
Current Status of the Use of Multifunctional Enzymes as Anti-Cancer Drug Targets



Abstract
:1. Introduction
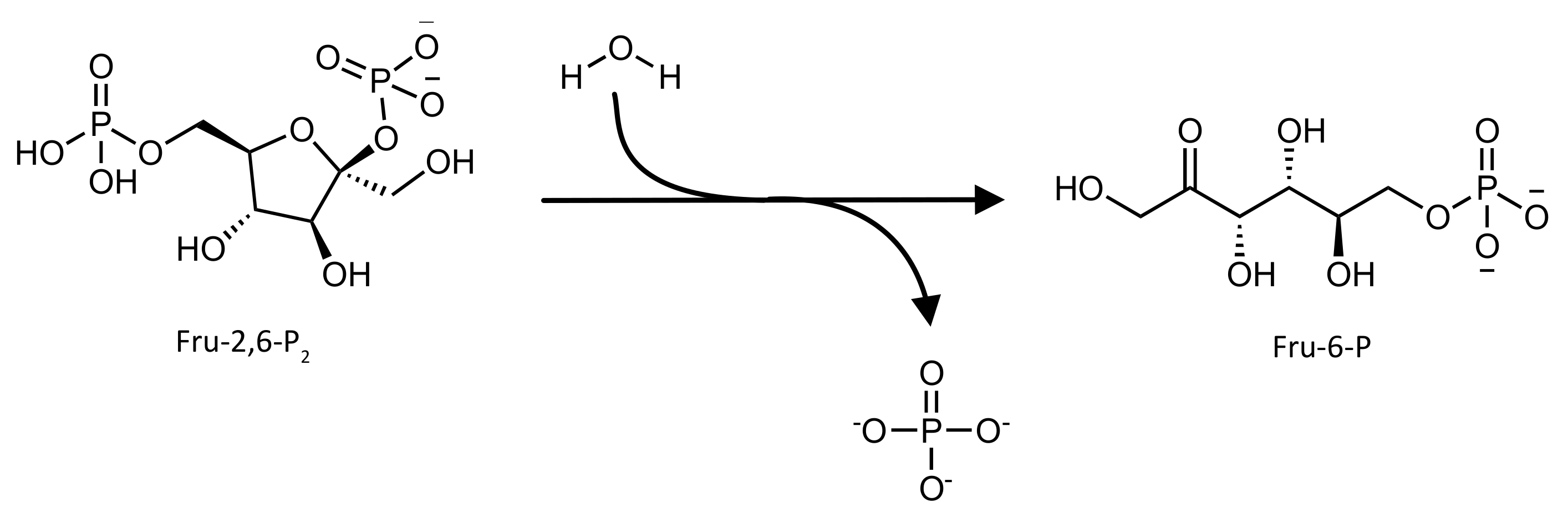
2. 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase (PFK-2/FBPase-2)
2.1. Biological Role
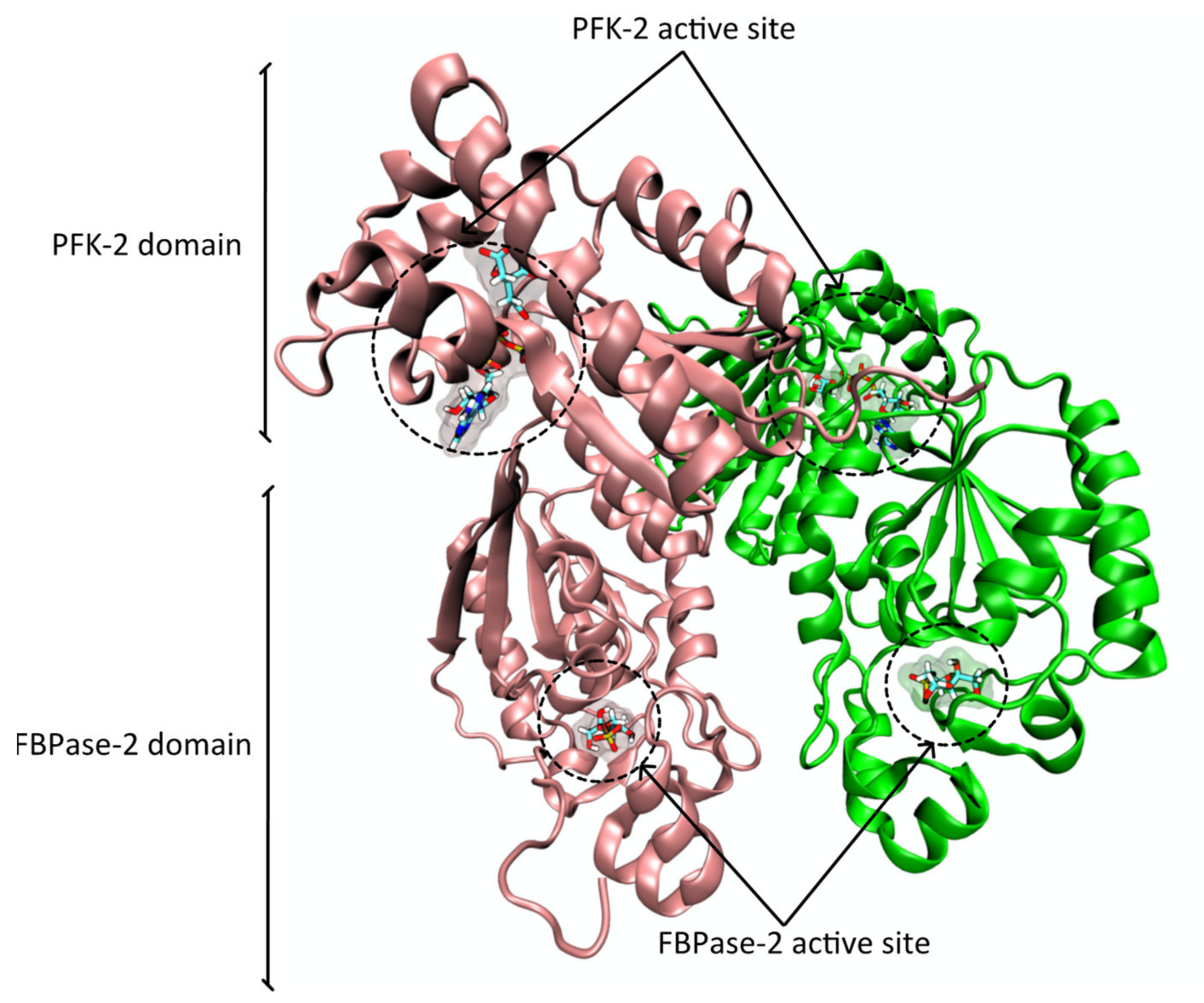
2.2. Protein Structure

2.3. Role in Cancer
2.4. Inhibitors
3. 5-aminoimidazole-4-carboxamide Ribonucleotide Formyltransferase/Inosine Monophosphate Cyclohydrolase (ATIC)
3.1. Biological Role
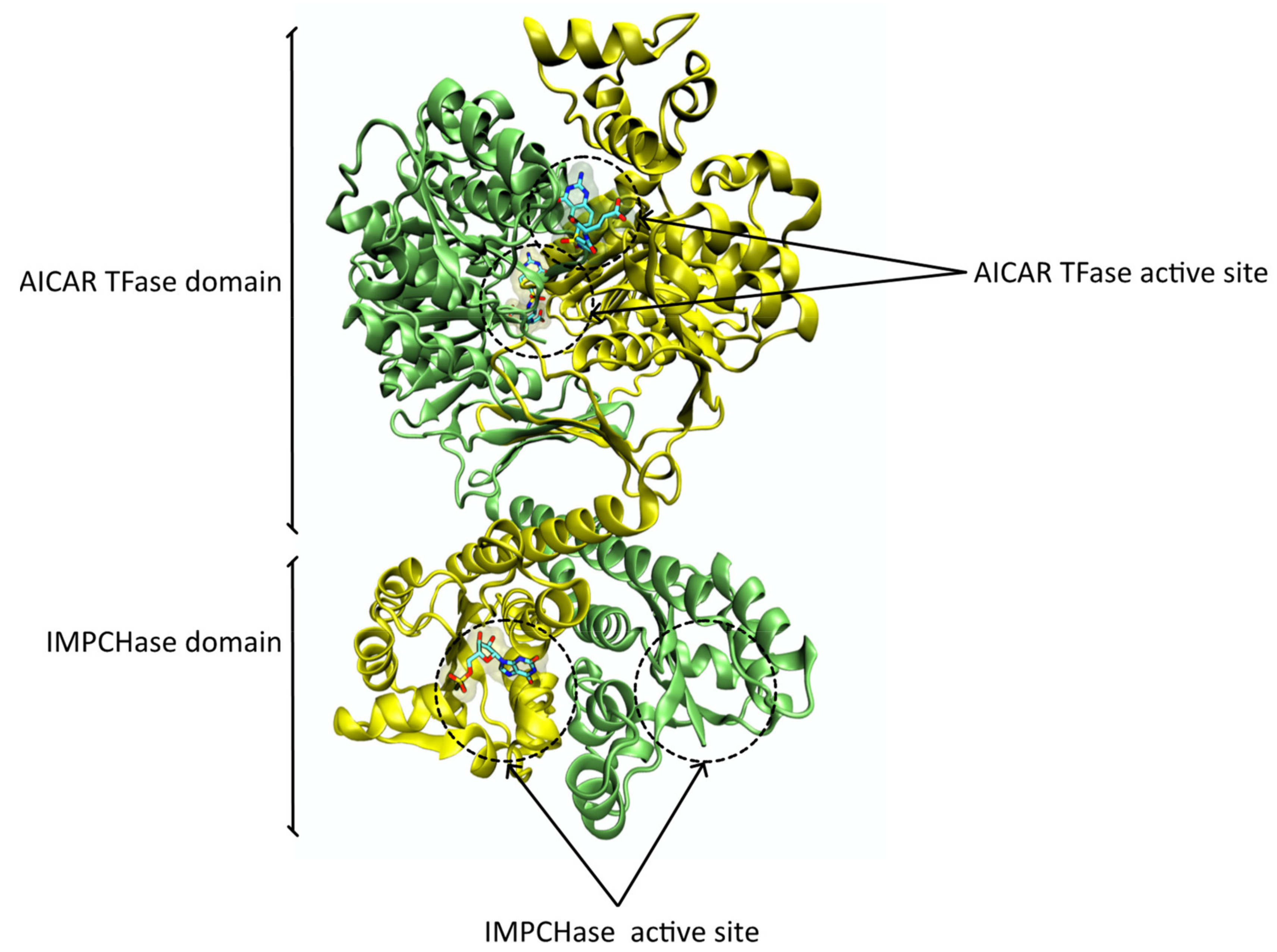
3.2. Protein Structure
3.3. Role in Cancer
3.4. Inhibitors
4. Leukotriene A4 Hydrolase (LTA4H)
4.1. Biological Role
4.2. Protein Structure
4.3. Role in Cancer
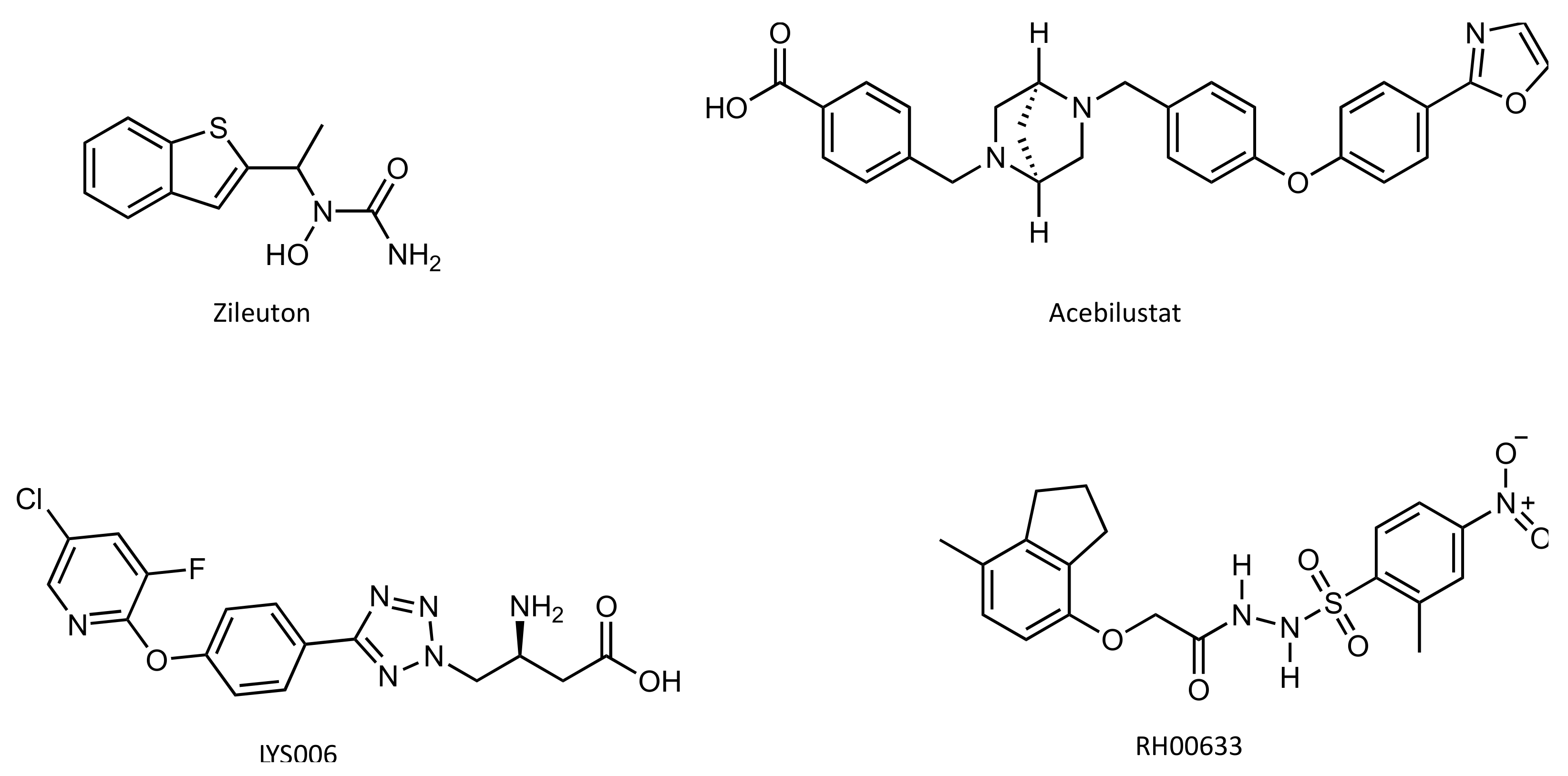
4.4. Inhibitors
5. Jumonji Domain-Containing Protein 6 (Jmjd6)
5.1. Biological Role
5.2. Protein Structure
5.3. Role in Cancer
5.4. Inhibitors
6. Current and Future Developments
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Counihan, J.L.; Grossman, E.A.; Nomura, D.K. Cancer Metabolism: Current Understanding and Therapies. Chem. Rev. 2018, 118, 6893–6923. [Google Scholar] [CrossRef] [PubMed]
- Salk, J.J.; Fox, E.J.; Loeb, L.A. Mutational Heterogeneity in Human Cancers: Origin and Consequences. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 51–75. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.; Singh, S.; Skvortsova, I.; Kumar, V. Promising Targets in Anti-cancer Drug Development: Recent Updates. Curr. Med. Chem. 2018, 24, 4729–4752. [Google Scholar] [CrossRef]
- Arruebo, M.; Vilaboa, N.; Sáez-Gutierrez, B.; Lambea, J.; Tres, A.; Valladares, M.; González-Fernández, Á. Assessment of the Evolution of Cancer Treatment Therapies. Cancers 2011, 3, 3279–3330. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal. Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Schramm, V.L. Transition States, Analogues, and Drug Development. ACS Chem. Biol. 2013, 8, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Rider, M.H.; Bertrand, L.; Vertommen, D.; Michels, P.A.; Rousseau, G.G.; Hue, L. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase: Head-to-head with a bifunctional enzyme that controls glycolysis. Biochem. J. 2004, 381, 561–579. [Google Scholar] [CrossRef] [Green Version]
- Hue, L.; Rider, M.H. Role of fructose 2,6-bisphosphate in the control of glycolysis in mammalian tissues. Biochem. J. 1987, 245, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Okar, D.A.; Lange, A.J. Fructose-2,6-bisphosphate and control of carbohydrate metabolism in eukaryotes. BioFactors 1999, 10, 1–14. [Google Scholar] [CrossRef]
- Pilkis, S.J.; El-Maghrabi, M.R.; Claus, T.H. Hormonal Regulation of Hepatic Gluconeogenesis and Glycolysis. Annu. Rev. Biochem. 1988, 57, 755–783. [Google Scholar] [CrossRef]
- Uyeda, K.; Furuya, E.; Luby, L.J. The effect of natural and synthetic D-fructose 2,6-bisphosphate on the regulatory kinetic properties of liver and muscle phosphofructokinases. J. Biol. Chem. 1981, 256, 8394–8399. [Google Scholar] [CrossRef]
- Wu, C.; Khan, S.A.; Peng, L.-J.; Lange, A.J. Roles for fructose-2,6-bisphosphate in the control of fuel metabolism: Beyond its allosteric effects on glycolytic and gluconeogenic enzymes. Adv. Enzym. Regul. 2006, 46, 72–88. [Google Scholar] [CrossRef]
- Pilkis, S.J.; Claus, T.H.; Kurland, I.J.; Lange, A.J. 6-Phosphofructo-2-Kinase/Fructose-2,6-Bisphosphatase: A Metabolic Signaling Enzyme. Annu. Rev. Biochem. 1995, 64, 799–835. [Google Scholar] [CrossRef]
- Okar, D.A.; Lange, A.J.; Manzano, À.; Navarro-Sabatè, A.; Riera, L.S.; Bartrons, R. PFK-2/FBPase-2: Maker and breaker of the essential biofactor fructose-2,6-bisphosphate. Trends Biochem. Sci. 2001, 26, 30–35. [Google Scholar] [CrossRef]
- Darville, M.I.; Crepin, K.M.; Hue, L.; Rousseau, G.G. 5’ flanking sequence and structure of a gene encoding rat 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. Proc. Natl. Acad. Sci. USA 1989, 86, 6543–6547. [Google Scholar] [CrossRef] [Green Version]
- Ros, S.; Schulze, A. Balancing glycolytic flux: The role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Manzano, A.; Rosa, J.L.; Ventura, F.; Pérez, J.X.; Nadal, M.; Estivill, X.; Ambrosio, S.; Gil, J.; Bartrons, R. Molecular cloning, expression, and chromosomal localization of a ubiquitously expressed human 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene (PFKFB3). Cytogenet. Genome Res. 1998, 83, 214–217. [Google Scholar] [CrossRef]
- Hasemann, C.A.; Istvan, E.S.; Uyeda, K.; Deisenhofer, J. The crystal structure of the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase reveals distinct domain homologies. Structure 1996, 4, 1017–1029. [Google Scholar] [CrossRef] [Green Version]
- Tauler, A.; Lange, A.J.; el-Maghrabi, M.R.; Pilkis, S.J. Expression of rat liver 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase and its kinase domain in Escherichia coli. Proc. Natl. Acad. Sci. 1989, 86, 7316–7320. [Google Scholar] [CrossRef] [Green Version]
- Tauler, A.; Rosenberg, A.H.; Colosia, A.; Studier, F.W.; Pilkis, S.J. Expression of the bisphosphatase domain of rat liver 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase in Escherichia coli. Proc. Natl. Acad. Sci. USA 1988, 85, 6642–6646. [Google Scholar] [CrossRef] [Green Version]
- Sakakibara, R.; Kato, M.; Okamura, N.; Nakagawa, T.; Komada, Y.; Tominaga, N.; Shimojo, M.; Fukasawa, M. Characterization of a Human Placental Fructose-6-Phosphate, 2-Kinase/Fructose-2,6-Bisphosphatase. J. Biochem. 1997, 122, 122–128. [Google Scholar] [CrossRef]
- Kotowski, K.; Rosik, J.; Machaj, F.; Supplitt, S.; Wiczew, D.; Jabłońska, K.; Wiechec, E.; Ghavami, S.; Dzięgiel, P. Role of PFKFB3 and PFKFB4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential as Therapeutic Targets. Cancers 2021, 13, 909. [Google Scholar] [CrossRef]
- Yi, M.; Ban, Y.; Tan, Y.; Xiong, W.; Li, G.; Xiang, B. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 and 4: A pair of valves for fine-tuning of glucose metabolism in human cancer. Mol. Metab. 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Crochet, R.B.; Kim, J.-D.; Lee, H.; Yim, Y.-S.; Kim, S.-G.; Neau, D.; Lee, Y.-H. Crystal structure of heart 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB2) and the inhibitory influence of citrate on substrate binding. Proteins: Struct. Funct. Bioinform. 2017, 85, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Flier, J.; Mueckler, M.; Usher, P.; Lodish, H. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science 1987, 235, 1492–1495. [Google Scholar] [CrossRef]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of Glucose Transporter 1 and Glycolytic Gene Expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [Green Version]
- Hockel, M.; Vaupel, P. Biological consequences of tumor hypoxia. Semin. Oncol. 2001, 28, 36–41. [Google Scholar] [CrossRef]
- Ahn, C.S.; Metallo, C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Nissler, K.; Petermann, H.; Wenz, I.; Brox, D. Fructose 2,6-bisphosphate metabolism in Ehrlich ascites tumour cells. J. Cancer Res. Clin. Oncol. 1995, 121, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Mojena, M.; Bosca, L.; Hue, L. Effect of glutamine on fructose 2,6-bisphosphate and on glucose metabolism in HeLa cells and in chick-embryo fibroblasts. Biochem, J. 1985, 232, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miralpeix, M.; Azcon-Bieto, J.; Bartrons, R.; Argiles, J.M. The impairment of respiration by glycolysis in the Lewis lung carcinoma. Cancer Lett. 1990, 50, 173–178. [Google Scholar] [CrossRef]
- Barker, J.; Khan, M.A.A.; Solomos, T. Mechanism of the Pasteur Effect. Nature 1964, 201, 1126–1127. [Google Scholar] [CrossRef]
- Minchenko, O.H.; Ogura, T.; Opentanova, I.L.; Minchenko, D.O.; Ochiai, A.; Caro, J.; Komisarenko, S.V.; Esumi, H. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene family overexpression in human lung tumor. Ukr. Biokhim. Zh. 2005, 77, 46–50. [Google Scholar]
- Obach, M.; Navarro-Sabaté, À.; Caro, J.; Kong, X.; Duran, J.; Gómez, M.; Perales, J.C.; Ventura, F.; Rosa, J.L.; Bartrons, R. 6-Phosphofructo-2-kinase (pfkfb3) Gene Promoter Contains Hypoxia-inducible Factor-1 Binding Sites Necessary for Transactivation in Response to Hypoxia. J. Biol. Chem. 2004, 279, 53562–53570. [Google Scholar] [CrossRef] [Green Version]
- Minchenko, O.; Opentanova, I.; Minchenko, D.; Ogura, T.; Esumi, H. Hypoxia induces transcription of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase-4 gene via hypoxia-inducible factor-1α activation. FEBS Lett. 2004, 576, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Minchenko, O.; Opentanova, I.; Caro, J. Hypoxic regulation of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene family (PFKFB-1-4) expression in vivo. FEBS Lett. 2003, 554, 264–270. [Google Scholar] [CrossRef]
- Hirata, T.; Watanabe, M.; Miura, S.; Ijichi, K.; Fukasawa, M.; Sakakibara, R. Inhibition of Tumor Cell Growth by A Specific 6-Phosphofructo-2-kinase Inhibitor,N-Bromoacetylethanolamine Phosphate, and Its Analogues. Biosci. Biotechnol. Biochem. 2014, 64, 2047–2052. [Google Scholar] [CrossRef]
- Dasgupta, S.; Rajapakshe, K.; Zhu, B.; Nikolai, B.C.; Yi, P.; Putluri, N.; Choi, J.M.; Jung, S.Y.; Coarfa, C.; Westbrook, T.F.; et al. Metabolic enzyme PFKFB4 activates transcriptional coactivator SRC-3 to drive breast cancer. Nature 2018, 556, 249–254. [Google Scholar] [CrossRef]
- Guo, Q.; Chen, Q.; Zhang, Y.; Zhou, W.; Li, X.; Li, C.; Zhang, Y.; Chen, H.; Liu, P.; Chu, Y.; et al. Click-Nucleic-Acid-Containing Codelivery System Inducing Collapse of Cellular Homeostasis for Tumor Therapy through Bidirectional Regulation of Autophagy and Glycolysis. ACS Appl. Mater. Interfaces 2020, 12, 57757–57767. [Google Scholar] [CrossRef]
- Ros, S.; Santos, C.R.; Moco, S.; Baenke, F.; Kelly, G.; Howell, M.; Zamboni, N.; Schulze, A. Functional Metabolic Screen Identifies 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 4 as an Important Regulator of Prostate Cancer Cell Survival. Cancer Discov. 2012, 2, 328–343. [Google Scholar] [CrossRef] [Green Version]
- Anwar, T.; Kumar, P.; Khan, A.U. Modern Tools and Techniques in Computer-Aided Drug Design. In Molecular Docking for Computer-Aided Drug Design; Elsevier: Amsterdam, The Netherlands, 2021; pp. 1–30. [Google Scholar] [CrossRef]
- Wang, Y.; Qu, C.; Liu, T.; Wang, C. PFKFB3 inhibitors as potential anticancer agents: Mechanisms of action, current developments, and structure-activity relationships. Eur. J. Med. Chem. 2020, 203, 112612. [Google Scholar] [CrossRef]
- Clem, B.; Telang, S.; Clem, A.; Yalcin, A.; Meier, J.; Simmons, A.; Rasku, M.A.; Arumugam, S.; Dean, W.L.; Eaton, J.; et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol. Cancer Ther. 2008, 7, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Houddane, A.; Bultot, L.; Novellasdemunt, L.; Johanns, M.; Gueuning, M.-A.; Vertommen, D.; Coulie, P.G.; Bartrons, R.; Hue, L.; Rider, M.H. Role of Akt/PKB and PFKFB isoenzymes in the control of glycolysis, cell proliferation and protein synthesis in mitogen-stimulated thymocytes. Cell. Signal. 2017, 34, 23–37. [Google Scholar] [CrossRef]
- Clem, B.F.; O’Neal, J.; Tapolsky, G.; Clem, A.L.; Imbert-Fernandez, Y.; Kerr, D.A.; Klarer, A.C.; Redman, R.; Miller, D.M.; Trent, J.O.; et al. Targeting 6-Phosphofructo-2-Kinase (PFKFB3) as a Therapeutic Strategy against Cancer. Mol. Cancer Ther. 2013, 12, 1461–1470. [Google Scholar] [CrossRef] [Green Version]
- Mondal, S.; Roy, D.; Sarkar Bhattacharya, S.; Jin, L.; Jung, D.; Zhang, S.; Kalogera, E.; Staub, J.; Wang, Y.; Xuyang, W.; et al. Therapeutic targeting of PFKFB3 with a novel glycolytic inhibitor PFK158 promotes lipophagy and chemosensitivity in gynecologic cancers. Int. J. Cancer 2019, 144, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Phase 1 Safety Study of ACT-PFK-158, 2HCl in Patients with Advanced Solid Malignancies. Available online: https://clinicaltrials.gov/ (accessed on 5 December 2021).
- Chesney, J.; Clark, J.; Lanceta, L.; Trent, J.O.; Telang, S. Targeting the sugar metabolism of tumors with a first-in-class 6-phosphofructo-2-kinase (PFKFB4) inhibitor. Oncotarget 2015, 6, 18001–18011. [Google Scholar] [CrossRef]
- Feng, C.; Li, Y.; Li, K.; Lyu, Y.; Zhu, W.; Jiang, H.; Wen, H. PFKFB4 is overexpressed in clear-cell renal cell carcinoma promoting pentose phosphate pathway that mediates Sunitinib resistance. J. Exp. Clin. Cancer Res. 2021, 40, 308. [Google Scholar] [CrossRef]
- Zhang, Y.; Morar, M.; Ealick, S.E. Structural biology of the purine biosynthetic pathway. Cell. Mol. Life Sci. 2008, 65, 3699–3724. [Google Scholar] [CrossRef] [Green Version]
- Yamaoka, T.; Kondo, M.; Honda, S.; Iwahana, H.; Moritani, M.; Ii, S.; Yoshimoto, K.; Itakura, M. Amidophosphoribosyltransferase Limits the Rate of Cell Growth-linked de Novo Purine Biosynthesis in the Presence of Constant Capacity of Salvage Purine Biosynthesis. J. Biol. Chem. 1997, 272, 17719–17725. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2016, 14, 11–31. [Google Scholar] [CrossRef]
- Pareek, V.; Pedley, A.M.; Benkovic, S.J. Human de novo purine biosynthesis. Crit. Rev. Biochem. Mol. Biol. 2020, 56, 1–16. [Google Scholar] [CrossRef]
- Cheong, C.-G.; Wolan, D.W.; Greasley, S.E.; Horton, P.A.; Beardsley, G.P.; Wilson, I.A. Crystal Structures of Human Bifunctional Enzyme Aminoimidazole-4-carboxamide Ribonucleotide Transformylase/IMP Cyclohydrolase in Complex with Potent Sulfonyl-containing Antifolates. J. Biol. Chem. 2004, 279, 18034–18045. [Google Scholar] [CrossRef] [Green Version]
- Greasley, S.E.; Horton, P.; Ramcharan, J.; Beardsley, G.P.; Benkovic, S.J.; Wilson, I.A. Crystal structure of a bifunctional transformylase and cyclohydrolase enzyme in purine biosynthesis. Nat. Struct. Biol. 2001, 8, 402–406. [Google Scholar] [CrossRef]
- Vergis, J.M.; Bulock, K.G.; Fleming, K.G.; Beardsley, G.P. Human 5-Aminoimidazole-4-carboxamide Ribonucleotide Transformylase/Inosine 5′-Monophosphate Cyclohydrolase. J. Biol. Chem. 2001, 276, 7727–7733. [Google Scholar] [CrossRef] [Green Version]
- Wolan, D.W.; Greasley, S.E.; Wall, M.J.; Benkovic, S.J.; Wilson, I.A. Structure of Avian AICAR Transformylase with a Multisubstrate Adduct Inhibitor β-DADF Identifies the Folate Binding Site. Biochemistry 2003, 42, 10904–10914. [Google Scholar] [CrossRef]
- Wall, M.; Shim, J.H.; Benkovic, S.J. Human AICAR Transformylase: Role of the 4-Carboxamide of AICAR in Binding and Catalysis. Biochemistry 2000, 39, 11303–11311. [Google Scholar] [CrossRef]
- Pedley, A.M.; Benkovic, S.J. A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem. Sci. 2017, 42, 141–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothbart, S.B.; Racanelli, A.C.; Moran, R.G. Pemetrexed Indirectly Activates the Metabolic Kinase AMPK in Human Carcinomas. Cancer Res. 2010, 70, 10299–10309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racanelli, A.C.; Rothbart, S.B.; Heyer, C.L.; Moran, R.G. Therapeutics by Cytotoxic Metabolite Accumulation: Pemetrexed Causes ZMP Accumulation, AMPK Activation, and Mammalian Target of Rapamycin Inhibition. Cancer Res. 2009, 69, 5467–5474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Jin, C.; Xu, M.; Zhou, L.; Li, D.; Yin, Y. Bifunctional enzyme ATIC promotes propagation of hepatocellular carcinoma by regulating AMPK-mTOR-S6 K1 signaling. Cell Commun. Signal. 2017, 15, 52. [Google Scholar] [CrossRef] [Green Version]
- Fales, K.R.; Njoroge, F.G.; Brooks, H.B.; Thibodeaux, S.; Torrado, A.; Si, C.; Toth, J.L.; Mc Cowan, J.R.; Roth, K.D.; Thrasher, K.J.; et al. Discovery of N-(6-Fluoro-1-oxo-1,2-dihydroisoquinolin-7-yl)-5-[(3R)-3-hydroxypyrrolidin-1-yl]thiophene-2-sulfonamide (LSN 3213128), a Potent and Selective Nonclassical Antifolate Aminoimidazole-4-carboxamide Ribonucleotide Formyltransferase (AICARFT) Inhibitor Effective at Tumor Suppression in a Cancer Xenograft Model. J. Med. Chem. 2017, 60, 9599–9616. [Google Scholar] [CrossRef]
- Brooks, H.B.; Meier, T.I.; Geeganage, S.; Fales, K.R.; Thrasher, K.J.; Konicek, S.A.; Spencer, C.D.; Thibodeaux, S.; Foreman, R.T.; Hui, Y.-H.; et al. Characterization of a novel AICARFT inhibitor which potently elevates ZMP and has anti-tumor activity in murine models. Sci. Rep. 2018, 8, 15458. [Google Scholar] [CrossRef]
- Furie, M.B. Recruitment of Leukocytes: Adhesion Molecules and Chemoattractants. In Pathobiology of Human Disease; Elsevier: Amsterdam, The Netherlands, 2014; pp. 275–288. [Google Scholar] [CrossRef]
- Di Gennaro, A.; Haeggström, J.Z. The Leukotrienes: Immune-Modulating Lipid Mediators of Disease. Adv. Immunol. 2012, 116, 51–92. [Google Scholar] [CrossRef]
- Abeles, A.M.; Pillinger, M.H.; Abramson, S.B. Inflammation and its mediators. In Rheumatology; Elsevier: Amsterdam, The Netherlands, 2015; pp. 169–182. [Google Scholar] [CrossRef]
- Tager, A.M.; Luster, A.D. BLT1 and BLT2: The leukotriene B4 receptors. Prostagland. Leukot. Essent. Fat. Acids 2003, 69, 123–134. [Google Scholar] [CrossRef]
- Gaggar, A.; Jackson, P.L.; Noerager, B.D.; O’Reilly, P.J.; McQuaid, D.B.; Rowe, S.M.; Clancy, J.P.; Blalock, J.E. A Novel Proteolytic Cascade Generates an Extracellular Matrix-Derived Chemoattractant in Chronic Neutrophilic Inflammation. J. Immunol. 2008, 180, 5662–5669. [Google Scholar] [CrossRef] [Green Version]
- Weathington, N.M.; van Houwelingen, A.H.; Noerager, B.D.; Jackson, P.L.; Kraneveld, A.D.; Galin, F.S.; Folkerts, G.; Nijkamp, F.P.; Blalock, J.E. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat. Med. 2006, 12, 317–323. [Google Scholar] [CrossRef]
- O’Reilly, P.; Jackson, P.L.; Noerager, B.; Parker, S.; Dransfield, M.; Gaggar, A.; Blalock, J.E. N-α-PGP and PGP, potential biomarkers and therapeutic targets for COPD. Respir. Res. 2009, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Wells, J.M.; O’Reilly, P.J.; Szul, T.; Sullivan, D.I.; Handley, G.; Garrett, C.; McNicholas, C.M.; Roda, M.A.; Miller, B.E.; Tal-Singer, R.; et al. An Aberrant Leukotriene A4Hydrolase–Proline-Glycine-Proline Pathway in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2014, 190, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Wells, J.M.; Jackson, P.L.; Viera, L.; Bhatt, S.P.; Gautney, J.; Handley, G.; King, R.W.; Xu, X.; Gaggar, A.; Bailey, W.C.; et al. A Randomized, Placebo-controlled Trial of Roflumilast. Effect on Proline-Glycine-Proline and Neutrophilic Inflammation in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2015, 192, 934–942. [Google Scholar] [CrossRef] [Green Version]
- Hardison, M.T.; Galin, F.S.; Calderon, C.E.; Djekic, U.V.; Parker, S.B.; Wille, K.M.; Jackson, P.L.; Oster, R.A.; Young, K.R.; Blalock, J.E.; et al. The Presence of a Matrix-Derived Neutrophil Chemoattractant in Bronchiolitis Obliterans Syndrome after Lung Transplantation. J. Immunol. 2009, 182, 4423–4431. [Google Scholar] [CrossRef]
- Numao, S.; Hasler, F.; Laguerre, C.; Srinivas, H.; Wack, N.; Jäger, P.; Schmid, A.; Osmont, A.; Röthlisberger, P.; Houguenade, J.; et al. Feasibility and physiological relevance of designing highly potent aminopeptidase-sparing leukotriene A4 hydrolase inhibitors. Sci. Rep. 2017, 7, 13591. [Google Scholar] [CrossRef] [Green Version]
- Haeggström, J.Z.; Wetterholm, A. Enzymes and receptors in the leukotriene cascade. Cell. Mol. Life Sci. 2002, 59, 742–753. [Google Scholar] [CrossRef]
- Stsiapanava, A.; Samuelsson, B.; Haeggström, J.Z. Capturing LTA4 hydrolase in action: Insights to the chemistry and dynamics of chemotactic LTB4 synthesis. Proc. Natl. Acad. Sci. USA 2017, 114, 9689–9694. [Google Scholar] [CrossRef] [Green Version]
- Haeggström, J.Z. Leukotriene A4 Hydrolase/Aminopeptidase, the Gatekeeper of Chemotactic Leukotriene B4 Biosynthesis. J. Biol. Chem. 2004, 279, 50639–50642. [Google Scholar] [CrossRef] [Green Version]
- Haeggström, J.Z.; Tholander, F.; Wetterholm, A. Structure and catalytic mechanisms of leukotriene A4 hydrolase. Prostaglandins Other Lipid Mediat. 2007, 83, 198–202. [Google Scholar] [CrossRef]
- Stsiapanava, A.; Olsson, U.; Wan, M.; Kleinschmidt, T.; Rutishauser, D.; Zubarev, R.A.; Samuelsson, B.; Rinaldo-Matthis, A.; Haeggstrom, J.Z. Binding of Pro-Gly-Pro at the active site of leukotriene A4 hydrolase/aminopeptidase and development of an epoxide hydrolase selective inhibitor. Proc. Natl. Acad. Sci. USA 2014, 111, 4227–4232. [Google Scholar] [CrossRef] [Green Version]
- Thunnissen, M.M.G.M.; Nordlund, P.; Haeggström, J.Z. Crystal structure of human leukotriene A(4) hydrolase, a bifunctional enzyme in inflammation. Nat. Struct. Biol. 2001, 8, 131–135. [Google Scholar] [CrossRef]
- Low, C.M.; Akthar, S.; Patel, D.F.; Löser, S.; Wong, C.-T.; Jackson, P.L.; Blalock, J.E.; Hare, S.A.; Lloyd, C.M.; Snelgrove, R.J. The development of novel LTA4H modulators to selectively target LTB4 generation. Sci. Rep. 2017, 7, 44449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tholander, F.; Muroya, A.; Roques, B.-P.; Fournié-Zaluski, M.-C.; Thunnissen, M.M.G.M.; Haeggström, J.Z. Structure-Based Dissection of the Active Site Chemistry of Leukotriene A4 Hydrolase: Implications for M1 Aminopeptidases and Inhibitor Design. Chem. Biol. 2008, 15, 920–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, C.-H.; Bode, A.M.; Pugliese, A.; Cho, Y.-Y.; Kim, H.-G.; Shim, J.-H.; Jeon, Y.-J.; Li, H.; Jiang, H.; Dong, Z. [6]-Gingerol Suppresses Colon Cancer Growth by Targeting Leukotriene A4Hydrolase. Cancer Res. 2009, 69, 5584–5591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Li, N.; Wang, S.; Wu, N.; Hong, J.; Jiao, X.; Krasna, M.J.; Beer, D.G.; Yang, C.S. Leukotriene A4 Hydrolase in Rat and Human Esophageal Adenocarcinomas and Inhibitory Effects of Bestatin. JNCI J. Natl. Cancer Inst. 2003, 95, 1053–1061. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wang, S.; Wu, N.; Yang, C. Leukotriene A4 Hydrolase as a Target for Cancer Prevention and Therapy. Curr. Cancer Drug Targets 2004, 4, 267–283. [Google Scholar] [CrossRef]
- Oi, N.; Yamamoto, H.; Langfald, A.; Bai, R.; Lee, M.-H.; Bode, A.M.; Dong, Z. LTA4H regulates cell cycle and skin carcinogenesis. Carcinogenesis 2017, 38, 728–737. [Google Scholar] [CrossRef] [Green Version]
- El-Hakim, I.E.; Langdon, J.D.; Zakrzewski, J.T.; Costello, J.F. Leukotriene B4 and oral cancer. Br. J. Oral Maxillofac. Surg. 1990, 28, 155–159. [Google Scholar] [CrossRef]
- Tong, W.-G.; Ding, X.-Z.; Talamonti, M.S.; Bell, R.H.; Adrian, T.E. LTB4 stimulates growth of human pancreatic cancer cells via MAPK and PI-3 kinase pathways. Biochem. Biophys. Res. Commun. 2005, 335, 949–956. [Google Scholar] [CrossRef]
- Ihara, A.; Wada, K.; Yoneda, M.; Fujisawa, N.; Takahashi, H.; Nakajima, A. Blockade of Leukotriene B4 Signaling Pathway Induces Apoptosis and Suppresses Cell Proliferation in Colon Cancer. J. Pharmacol. Sci. 2007, 103, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Sun, Z.; Chan, D.; Cartwright, C.A.; Vijjeswarapu, M.; Ding, J.; Chen, X.; Newman, R.A. Zyflamend® reduces LTB 4 formation and prevents oral carcinogenesis in a 7,12-dimethylbenz[α]anthracene (DMBA)-induced hamster cheek pouch model. Carcinogenesis 2008, 29, 2182–2189. [Google Scholar] [CrossRef] [Green Version]
- Polyak, K.; Kato, J.Y.; Solomon, M.J.; Sherr, C.J.; Massague, J.; Roberts, J.M.; Koff, A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994, 8, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Shapira, M.; Ben-Izhak, O.; Linn, S.; Futerman, B.; Minkov, I.; Hershko, D.D. The prognostic impact of the ubiquitin ligase subunits Skp2 and Cks1 in colorectal carcinoma. Cancer 2005, 103, 1336–1346. [Google Scholar] [CrossRef]
- He, W.; Wang, X.; Chen, L.; Guan, X. A Crosstalk Imbalance Between p27Kip1 and Its Interacting Molecules Enhances Breast Carcinogenesis. Cancer Biother. Radiopharm. 2012, 27, 399–402. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.-F.; Chen, T.-J.; Lin, C.-Y.; Chen, L.-T.; Lin, L.-C.; Hsing, C.-H.; Lee, S.-W.; Sheu, M.-J.; Lee, H.-H.; Shiue, Y.-L.; et al. SKP2 overexpression is associated with a poor prognosis of rectal cancer treated with chemoradiotherapy and represents a therapeutic target with high potential. Tumor Biol. 2013, 34, 1107–1117. [Google Scholar] [CrossRef]
- Hershko, D.D. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer 2008, 112, 1415–1424. [Google Scholar] [CrossRef]
- Timmerbeul, I.; Garrett-Engele, C.M.; Kossatz, U.; Chen, X.; Firpo, E.; Grunwald, V.; Kamino, K.; Wilkens, L.; Lehmann, U.; Buer, J.; et al. Testing the importance of p27 degradation by the SCFskp2 pathway in murine models of lung and colon cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 14009–14014. [Google Scholar] [CrossRef] [Green Version]
- Israel, E. The Effect of Inhibition of 5-Lipoxygenase by Zileuton in Mild-to-Moderate Asthma. Ann. Intern. Med. 1993, 119, 1059–1066. [Google Scholar] [CrossRef]
- Röhn, T.A.; Numao, S.; Otto, H.; Loesche, C.; Thoma, G. Drug discovery strategies for novel leukotriene A4 hydrolase inhibitors. Expert Opin. Drug Discov. 2021, 16, 1483–1495. [Google Scholar] [CrossRef]
- Haeggström, J.Z. Leukotriene biosynthetic enzymes as therapeutic targets. J. Clin. Investig. 2018, 128, 2680–2690. [Google Scholar] [CrossRef]
- Elborn, J.S.; Horsley, A.; MacGregor, G.; Bilton, D.; Grosswald, R.; Ahuja, S.; Springman, E.B. Phase I Studies of Acebilustat: Biomarker Response and Safety in Patients with Cystic Fibrosis. Clin. Transl. Sci. 2017, 10, 28–34. [Google Scholar] [CrossRef]
- Markert, C.; Thoma, G.; Srinivas, H.; Bollbuck, B.; Luond, R.M.; Miltz, W.; Walchli, R.; Wolf, R.; Hinrichs, J.; Bergsdorf, C.; et al. Discovery of LYS006, a Potent and Highly Selective Inhibitor of Leukotriene A4 Hydrolase. J. Med. Chem. 2021, 64, 1889–1903. [Google Scholar] [CrossRef]
- EMPIRE CF: A Phase 2 Study to Evaluate the Efficacy, Safety, and Tolerability of CTX-4430 in Adult Cystic Fibrosis (CF) Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT02443688 (accessed on 5 December 2021).
- CTX-4430 for the Treatment of Moderate to Severe Facial Acne Vulgaris. Available online: https://clinicaltrials.gov/ct2/show/NCT02385760 (accessed on 5 December 2021).
- Study of Efficacy and Safety of Investigational Treatments in Patients with Moderate to Severe Hidradenitis Suppurativa. Available online: https://clinicaltrials.gov/ct2/show/NCT03827798 (accessed on 5 December 2021).
- Study to Assess the Efficacy and Safety of LYS006 in Patients with Moderate to Severe Inflammatory Acne. Available online: https://clinicaltrials.gov/ct2/show/NCT03497897 (accessed on 5 December 2021).
- Study of Efficacy, Safety, and Tolerability of LYS006, in Patients with Mild to Moderate Ulcerative Colitis. Available online: https://clinicaltrials.gov/ct2/show/NCT04074590 (accessed on 5 December 2021).
- Study of Various Treatments in Non-alcoholic Fatty Liver Disease (NAFLD) Patients Who Have Aspects of Non-alcoholic Steatohepatitis (NASH) (NEXSCOT). Available online: https://clinicaltrials.gov/ct2/show/NCT04147195 (accessed on 5 December 2021).
- Audat, S.A.; Al-Shar’i, N.A.; Al-Oudat, B.A.; Bryant-Friedrich, A.; Bedi, M.F.; Zayed, A.L.; Al-Balas, Q.A. Identification of Human Leukotriene A4 Hydrolase Inhibitors Using Structure-Based Pharmacophore Modeling and Molecular Docking. Molecules 2020, 25, 2871. [Google Scholar] [CrossRef]
- Snelgrove, R.J.; Jackson, P.L.; Hardison, M.T.; Noerager, B.D.; Kinloch, A.; Gaggar, A.; Shastry, S.; Rowe, S.M.; Shim, Y.M.; Hussell, T.; et al. A Critical Role for LTA4H in Limiting Chronic Pulmonary Neutrophilic Inflammation. Science 2010, 330, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Akthar, S.; Patel, D.F.; Beale, R.C.; Peiró, T.; Xu, X.; Gaggar, A.; Jackson, P.L.; Blalock, J.E.; Lloyd, C.M.; Snelgrove, R.J. Matrikines are key regulators in modulating the amplitude of lung inflammation in acute pulmonary infection. Nat. Commun. 2015, 6, 8423. [Google Scholar] [CrossRef] [Green Version]
- Ramazi, S.; Allahverdi, A.; Zahiri, J. Evaluation of post-translational modifications in histone proteins: A review on histone modification defects in developmental and neurological disorders. J. Biosci. 2020, 45, 135. [Google Scholar] [CrossRef]
- Ramazi, S.; Zahiri, J. Post-translational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef]
- Mann, M.; Jensen, O.N. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 2003, 21, 255–261. [Google Scholar] [CrossRef]
- Xu, Y.; Chou, K.-C. Recent Progress in Predicting Posttranslational Modification Sites in Proteins. Curr. Top. Med. Chem. 2015, 16, 591–603. [Google Scholar] [CrossRef]
- Zurlo, G.; Guo, J.; Takada, M.; Wei, W.; Zhang, Q. New Insights into Protein Hydroxylation and Its Important Role in Human Diseases. Biochim. Biophys. Acta Rev. Cancer 2016, 1866, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.Y.; Teyssier, C.; Strahl, B.D.; Stallcup, M.R. Role of Protein Methylation in Regulation of Transcription. Endocr. Rev. 2005, 26, 147–170. [Google Scholar] [CrossRef]
- Aletta, J.M.; Cimato, T.R.; Ettinger, M.J. Protein methylation: A signal event in post-translational modification. Trends Biochem. Sci. 1998, 23, 89–91. [Google Scholar] [CrossRef]
- Smith, W.A.; Schurter, B.T.; Wong-Staal, F.; David, M. Arginine Methylation of RNA Helicase A Determines Its Subcellular Localization. J. Biol. Chem. 2004, 279, 22795–22798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mowen, K.A.; Schurter, B.T.; Fathman, J.W.; David, M.; Glimcher, L.H. Arginine methylation of NIP45 modulates cytokine gene expression in effector T lymphocytes. Mol. Cell 2004, 15, 559–571. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.E.; Silver, P.A. State of the Arg. Cell 2001, 106, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Guccione, E.; Richard, S. The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 642–657. [Google Scholar] [CrossRef]
- Gayatri, S.; Bedford, M.T. Readers of histone methylarginine marks. Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1839, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Hughes, R.M.; Waters, M.L. Arginine Methylation in a β-Hairpin Peptide: Implications for Arg−π Interactions, ΔCp°, and the Cold Denatured State. J. Am. Chem. Soc. 2006, 128, 12735–12742. [Google Scholar] [CrossRef]
- Wu, Q.; Schapira, M.; Arrowsmith, C.H.; Barsyte-Lovejoy, D. Protein arginine methylation: From enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discov. 2021, 20, 509–530. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, Y.; Shi, X. Emerging roles of lysine methylation on non-histone proteins. Cell. Mol. Life Sci. 2015, 72, 4257–4272. [Google Scholar] [CrossRef]
- Chang, B.; Chen, Y.; Zhao, Y.; Bruick, R.K. JMJD6 Is a Histone Arginine Demethylase. Science 2007, 318, 444–447. [Google Scholar] [CrossRef] [Green Version]
- Wesche, J.; Kühn, S.; Kessler, B.M.; Salton, M.; Wolf, A. Protein arginine methylation: A prominent modification and its demethylation. Cell. Mol. Life Sci. 2017, 74, 3305–3315. [Google Scholar] [CrossRef]
- Webby, C.J.; Wolf, A.; Gromak, N.; Dreger, M.; Kramer, H.; Kessler, B.; Nielsen, M.L.; Schmitz, C.; Butler, D.S.; Yates, J.R.; et al. Jmjd6 Catalyses Lysyl-Hydroxylation of U2AF65, a Protein Associated with RNA Splicing. Science 2009, 325, 90–93. [Google Scholar] [CrossRef]
- Vangimalla, S.S.; Ganesan, M.; Kharbanda, K.K.; Osna, N.A. Bifunctional Enzyme JMJD6 Contributes to Multiple Disease Pathogenesis: New Twist on the Old Story. Biomolecules 2017, 7, 41. [Google Scholar] [CrossRef]
- Meng, Y.; Li, H.; Liu, C.; Zheng, L.; Shen, B. Jumonji domain-containing protein family: The functions beyond lysine demethylation. J. Mol. Cell Biol. 2018, 10, 371–373. [Google Scholar] [CrossRef]
- Hewitson, K.S.; Granatino, N.; Welford, R.W.D.; McDonough, M.A.; Schofield, C.J. Oxidation by 2-oxoglutarate oxygenases: Non-haem iron systems in catalysis and signalling. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2005, 363, 807–828. [Google Scholar] [CrossRef]
- Liu, Y.; Long, Y.-H.; Wang, S.-Q.; Zhang, Y.-Y.; Li, Y.-F.; Mi, J.-S.; Yu, C.-H.; Li, D.-Y.; Zhang, J.-H.; Zhang, X.-J. JMJD6 regulates histone H2A.X phosphorylation and promotes autophagy in triple-negative breast cancer cells via a novel tyrosine kinase activity. Oncogene 2018, 38, 980–997. [Google Scholar] [CrossRef]
- Lee, S.; Liu, H.; Hill, R.; Chen, C.; Hong, X.; Crawford, F.; Kingsley, M.; Zhang, Q.; Liu, X.; Chen, Z.; et al. JMJD6 cleaves MePCE to release positive transcription elongation factor b (P-TEFb) in higher eukaryotes. eLife 2020, 9, e53930. [Google Scholar] [CrossRef]
- Yang, J.; Chen, S.; Yang, Y.; Ma, X.; Shao, B.; Yang, S.; Wei, Y.; Wei, X. Jumonji domain-containing protein 6 protein and its role in cancer. Cell Prolif. 2020, 53. [Google Scholar] [CrossRef] [Green Version]
- Mantri, M.; Krojer, T.; Bagg, E.A.; Webby, C.J.; Butler, D.S.; Kochan, G.; Kavanagh, K.L.; Oppermann, U.; McDonough, M.A.; Schofield, C.J. Crystal Structure of the 2-Oxoglutarate- and Fe(II)-Dependent Lysyl Hydroxylase JMJD6. J. Mol. Biol. 2010, 401, 211–222. [Google Scholar] [CrossRef]
- Clifton, I.J.; McDonough, M.A.; Ehrismann, D.; Kershaw, N.J.; Granatino, N.; Schofield, C.J. Structural studies on 2-oxoglutarate oxygenases and related double-stranded β-helix fold proteins. J. Inorg. Biochem. 2006, 100, 644–669. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer Epigenetics: From Mechanism to Therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unoki, M.; Masuda, A.; Dohmae, N.; Arita, K.; Yoshimatsu, M.; Iwai, Y.; Fukui, Y.; Ueda, K.; Hamamoto, R.; Shirakawa, M.; et al. Lysyl 5-Hydroxylation, a Novel Histone Modification, by Jumonji Domain Containing 6 (JMJD6). J. Biol. Chem. 2013, 288, 6053–6062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böse, J.; Gruber, A.D.; Helming, L.; Schiebe, S.; Wegener, I.; Hafner, M.; Beales, M.; Köntgen, F.; Lengeling, A. The phosphatidylserine receptor has essential functions during embryogenesis but not in apoptotic cell removal. J. Biol. 2004, 3, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeckel, J.N.; Guarani, V.; Koyanagi, M.; Roexe, T.; Lengeling, A.; Schermuly, R.T.; Gellert, P.; Braun, T.; Zeiher, A.; Dimmeler, S. Jumonji domain-containing protein 6 (Jmjd6) is required for angiogenic sprouting and regulates splicing of VEGF-receptor 1. Proc. Natl. Acad. Sci. USA 2011, 108, 3276–3281. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Si, W.; Liu, X.; He, L.; Ren, J.; Yang, Z.; Yang, J.; Li, W.; Liu, S.; Pei, F.; et al. JMJD6 promotes melanoma carcinogenesis through regulation of the alternative splicing of PAK1, a key MAPK signaling component. Mol. Cancer 2017, 16, 175. [Google Scholar] [CrossRef]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef]
- Miller, T.E.; Liau, B.B.; Wallace, L.C.; Morton, A.R.; Xie, Q.; Dixit, D.; Factor, D.C.; Kim, L.J.Y.; Morrow, J.J.; Wu, Q.; et al. Transcription elongation factors represent in vivo cancer dependencies in glioblastoma. Nature 2017, 547, 355–359. [Google Scholar] [CrossRef]
- Wong, M.; Sun, Y.; Xi, Z.; Milazzo, G.; Poulos, R.C.; Bartenhagen, C.; Bell, J.L.; Mayoh, C.; Ho, N.; Tee, A.E.; et al. JMJD6 is a tumorigenic factor and therapeutic target in neuroblastoma. Nat. Commun. 2019, 10, 3319. [Google Scholar] [CrossRef] [Green Version]
- Manfredi, J.; Wang, F.; He, L.; Huangyang, P.; Liang, J.; Si, W.; Yan, R.; Han, X.; Liu, S.; Gui, B.; et al. JMJD6 Promotes Colon Carcinogenesis through Negative Regulation of p53 by Hydroxylation. PLoS Biol. 2014, 12, e1001819. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Tie, Y.; Fang, Z.; Wu, X.; Yi, T.; Huang, S.; Liang, X.; Qian, Y.; Wang, X.; Pi, R.; et al. Jumonji domain-containing 6 (JMJD6) identified as a potential therapeutic target in ovarian cancer. Signal. Transduct. Target. Ther. 2019, 4, 24. [Google Scholar] [CrossRef] [Green Version]
- Ran, T.; Xiao, R.; Huang, Q.; Yuan, H.; Lu, T.; Liu, W. In Silico Discovery of JMJD6 Inhibitors for Cancer Treatment. ACS Med. Chem. Lett. 2019, 10, 1609–1613. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, R.; Liu, Y.; Fang, Z.; Zhang, H.; Fan, Y.; Yang, S.; Xiang, R. Discovery of a new class of JMJD6 inhibitors and structure–activity relationship study. Bioorganic Med. Chem. Lett. 2021, 44, 128109. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Specific Target | Inhibitors | Evaluated in vitro | Evaluated in vivo | Clinical Trials in Cancer Patients |
|---|---|---|---|---|---|
| PFK-2/FBPase-2 | PFK-2 activity of PFKB3 | 3PO [46] | yes [46] | yes [46] | no |
| PFK15 [48] | yes [48] | yes [48] | no | ||
| PFK158 [48] | yes [49] | yes [49] | NCT02044861 [50] | ||
| PFK-2 activity of PFKFB4 | 5MPN [51] | yes [51] | yes [51] | no | |
| ATIC | AICAR TFase | BW1540 [57] | no | no | no |
| AICAR TFase | BW2315 [57] | no | no | no | |
| AICAR TFase | LSN3213128 [66] | yes [66,67] | yes [66,67] | no | |
| LTA4H | epoxide hydrolase and aminopeptidase activities | Acebilustat | no available data | no available data | no (1) |
| epoxide hydrolase and aminopeptidase | LYS006 [105] | yes [105] | yes [105] | no (1) | |
| epoxide hydrolase | RH00633 [112] | yes [112] | no | no | |
| Jmjd6 | Demethylase and hydroxylase | SKLB325 [150] | yes [150] | yes [150] | no |
| Demethylase and hydroxylase | WL12 [151] | yes [151] | no | no | |
| Demethylase and hydroxylase | 7p [152] | no | no | no |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teixeira, C.S.S.; Sousa, S.F. Current Status of the Use of Multifunctional Enzymes as Anti-Cancer Drug Targets. Pharmaceutics 2022, 14, 10. https://doi.org/10.3390/pharmaceutics14010010
Teixeira CSS, Sousa SF. Current Status of the Use of Multifunctional Enzymes as Anti-Cancer Drug Targets. Pharmaceutics. 2022; 14(1):10. https://doi.org/10.3390/pharmaceutics14010010
Chicago/Turabian StyleTeixeira, Carla S. S., and Sérgio F. Sousa. 2022. "Current Status of the Use of Multifunctional Enzymes as Anti-Cancer Drug Targets" Pharmaceutics 14, no. 1: 10. https://doi.org/10.3390/pharmaceutics14010010
APA StyleTeixeira, C. S. S., & Sousa, S. F. (2022). Current Status of the Use of Multifunctional Enzymes as Anti-Cancer Drug Targets. Pharmaceutics, 14(1), 10. https://doi.org/10.3390/pharmaceutics14010010