
Granzyme B PET Imaging in Response to In Situ Vaccine Therapy Combined with αPD1 in a Murine Colon Cancer Model


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Information
2.2. Radiochemistry of [18F]AlF-mNOTA-GZP
2.3. CpG-Animal Model
2.4. Small Animal PET-CT Imaging
2.5. Fluorescence-Assisted Cell Sorting (FACS)
2.6. Statistical Analysis
3. Results
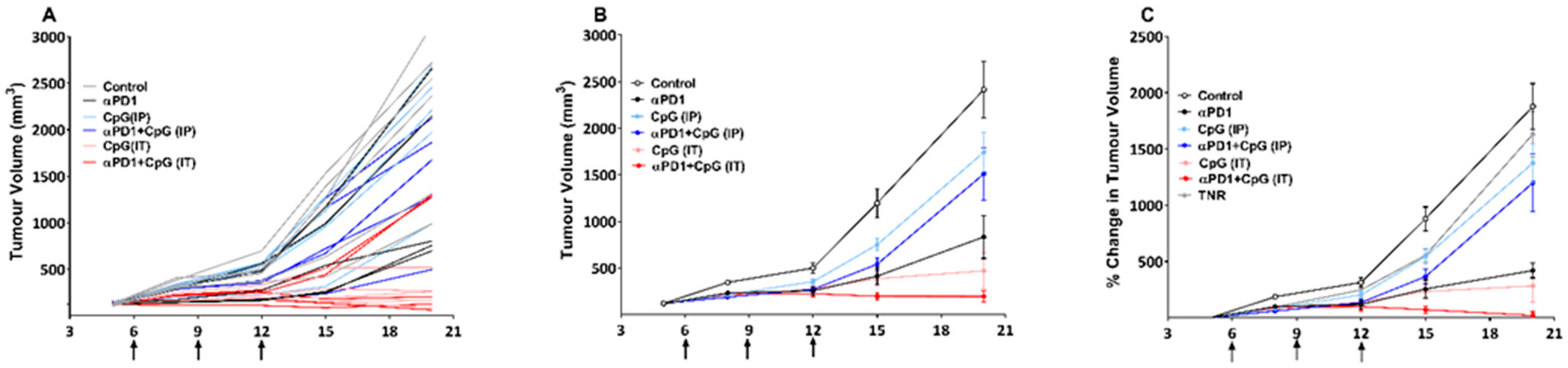
3.1. Assessment of Treatment Efficacy Using Tumour Growth Volume
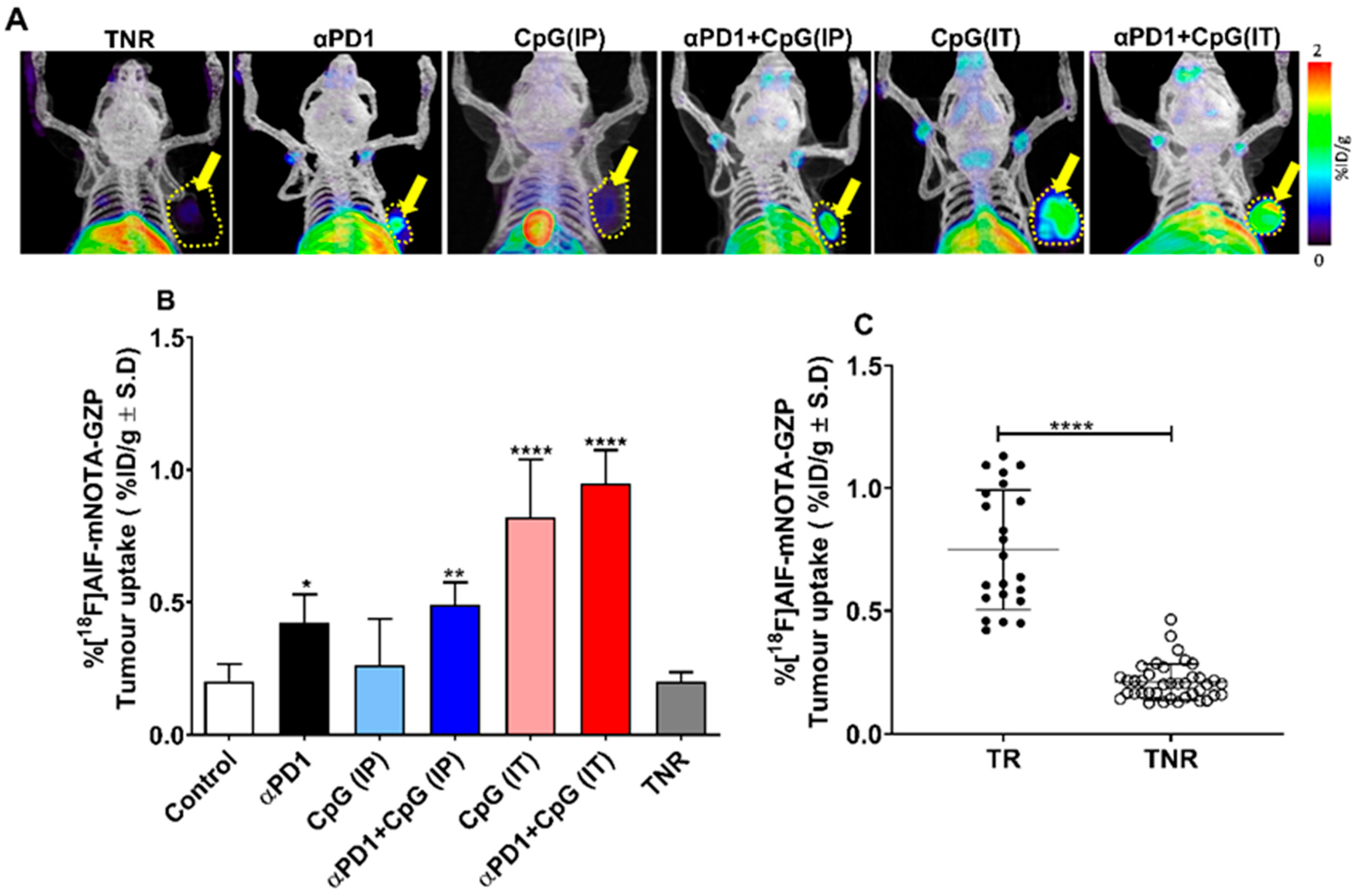
3.2. Assessment of Treatment Efficacy Using [18F]AlF-mNOTA-GZP PET Imaging
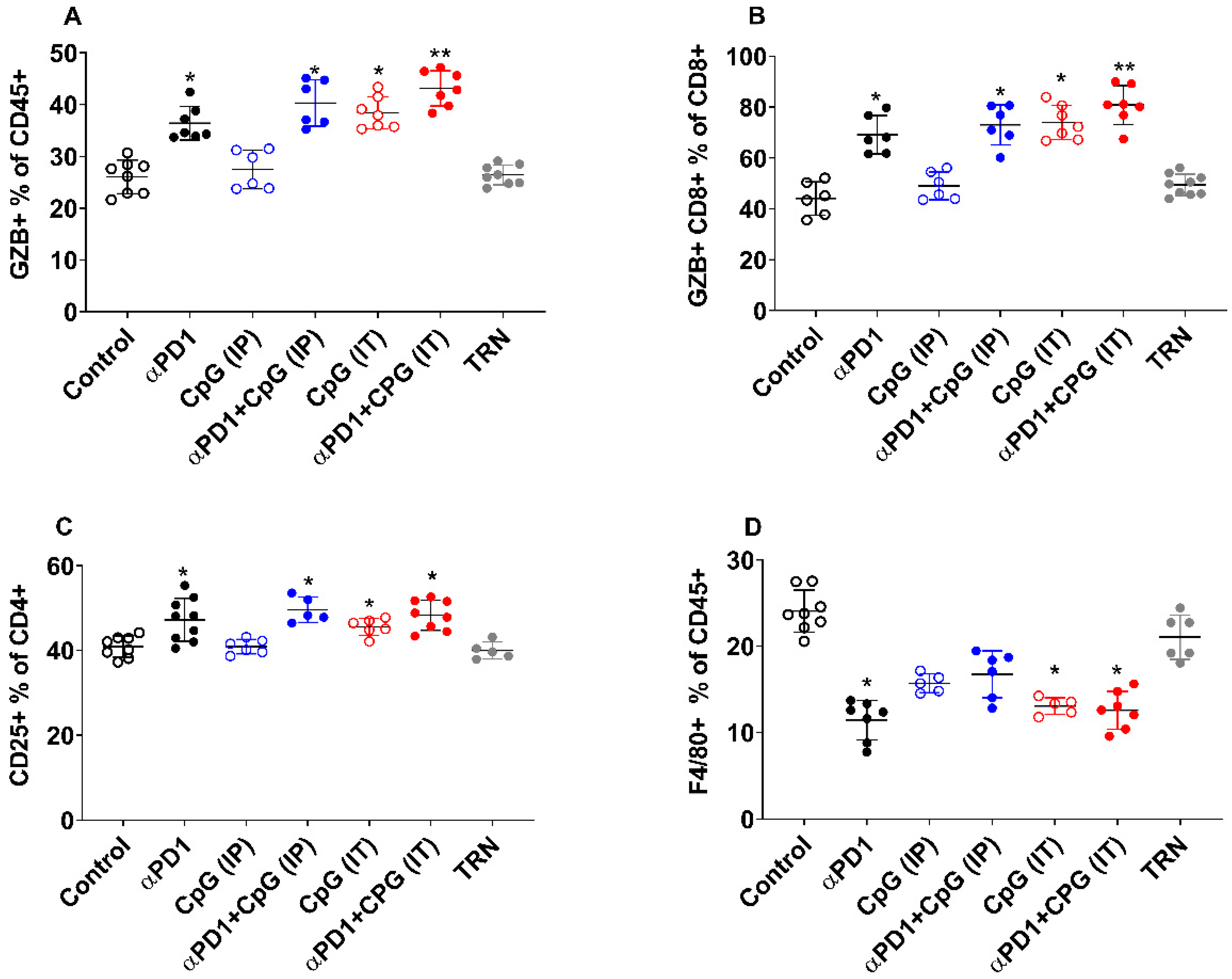
3.3. Immune Cell Profiling by FACS and Its Correlation to [18F]AlF-mNOTA-GZP Tumour Uptake
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McCune, J.S. Rapid Advances in Immunotherapy to Treat Cancer. Clin. Pharmacol. Ther. 2018, 103, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Fan, A.; Wang, B.; Wang, X.; Nie, Y.; Fan, D.; Zhao, X.; Lu, Y. Immunotherapy in colorectal cancer: Current achievements and future perspective. Int. J. Biol. Sci. 2021, 17, 3837–3849. [Google Scholar] [CrossRef] [PubMed]
- Heckelsmiller, K.; Rall, K.; Beck, S.; Schlamp, A.; Seiderer, J.; Jahrsdörfer, B.; Krug, A.; Rothenfusser, S.; Endres, S.; Hartmann, G. Peritumoral CpG DNA Elicits a Coordinated Response of CD8 T Cells and Innate Effectors to Cure Established Tumors in a Murine Colon Carcinoma Model. J. Immunol. 2002, 169, 3892. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Song, W.; Czerwinski, D.K.; Varghese, B.; Uematsu, S.; Akira, S.; Krieg, A.M.; Levy, R. Lymphoma Immunotherapy with CpG Oligodeoxynucleotides Requires TLR9 Either in the Host or in the Tumor Itself. J. Immunol. 2007, 179, 2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczanowska, S.; Joseph, A.M.; Davila, E. TLR agonists: Our best frenemy in cancer immunotherapy. J. Leukoc. Biol. 2013, 93, 847–863. [Google Scholar] [CrossRef] [Green Version]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef]
- Marshall, J.D.; Heeke, D.S.; Abbate, C.; Yee, P.; Van Nest, G. Induction of interferon-γ from natural killer cells by immunostimulatory CpG DNA is mediated through plasmacytoid-dendritic-cell-produced interferon-α and tumour necrosis factor-α. Immunology 2006, 117, 38–46. [Google Scholar] [CrossRef]
- Bode, C.; Zhao, G.; Steinhagen, F.; Kinjo, T.; Klinman, D.M. CpG DNA as a vaccine adjuvant. Expert Rev. Vaccines 2011, 10, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Lou, Y.; Liu, C.; Lizée, G.; Peng, W.; Xu, C.; Ye, Y.; Rabinovich, B.A.; Hailemichael, Y.; Gelbard, A.; Zhou, D.; et al. Antitumor activity mediated by CpG: The route of administration is critical. J. Immunother. 2011, 34, 279–288. [Google Scholar] [CrossRef]
- Kell, S.A.; Kachura, M.A.; Renn, A.; Traquina, P.; Coffman, R.L.; Campbell, J.D. Preclinical development of the TLR9 agonist DV281 as an inhaled aerosolized immunotherapeutic for lung cancer: Pharmacological profile in mice, non-human primates, and human primary cells. Int. Immunopharmacol. 2019, 66, 296–308. [Google Scholar] [CrossRef]
- Hammerich, L.; Binder, A.; Brody, J.D. In situ vaccination: Cancer immunotherapy both personalized and off-the-shelf. Mol. Oncol. 2015, 9, 1966–1981. [Google Scholar] [CrossRef] [Green Version]
- Mason, K.; Hunter, N. CpG plus radiotherapy: A review of preclinical works leading to clinical trial. Front. Oncol. 2012, 2, 101. [Google Scholar] [CrossRef] [Green Version]
- Frank, M.J.; Reagan, P.M.; Bartlett, N.L.; Gordon, L.I.; Friedberg, J.W.; Czerwinski, D.K.; Long, S.R.; Hoppe, R.T.; Janssen, R.; Candia, A.F.; et al. In Situ Vaccination with a TLR9 Agonist and Local Low-Dose Radiation Induces Systemic Responses in Untreated Indolent Lymphoma. Cancer Discov. 2018, 8, 1258–1269. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Medina, T.; Kummar, S.; Amin, A.; Kalbasi, A.; Drabick, J.J.; Barve, M.; Daniels, G.A.; Wong, D.J.; Schmidt, E.V.; et al. SD-101 in Combination with Pembrolizumab in Advanced Melanoma: Results of a Phase Ib, Multicenter Study. Cancer Discov. 2018, 8, 1250–1257. [Google Scholar] [CrossRef] [Green Version]
- Gallotta, M.; Assi, H.; Degagné, É.; Kannan, S.K.; Coffman, R.L.; Guiducci, C. Inhaled TLR9 Agonist Renders Lung Tumors Permissive to PD-1 Blockade by Promoting Optimal CD4+ and CD8+ T cell Interplay. Cancer Res. 2018, 78, 4943–4956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanpouille-Box, C.; Pilones, K.A.; Wennerberg, E.; Formenti, S.C.; Demaria, S. In situ vaccination by radiotherapy to improve responses to anti-CTLA-4 treatment. Vaccine 2015, 33, 7415–7422. [Google Scholar] [CrossRef] [Green Version]
- Goggi, J.L.; Hartimath, S.V.; Hwang, Y.; Tan, Y.X.; Khanapur, S.; Ramasamy, B.; Jiang, L.; Yong, F.F.; Cheng, P.; Tan, P.W.; et al. Examining Immunotherapy Response Using Multiple Radiotracers. Mol. Imaging Biol. 2020, 22, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Goggi, J.L.; Tan, Y.X.; Hartimath, S.V.; Jieu, B.; Hwang, Y.Y.; Jiang, L.; Boominathan, R.; Cheng, P.; Yuen, T.Y.; Chin, H.X.; et al. Granzyme B PET Imaging of Immune Checkpoint Inhibitor Combinations in Colon Cancer Phenotypes. Mol. Imaging Biol. 2020, 22, 1392–1402. [Google Scholar] [CrossRef] [PubMed]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Aslan, K.; Turco, V.; Blobner, J.; Sonner, J.K.; Liuzzi, A.R.; Núñez, N.G.; De Feo, D.; Kickingereder, P.; Fischer, M.; Green, E.; et al. Heterogeneity of response to immune checkpoint blockade in hypermutated experimental gliomas. Nat. Commun. 2020, 11, 931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Dudzinski, S.; Beckermann, K.E.; Young, K.; McKinley, E.; McIntyre, J.O.; Rathmell, J.C.; Xu, J.; Gore, J.C. MRI of tumor T cell infiltration in response to checkpoint inhibitor therapy. J. Immunother. Cancer 2020, 8, e000328. [Google Scholar] [CrossRef] [PubMed]
- Larimer, B.M.; Wehrenberg-Klee, E.; Dubois, F.; Mehta, A.; Kalomeris, T.; Flaherty, K.; Boland, G.; Mahmood, U. Granzyme B PET Imaging as a Predictive Biomarker of Immunotherapy Response. Cancer Res. 2017, 77, 2318–2327. [Google Scholar] [CrossRef] [Green Version]
- Rashidian, M.; LaFleur, M.W.; Verschoor, V.L.; Dongre, A.; Zhang, Y.; Nguyen, T.H.; Kolifrath, S.; Aref, A.R.; Lau, C.J.; Paweletz, C.P.; et al. Immuno-PET identifies the myeloid compartment as a key contributor to the outcome of the antitumor response under PD-1 blockade. Proc. Natl. Acad. Sci. USA 2019, 116, 16971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieg, A.M. Therapeutic potential of Toll-like receptor 9 activation. Nat. Rev. Drug Discov. 2006, 5, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Link, B.K.; Ballas, Z.K.; Weisdorf, D.; Wooldridge, J.E.; Bossler, A.D.; Shannon, M.; Rasmussen, W.L.; Krieg, A.M.; Weiner, G.J. Oligodeoxynucleotide CpG 7909 Delivered as Intravenous Infusion Demonstrates Immunologic Modulation in Patients With Previously Treated Non-Hodgkin Lymphoma. J. Immunother. 2006, 29, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.D.; Fearon, K.; Abbate, C.; Subramanian, S.; Yee, P.; Gregorio, J.; Coffman, R.L.; Van Nest, G. Identification of a novel CpG DNA class and motif that optimally stimulate B cell and plasmacytoid dendritic cell functions. J. Leukoc. Biol. 2003, 73, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Milhem, M.M.; Long, G.V.; Hoimes, C.J.; Amin, A.; Lao, C.D.; Conry, R.M.; Hunt, J.; Daniels, G.A.; Almubarak, M.; Shaheen, M.F.; et al. Phase 1b/2, open label, multicenter, study of the combination of SD-101 and pembrolizumab in patients with advanced melanoma who are naïve to anti-PD-1 therapy. J. Clin. Oncol. 2019, 37, 9534. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/show/NCT03007732 (accessed on 14 February 2020).
- Available online: https://clinicaltrials.gov/ct2/history/NCT02927964?V_1=View (accessed on 30 June 2020).
- Neves, A.A.; Brindle, K.M. Assessing responses to cancer therapy using molecular imaging. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2006, 1766, 242–261. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Hoos, A.; Day, S.; Weber, J.S.; Hamid, O.; Lebbé, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the Evaluation of Immune Therapy Activity in Solid Tumors: Immune-Related Response Criteria. Clin. Cancer Res. 2009, 15, 7412. [Google Scholar] [CrossRef] [Green Version]
- Yirgin, I.K.; Erturk, S.M.; Dogan, I.; Vatansever, S. Are radiologists ready to evaluate true response to immunotherapy? Insights Imaging 2021, 12, 29. [Google Scholar] [CrossRef]
- Hodi, F.S.; Hwu, W.-J.; Kefford, R.; Weber, J.S.; Daud, A.; Hamid, O.; Patnaik, A.; Ribas, A.; Robert, C.; Gangadhar, T.C.; et al. Evaluation of Immune-Related Response Criteria and RECIST v1.1 in Patients with Advanced Melanoma Treated With Pembrolizumab. J. Clin. Oncol. 2016, 34, 1510–1517. [Google Scholar] [CrossRef]
- Saillard, M.; Cenerenti, M.; Romero, P.; Jandus, C. Impact of Immunotherapy on CD4 T Cell Phenotypes and Function in Cancer. Vaccines 2021, 9, 454. [Google Scholar] [CrossRef]
- Alam, I.S.; Mayer, A.T.; Sagiv-Barfi, I.; Wang, K.; Vermesh, O.; Czerwinski, D.K.; Johnson, E.M.; James, M.L.; Levy, R.; Gambhir, S.S. Imaging activated T cells predicts response to cancer vaccines. J. Clin. Investig. 2018, 128, 2569–2580. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Uptake of [18F]AlF-mNOTA-GZP in CT-26 Tumour | |
|---|---|
| Control | 0.20 ± 0.06 |
| Treatment Responder (TR) | |
| αPD1 | 0.42 ± 0.10 * |
| CpG-ODN (IP) | 0.26 ± 0.17 |
| αPD1 + CpG-ODN (IP) | 0.49 ± 0.08 ** |
| CpG-ODN (IT) | 0.82 ± 0.21 **** |
| αPD1 + CpG-ODN (IT) | 0.95 ± 0.12 **** |
| Treatment Non-Responder (TNR) | 0.20 ± 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hartimath, S.V.; Ramasamy, B.; Xuan, T.Y.; Rong, T.J.; Khanapur, S.; Cheng, P.; Hwang, Y.Y.; Robins, E.G.; Goggi, J.L. Granzyme B PET Imaging in Response to In Situ Vaccine Therapy Combined with αPD1 in a Murine Colon Cancer Model. Pharmaceutics 2022, 14, 150. https://doi.org/10.3390/pharmaceutics14010150
Hartimath SV, Ramasamy B, Xuan TY, Rong TJ, Khanapur S, Cheng P, Hwang YY, Robins EG, Goggi JL. Granzyme B PET Imaging in Response to In Situ Vaccine Therapy Combined with αPD1 in a Murine Colon Cancer Model. Pharmaceutics. 2022; 14(1):150. https://doi.org/10.3390/pharmaceutics14010150
Chicago/Turabian StyleHartimath, Siddesh V., Boominathan Ramasamy, Tan Yun Xuan, Tang Jun Rong, Shivashankar Khanapur, Peter Cheng, You Yi Hwang, Edward G. Robins, and Julian L. Goggi. 2022. "Granzyme B PET Imaging in Response to In Situ Vaccine Therapy Combined with αPD1 in a Murine Colon Cancer Model" Pharmaceutics 14, no. 1: 150. https://doi.org/10.3390/pharmaceutics14010150
APA StyleHartimath, S. V., Ramasamy, B., Xuan, T. Y., Rong, T. J., Khanapur, S., Cheng, P., Hwang, Y. Y., Robins, E. G., & Goggi, J. L. (2022). Granzyme B PET Imaging in Response to In Situ Vaccine Therapy Combined with αPD1 in a Murine Colon Cancer Model. Pharmaceutics, 14(1), 150. https://doi.org/10.3390/pharmaceutics14010150