTGTGAGCCTCCTGTCGAA. The combinatorial oligonucleotide library and amplification primers were HPLC purified and obtained by the Oligonucleotide and Peptide synthesis unit of the Queen’s Medical Centre, University of Nottingham, UK.
2.3. Biotinylation of the MUC1/Y Peptides
The MUC1/Y peptides (10mer and 20mer) were labelled with biotin–xx using a FluoReporter® biotin-XX Protein Labelling Kit (Invitrogen Molecular Probes, Paisley, UK). The biotinylation of the peptides was conducted according to the manufacturer´s protocol. The peptide and the biotin–xx SE (1:2 molar ratio, respectively) were incubated at room temperature for 2 h with gentle shaking. The labelled peptide reaction mixture was purified on a PD-10 (GE Healthcare, Chalfont St Giles, UK) desalting column using PBS (pH 7.2) to elute the fractions.
2.4. Selection of Aptamers
Aptamers were selected using a mixture of one-round selections on PCR tubes and a consecutive selection of 8 rounds using affinity chromatography, as follows:
2.4.1. Single Round Selection on Streptavidin Coated Tubes
This single round selection protocol employs immobilized biotinylated peptides on streptavidin-coated PCR tubes. The streptavidin-coated PCR tubes have a binding capacity of 15 ng/tube for biotin. The immobilization of the peptides in each streptavidin coated PCR tube was conducted in a 1:2 ratio of the biotin binding capacity (15 ng) to the biotinylated peptides (30 ng) and incubated for 3 min at 37 °C. Upon completion of the incubation period, excess biotinylated peptides were removed. The aptamer library was incubated with the immobilized MUC1/Y 10mer and 20mer peptide at room temperature for an hour with gentle shaking. Upon completion of the incubation period, excess aptamer library was removed, followed by a single wash with a binding solution. Two variations were used in the elution protocol, based on increased salt concentration or increased temperature, to effect denaturation of the aptamer-peptide complex. Salt Elution: The elution of the aptamers was performed via washing each tube individually with increasing salt concentrations, using an initial wash with 0.2 M NaCl and further elution washes using 0.1 M increments of NaCl concentration to a maximum of 1.5 M NaCl. Temperature Elution: The aptamers were eluted by washing each tube individually with 100 µL sterile water at increasing temperatures following an initial wash with sterile water at 25 °C for 2 min. Subsequently, the incubated water was removed and transferred to a fresh tube. Further elution wash steps were conducted in steps from 25 °C to a maximum of 95 °C temperature, using 5 °C increments.
2.4.2. Affinity Chromatography-Based Selection
The 20mer peptide was immobilised onto a Sepharose resin and packed into a 1 mL column. The amplified aptamer library in potassium phosphate buffer (KPB) was incubated with the peptide for 1 h at 37 °C (rotating continuously). The column was subsequently washed with KPB to eliminate unbound species, and bound aptamers were eluted in KPB with increasing salt concentrations from 1–1.5 M NaCl (0.1 M increments) and then a final wash with 3 M NaSCN. Following desalting, the fractions were amplified using unidirectional PCR and reloaded into the column in a series of 8 rounds of selection and amplification. At the last round, fractions were desalted, amplified, and visualised on a 2% agarose gel. Aptamers were observed up to the highest end of the gradient, the 3 M NaSCN fraction, and these were subsequently cloned and sequenced. Three aptamers resulted from this second selection step [
11].
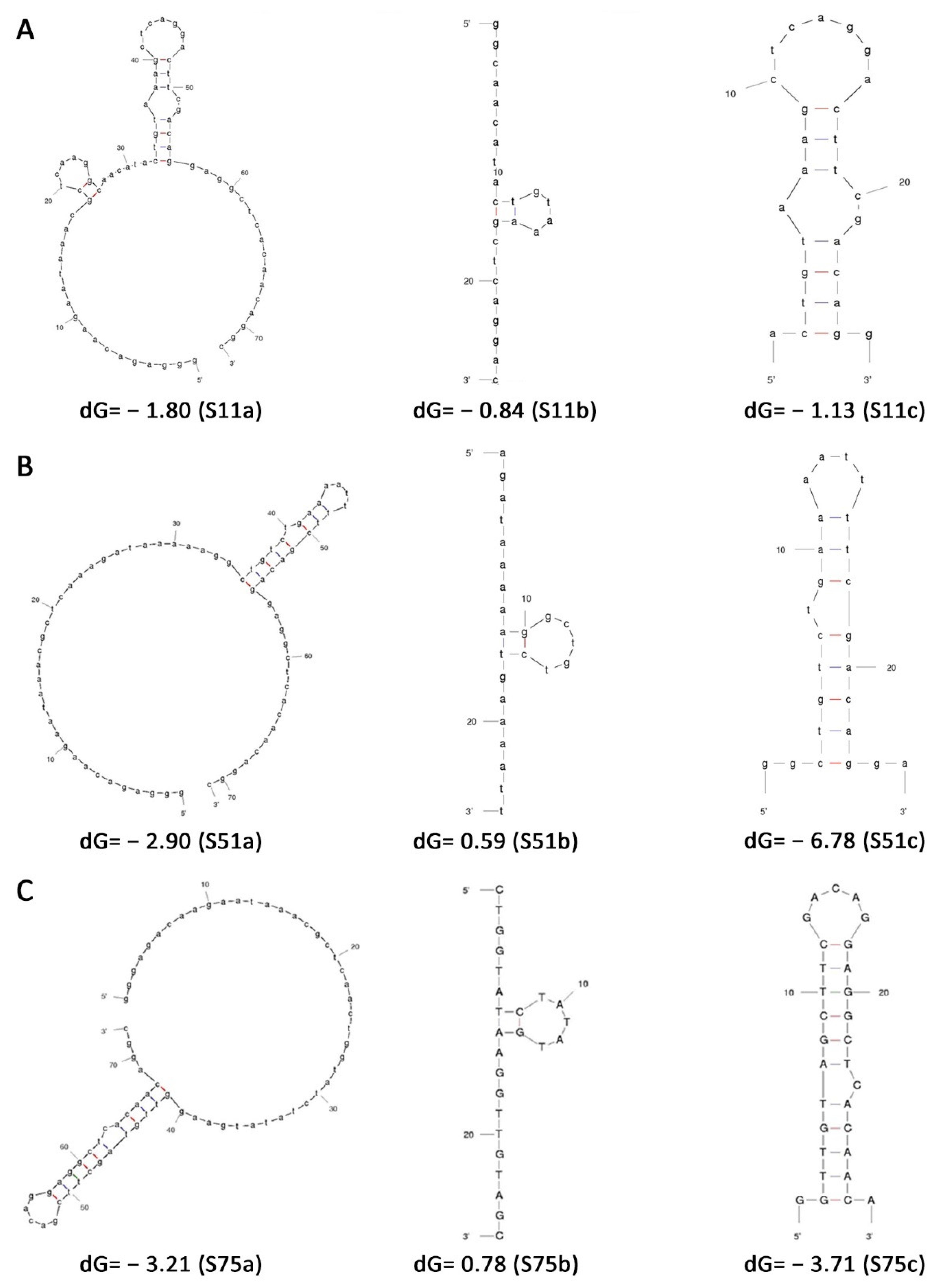
All selected aptamers were analysed for secondary structure, based on the mFold program, available online free-of-cost at the MFold Web Server of the University of Albany [
20]. All the structures were predicted at 37 °C with 100 mM NaCl and 5 mM MgCl
2.
2.5. Ru (II) Complex Displacement Assay
2.5.1. Ru (II) Complex Binding to the Aptamer (in Binding Solution)
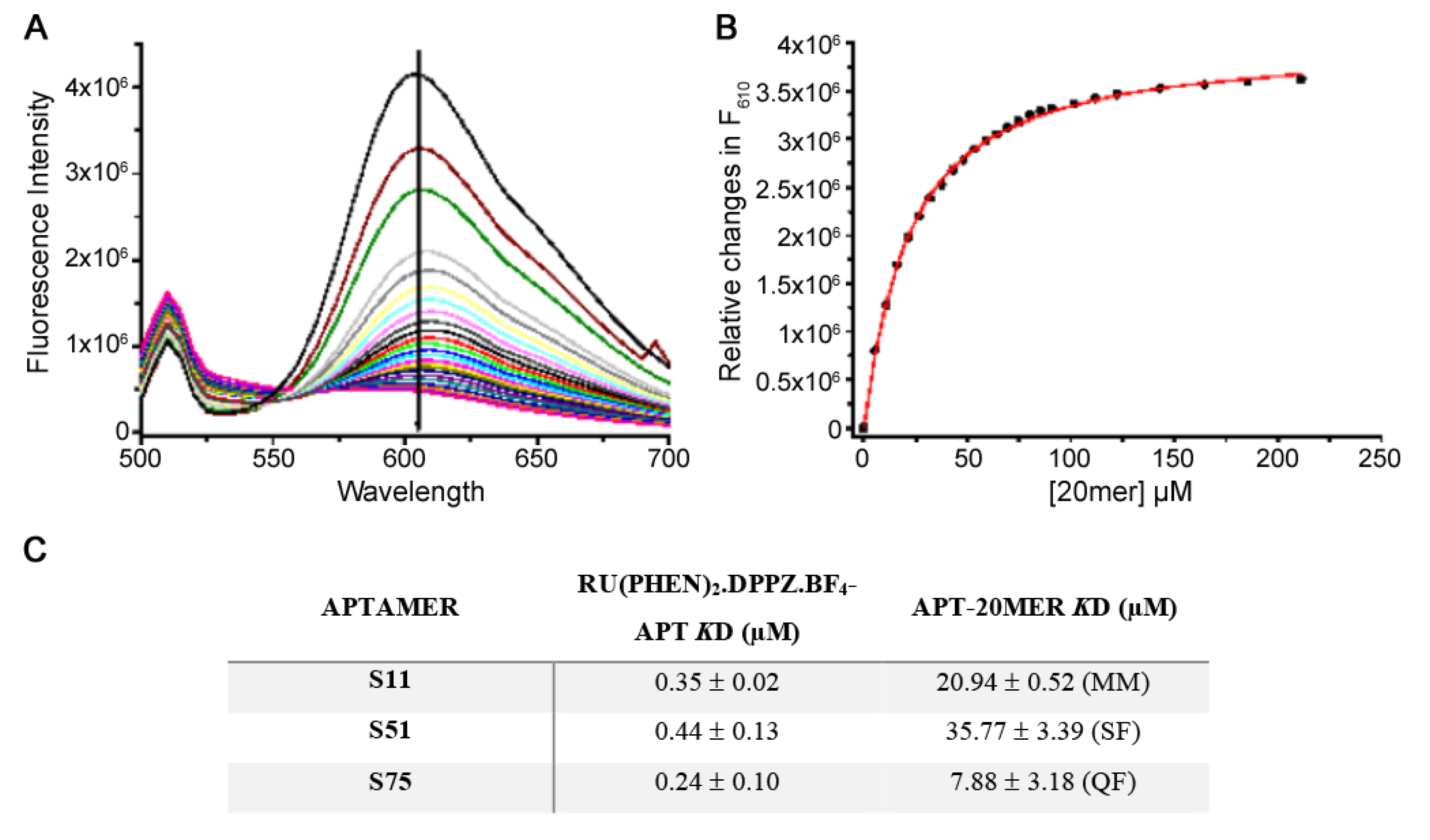
A stock solution of Ru (II) complex was titrated into 50 nM aptamer. Upon each aliquot, the emission spectrum (excitation wavelength: 434 nm) of the solution was recorded (until no spectral changes could be observed upon further additions). In order to quantify the binding, fluorescence changes at 610 nm were plotted as a function of Ru(phen)2.dppz.BF4 concentration. The data were best fit to a sigmoidal function from which a value for Kd was determined.
2.5.2. Peptide (20mer) Binding to the Aptamer and Displacement of the Ru (II) Complex
The individual aptamers (50 nM) were mixed with near saturating concentrations of the dye (determined from titration experiments: 900 nM for S11, 1.5 µM for S51 and 2 µM for S75) and incubated until no fluorescent changes could be observed (~30 min). Subsequently, a stock solution of the 20mer peptide was titrated into the aptamer-dye solution and the emission spectrum was recorded (until no further changes could be observed). The relative fluorescence decrease at 610 nm was plotted as a function of peptide concentration, and data could fit to a sigmoidal, hyperbolic or quadratic function equally well. The fluorescence of the 20mer was recorded to see if it emits at the same wavelength used for the assay (i.e., between 550–650 nm). The 20mer only slightly emits at 610 nm, which is insignificant compared with the fluorescence changes observed during the ruthenium assay titrations. The fluorescence of the peptide only becomes apparent when the aptamer becomes saturated with the peptide in excess, and all the dye is displaced. Although it is reported that the dye itself has no fluorescence when in a solution, we observed a small and relatively insignificant signal compared with that of the dye when it is intercalated into DNA; this was considered when assessing data from experiments following the titration of the dye into the aptamer, by subtracting the emission of the free dye from the intercalating fluorescence of the dye-aptamer complex.
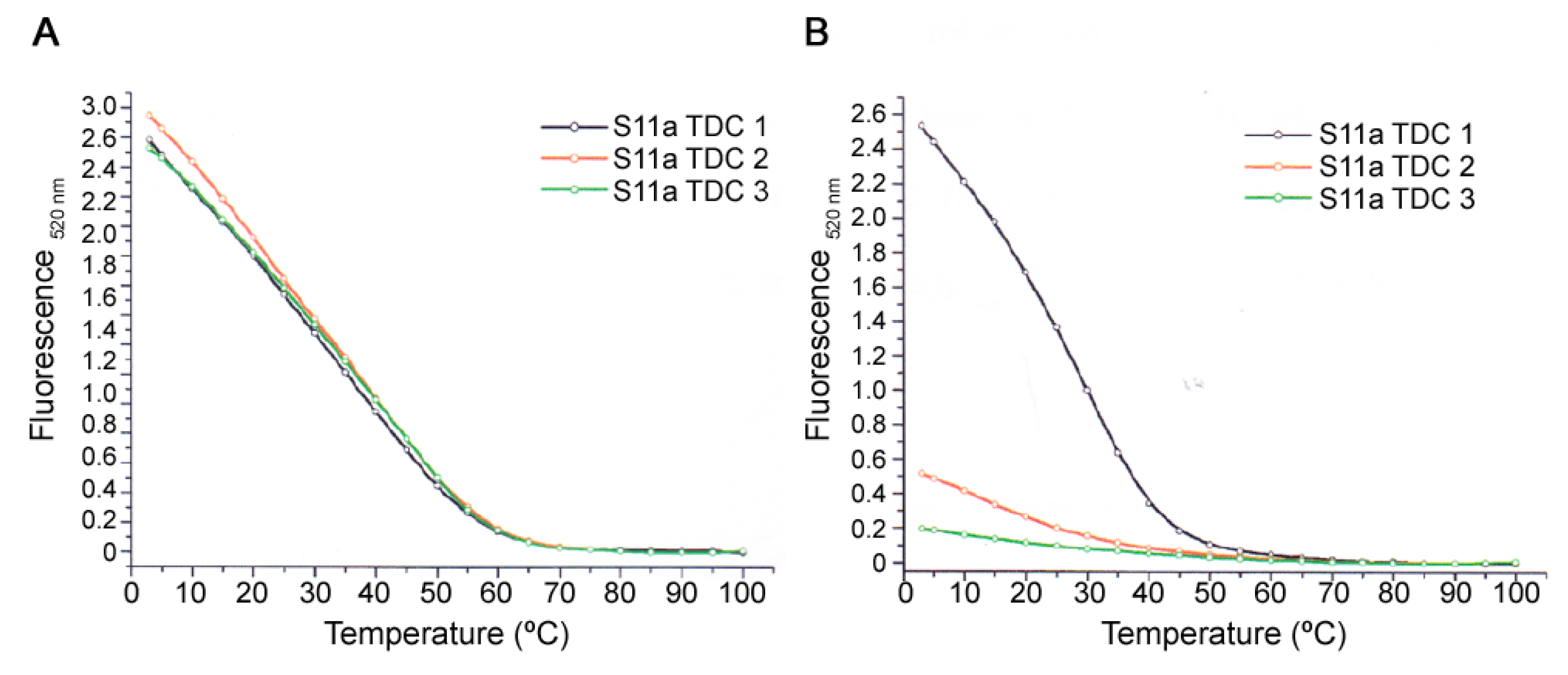
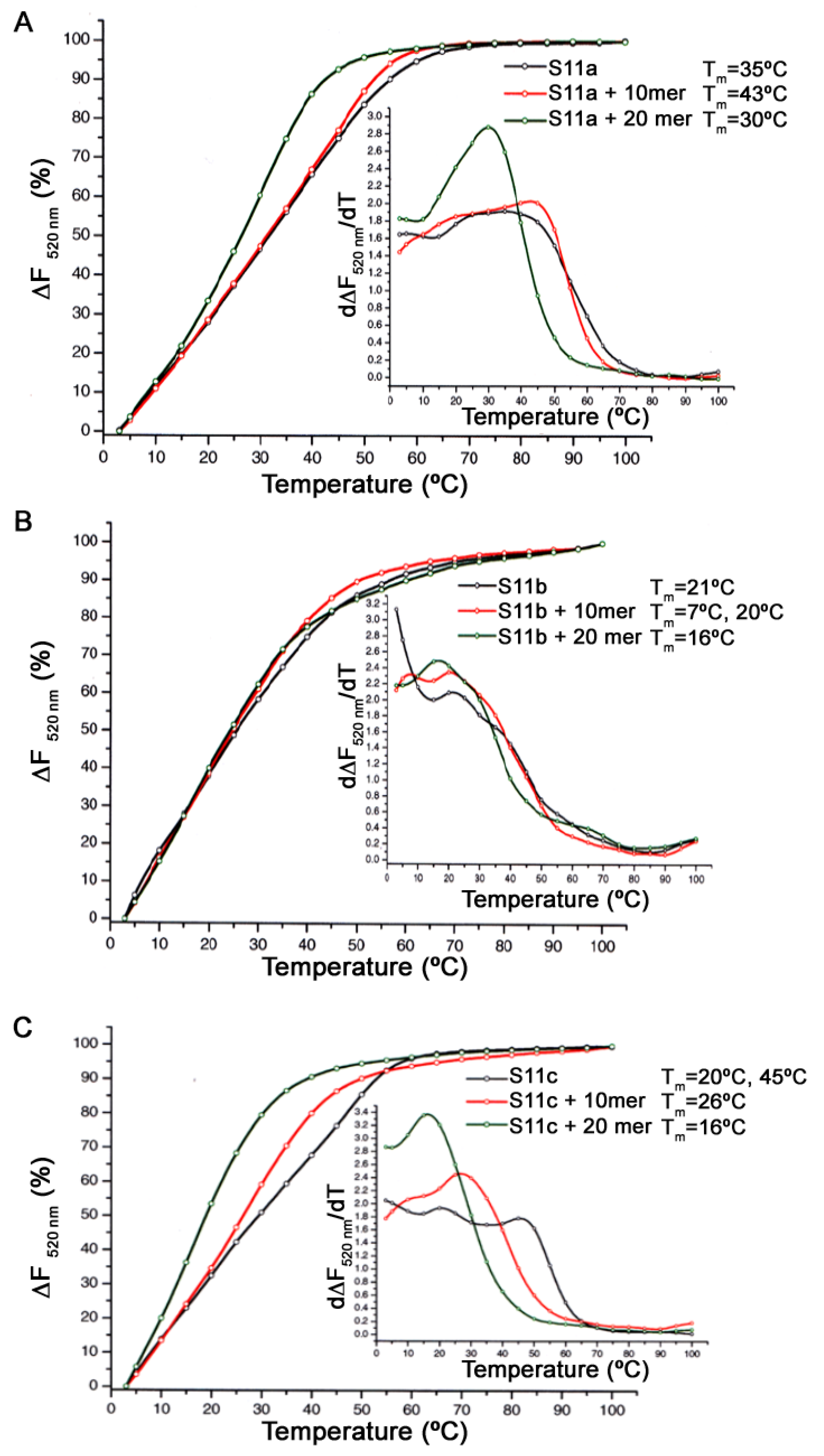
2.6. Thermal Denaturation Assay
A mixture composed of the aptamer (0.180 mM) and either the 10mer or 20mer peptide (180 mM) in a 1:1000 ratio aptamer to the peptide in binding solution was incubated for 1 h at ambient temperature. Subsequently, the 10× SYBR green I nucleic acid gel stain (2.5 mL) was added to the reaction mixture and kept on ice (4 °C) for 30 min. The aptamer-peptide mixture was heated from 3 °C to 100 °C at a 1 °C/min rate (denaturation step), followed by a cooling down process from 100 °C to 3 °C at a 1 °C/min rate (renaturation step). The fluorescence emission was recorded every minute during the denaturation step; for every 10 °C, increments from 80 °C to 10 °C were also recorded and thereafter, at 5 °C and 3 °C. The denaturation-renaturation cycles were repeated in triplicates and ran on a DNA Engine Opticon 2 Real-Time PCR (MJ Research, Waltham, MA, USA.) at 520 nm using excitation at 497 nm. The final analysis of the data was conducted using the software Origin 6.0. (Origin Lab Corporation, Northampton, UK) The SYBR green I nucleic acid gel stain was purchased from Sigma–Aldrich (Dorset, UK).
2.7. Serum Stability Assay
Serum stability assays were performed for each aptamer in mouse and human serum, purchased from Sigma–Aldrich (Dorset, UK), using a DNase solution as the control. The assay consisted of incubating a 3 µM solution of each aptamer in serum (final concentration of 93%) or with 0.1 U/mL DNase at 37 °C. At designated time points, the reaction was quenched and frozen immediately in liquid nitrogen. PAGE analysis was used to visualise the degradation of the aptamers in time. The stability assays in human serum were initially performed over 4 h but were subsequently repeated over 24 h to acquire a complete picture of the aptamer stability.
2.8. Cell Culture
MCF-7 (ATCC: HTB-22, breast cancer cell line), DU145 (ATCC: HTB-81, prostate cancer cell line), Calu-6 (ATCC: HTB-56, lung cancer cell line), A498 (ATCC: HTB-44, kidney cancer cell line), SK-OV-3 (ATCC: HTB-77, ovarian cancer cell line), HT-29 (ATCC: HTB-38, colon cancer cell line), PC-3 (ATCC: CRL-1435), OVCAR-3 (ATCC: HTB-161, ovarian cancer cell line), A549 (ATCC: CCL-185, lung cancer cell line), and MDA-MB-231 (ATCC: HTB-26, breast cancer cell line) were grown at 37 °C and 5% CO2 in minimal essential medium Eagle (MEM) cell culture medium supplemented with 10% fetal bovine serum and, 1% L-glutamine. Trypsin-EDTA, minimal essential medium Eagle (MEM), L-Glutamine, fetal bovine serum and cell culture flasks (75 cm2, surface treated with filter cap) were purchased from PAA Laboratories (Somerset, UK). Hanks balanced salt solution was obtained from Sigma–Aldrich (Dorset, UK).
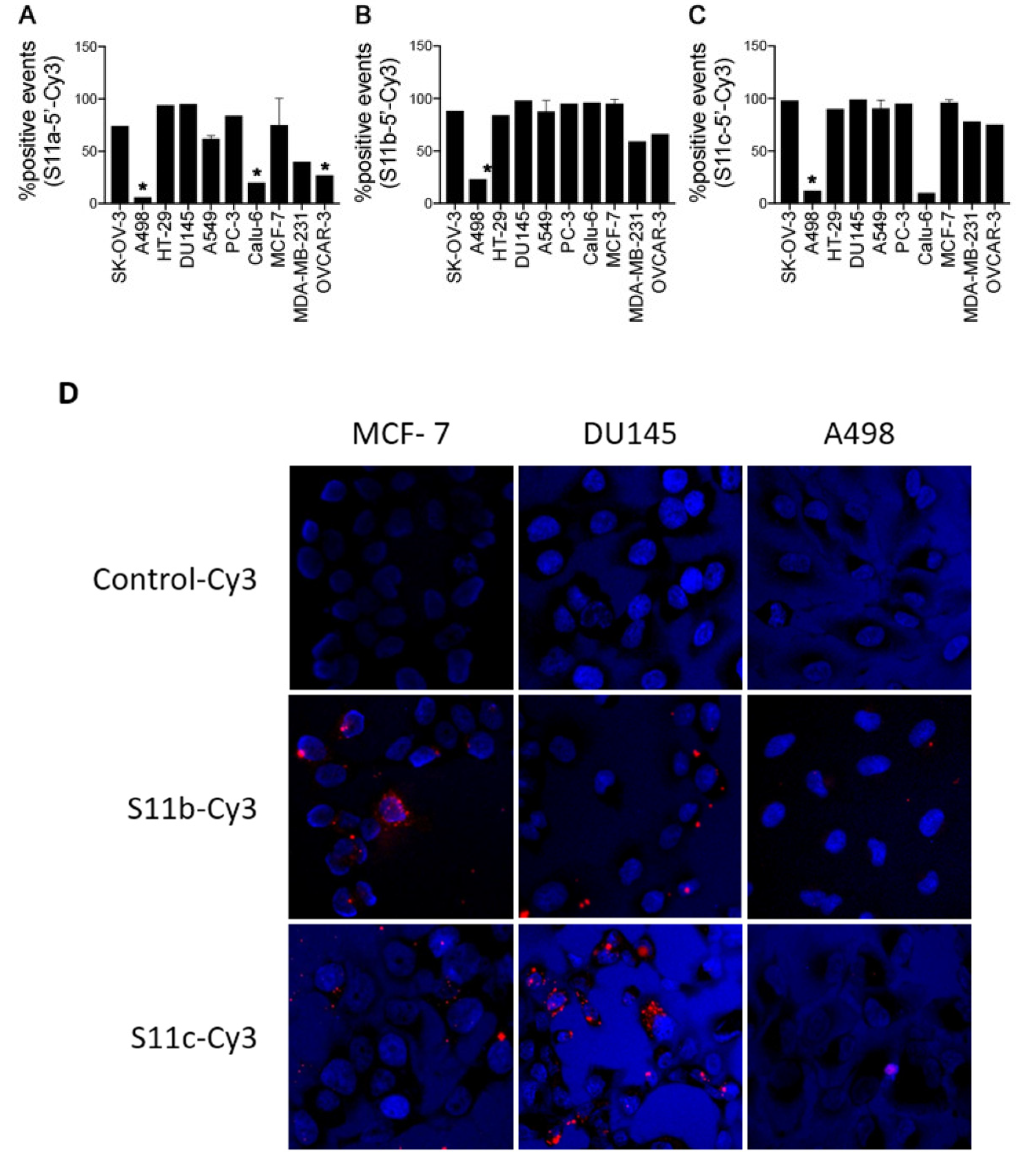
2.9. Fluorescence Activated Cell Sorting (FACs)
All cells used in the FACS analysis were washed with PBS. Fluorescently labelled (Cy3) aptamers in PBS were placed at 95 °C for 5 min and were subsequently cooled on the bench slowly. Cy3 fluorescently labelled aptamers (2 µM) were incubated with 500,000 cells in 200 µL of PBS at 4 °C for 1 h. After incubation, the cells were washed by centrifugation for 5 min at 1000 rpm with PBS (400 µL × 3) and resuspended in PBS (400 µL), aiming for a total cell count of either 5000 or 10,000 cells, for analysis on the flow cytometer. The analysis was performed on a FACS Calibur, Becton Dickinson flow cytometer (BD, San Jose, CA, USA) with a maximum sort rate of 10,000 total cells/second. Purchases of 5’ Cy3 labelled 25 oligonucleotides were from Integrated DNA Technologies (Budapest, Hungary). PBS tablets were purchased from Sigma–Aldrich (Dorset, UK).
2.10. Fluorescence Microscopy
MCF-7, DU145, CALU-6 and A498 cells (50,000 cells) were plated in duplicates on round glass coverslips, placed inside each well of a 24-well cell culture plate and grown overnight. The adhered cells were washed with PBS (100 µL), followed by incubation with aptamer (0.5 µM) in serum-free MEM (300 µL) per well for 3 h at 37 °C. Subsequently, the cells were washed with PBS (200 µL × 3), followed by the addition of 4% paraformaldehyde (PFA) (200 µL) and incubated for 15 min at room temperature. The cells were then washed with PBS (200 µL × 2), treated with 0.1% Triton (200 µL) for 10 min at room temperature, followed by a PBS wash (200 µL × 2). The cells were then incubated with Hoechst 33258 (75 µL of 1 mg/mL) for 15 min at room temperature, followed by a PBS wash (200 µL × 3). The glass inserts were mounted onto glass microscope slides using CFPVOH (polyvinyl alcohol) and AF100 anti-fadent (10:1 ratio). The slides were visualised under a BX6l motorized Olympus DP72 fluorescence microscope digital camera, using Cy3 and DAPI filter with 40× magnification. The images were analysed using the software ImageJ (U.S. National Institutes of Health, Bethesda, MD, USA).
2.11. Cell Viability Studies
SRB assays were performed using DU145, MCF-7 and A498 (negative control) tumour cell lines in 96-well cell culture plates. Cells were plated at 1000 cells/well for DU145 and MCF-7, and 2000 cells/well for the A498 cell line and were incubated at 37 °C overnight (the cell densities varied in the optimisation experiments, as stated in the results). Aptamers (in PBS) were at concentrations starting from 100 µM with subsequent two-fold dilutions across the plate (in media) and were incubated with the cells (in triplicate measurements) at 37 °C for 3 or 6 days (depending on the experiment). Prior to incubation, the aptamers were placed at 95 °C for 5 min and left to cool on the bench and filtered through a 0.2 µm filter unit. After the incubation period, the cells were fixed by removing the growth media and adding 10% TCA in fresh media, leaving the plates at 4 °C for 1 h. Plates were then washed with distilled water and left until completely dry. Subsequently, 0.4% SRB in 1% acetic acid (100 µL) was added to the wells and incubated for 30 min, followed by washing with distilled water, drying, and the addition of 100 µL of 20 mM Tris-HCl, pH 10.5. The absorbance at 564 nm for individual plates was immediately recorded.
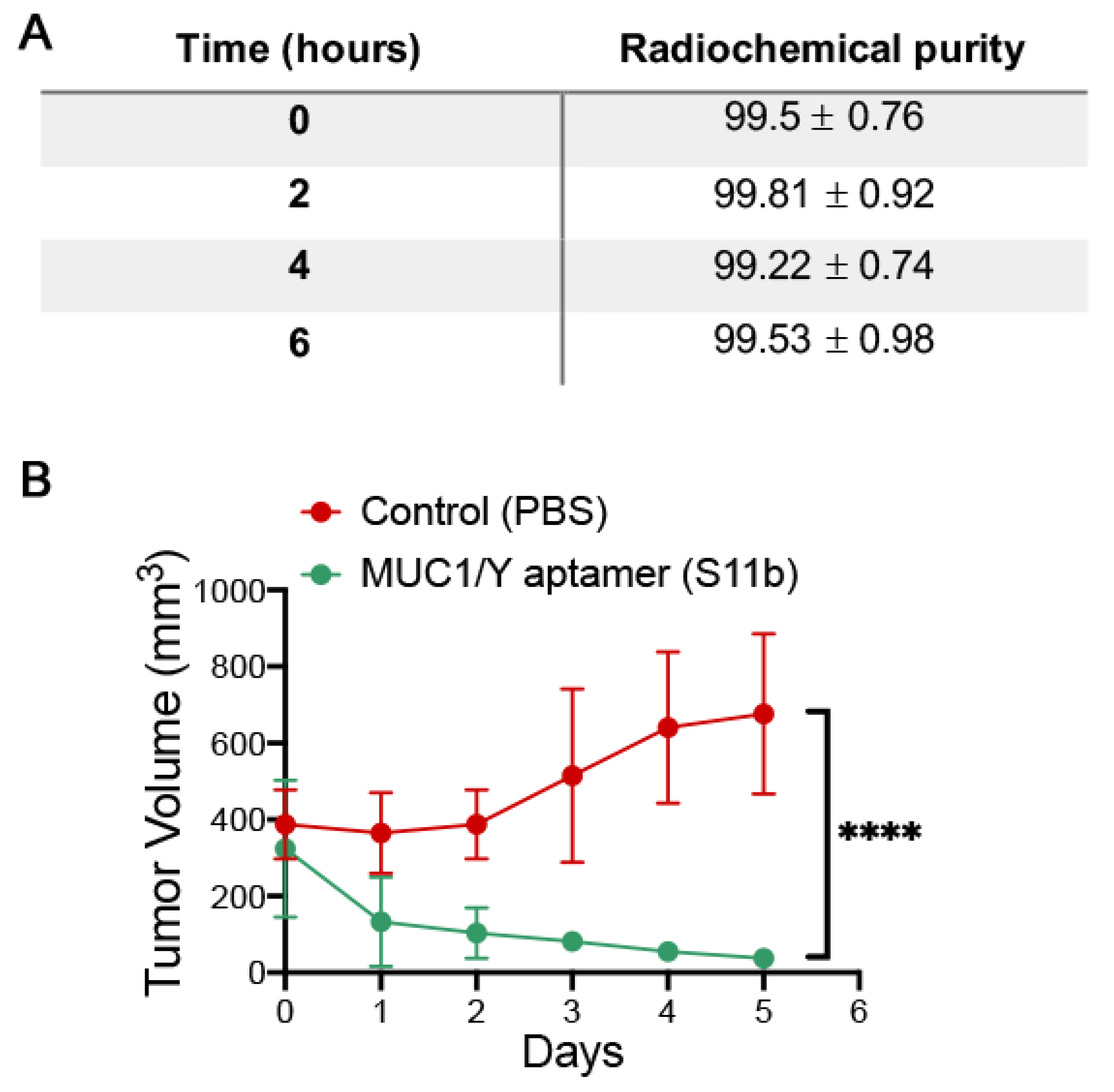
2.12. In Vivo Studies
In order to evaluate the clinical application of the S11b DNA Aptamer against the MUC1/Y, preclinical studies in MDA-MB-231 tumour-bearing mice were conducted. NOD/scid mice were bred at the animal facility of IPEN (Brazil); all experiments complied with the relevant laws and were approved by the local animal ethics committee protocol approval 181/17 (date of approval 19 January 2017). Mice were housed in standard ventilated cages with controlled 12:12 h light/dark cycles, and provided unlimited access to water and standard rodent feed. All animal handling followed institutional guidelines.
For tumour induction, 20 eight-week-old female NOD/scid mice were subcutaneously injected with 2 × 106 MDA-MB-231 cells and growth was accompanied for 5 weeks. Mice were observed three times per week for evidence of distress, ascites, paralysis, or excessive weight loss. When the tumour reached approximately 350 mm3, mice were divided into 2 groups:
Group 1: A total of ten MDA-MB-231 tumour-bearing mice were treated intraperitoneally for 5 consecutive days with 100 mg/Kg of S11b DNA aptamer.
Group 2: Control group—A total of ten MDA-MB-231 tumour-bearing mice were treated intraperitoneally for 5 consecutive days with a saline solution (0.9% NaCl).
2.13. Pharmacokinetic of S11b DNA Aptamer against the MUC1/Y
The pharmacokinetics (PK) parameters: concentration at zero time, elimination constant (K), the volume of distribution, elimination of half-life and blood clearance were evaluated in healthy Wistar rats (n = 12). In order to perform the PK analysis, 200 µg of radiolabelled S11b DNA aptamer was injected retro-orbitally into healthy Wistar rats, and at times of 1, 2, 4, 8, and 24 h, 100 µL of blood was collected from the tail vein. The conversion of radioactive activitye to mass of S11b DNA aptamer against the MUC1/Y was calculated considering the initial mass (200 µg) administered and the labelling quality control data using the radioactive decay formula: .
2.14. Radiolabelling of S11b DNA Aptamer against the MUC1/Y with 99mTc
The radiolabelling process was done using 150 µg of S11b DNA aptamer against the MUC1/Y incubated in stannous chloride (SnCl2) solutions (80 µL/mL) (Sigma–Aldrich, St Louis, MO, USA) for 20 min at room temperature. Then, this solution was incubated with 100 µCi (approximately 300 µL) of technetium-99m (99mTc) for another 10 min. In order to establish the quality control of the labelling process, radio thin layer chromatography (RTLC) was performed using Whatman paper no.1 as the stationary phase, 200 µL of 99mTc- S11b DNA aptamer against the MUC1/Y as spot point and acetone (Sigma–Aldrich, St Louis, MO, USA) as the mobile phase. The radioactivity of the strips was verified in a γ-counter (Perkin Elmer Wizard® 2470, Shelton, CT, USA). The RTLC was performed in triplicate for each time point (0, 2, 4, and 6 h).
4. Discussion
The MUC1 gene encodes for a transmembrane glycoprotein overexpressed and aberrantly glycosylated in several types of epithelial cancers, which plays an important role in the progression of the disease [
27]. Tumour-associated MUC1 differs from that expressed in healthy cells by presenting increased sialylation and loss of core 1 O-glycans and abnormal cellular distribution [
28,
29]. These features are responsible for tumour cell loss of polarity, redistribution of cell surface growth receptors such as EGFR and hyperactivation of a critical signalling pathway that drives tumour progression [
30].
Several isoforms of MUC1, such as MUC1/A, MUC1/B, MUC1/C, MUC1/D, MUC1/X (or MUC1/Z), MUC1/Y, and MUC1/ZD, are discovered in humans as a result of alternative splicing, exon skipping, and intron retention. MUC1/Y, in particular, was found to be highly expressed in breast, ovarian, and prostate cancer cells [
9,
23]. MUC1/Y has been implicated in several cellular functions, such as the overexpression of cytokines and the modulation of immune responses, through its binding with MUC1/SEC. Furthermore, MUC1/Y has been shown to induce the transcription of proinflammatory cytokines via NF-kB [
8,
9,
31].
In this work, we selected aptamers against the MUC1/Y alternative splice variant and characterized them for their stability, binding affinity, capacity to bind to cell lines expressing MUC1/Y, and as tools to determine the presence of MUC1/Y in cell lines with unknown expression patterns. Furthermore, with the intention of developing such aptamers as potential anticancer agents, we tested their capacity to inhibit tumour cell growth in vitro and in vivo. As a result, we demonstrated for the first time a potential role of MUC1/Y in cancer growth and proliferation in a mouse model of breast cancer and its potential as a target for anticancer therapeutics. Even though we used only the exposed peptide at the splice region of the MUC1/Y as the target and selected aptamers showed a relatively low affinity in terms of their Ru (II) complex displacement constant towards the peptide, they were capable of binding to cancer cell lines in flow cytometry and fluorescent microscopy experiments. Furthermore, these aptamers could reduce tumour cell growth both in vitro and in vivo in a MDA-MB-231 tumour-bearing mice model.
A previous demonstration showed that the interaction of MUC1/Y with MUC1/SEC could enhance second messenger proteins capable of eliciting pro-tumoural cellular responses [
9]; this may indicate that blocking MUC1/Y inhibits its interaction with MUC1/SEC and its binding to tumour cells, inhibiting their proliferation. Additionally, blocking the interaction of MUC1/Y with MUC1/SEC could have an anti-inflammatory effect, reducing tumour-associated inflammation through the blocking of proinflammatory cytokine expression.
Among all therapeutic molecules, DNA aptamers have increasingly drawn the attention of researchers due to their ability to stably bind a wide variety of molecules. DNA aptamers can be more specific than small molecules and peptides, but they are smaller and more flexible than antibodies or antibody fragments. Aptamers have a higher tissue penetration capacity than antibodies because of their smaller size; they are more effectively eliminated from the bloodstream and promote little or no immunogenicity [
32,
33]. MacugenTM remains the only aptamer approved by FDA (2004) to date with a therapeutic function, and its use is limited because of its choice of target, compared with Avastin. However, aptamers remain molecules of high therapeutic potential, with the nature of a biologic but production of a synthetic molecule. Our present study highlights their potential in the treatment of MUC1/Y-expressing tumours, as well as unveils the role of MUC1/Y in cancer growth and its potential as a target for cancer therapy.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}