

Positron Emission Tomography Probes for Imaging Cytotoxic Immune Cells

Abstract
:1. Introduction
2. Immuno-Oncology
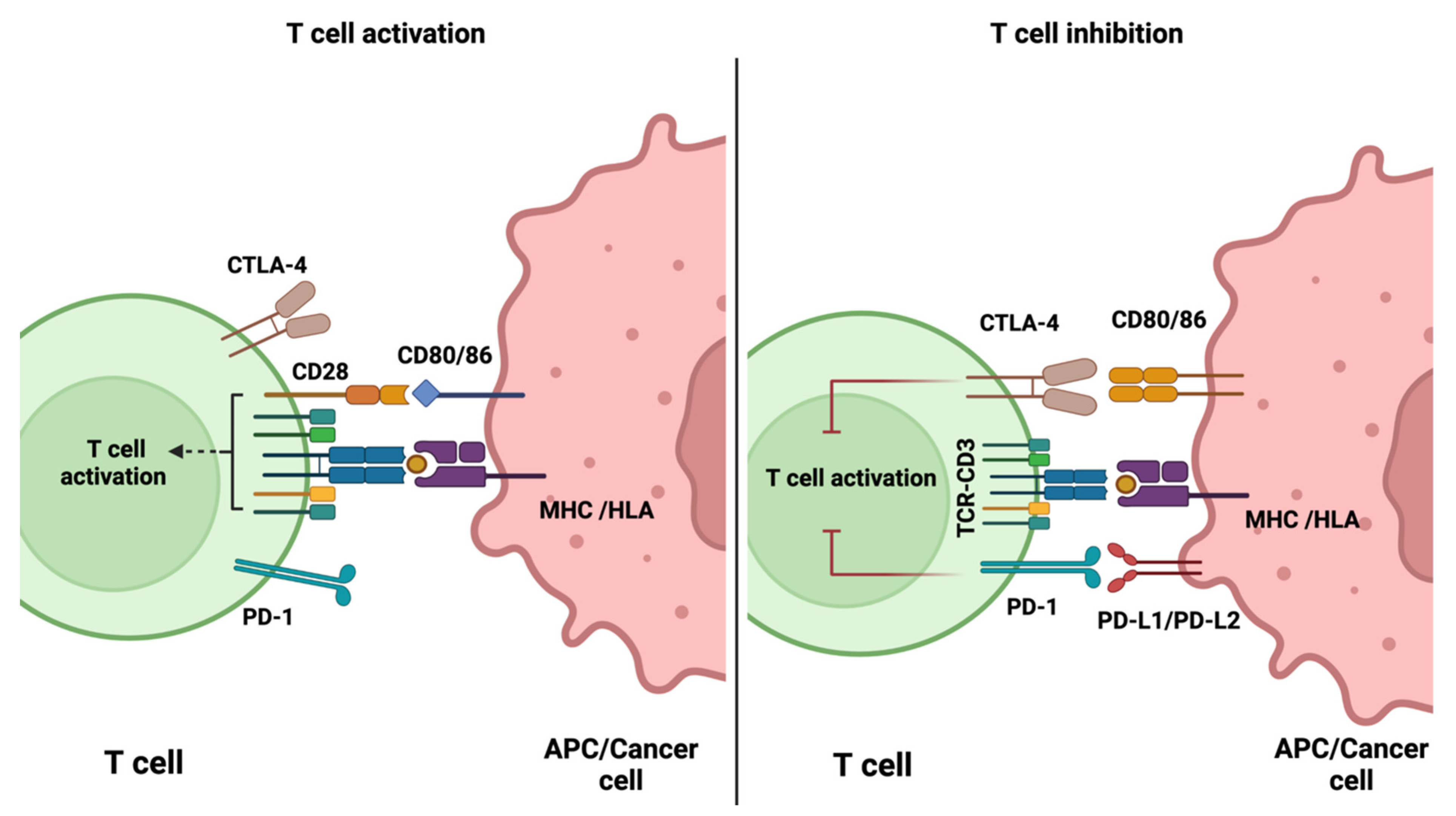
2.1. Immune Checkpoint Inhibitors
2.2. CAR-T Cell Therapy
2.3. γδ T Cell Therapy
2.4. Cytokines
2.4.1. Challenges Associated with Cancer Immunotherapy
2.4.2. PET Imaging of Immune Cells
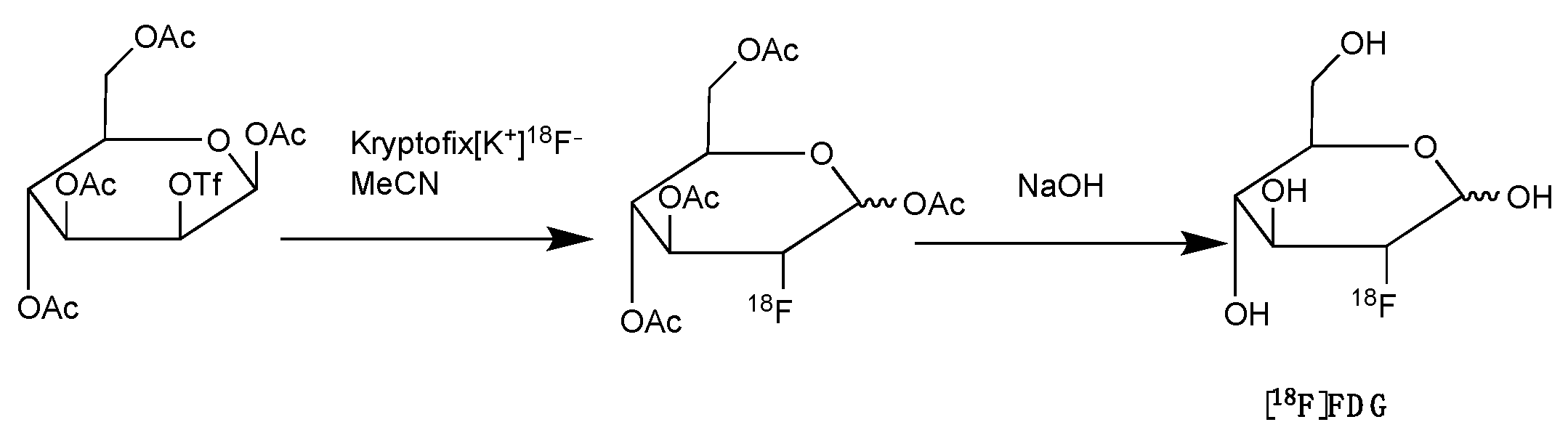
2.5. Fluorine-18 Labelled Fluorodeoxyglucose ([18F]FDG)
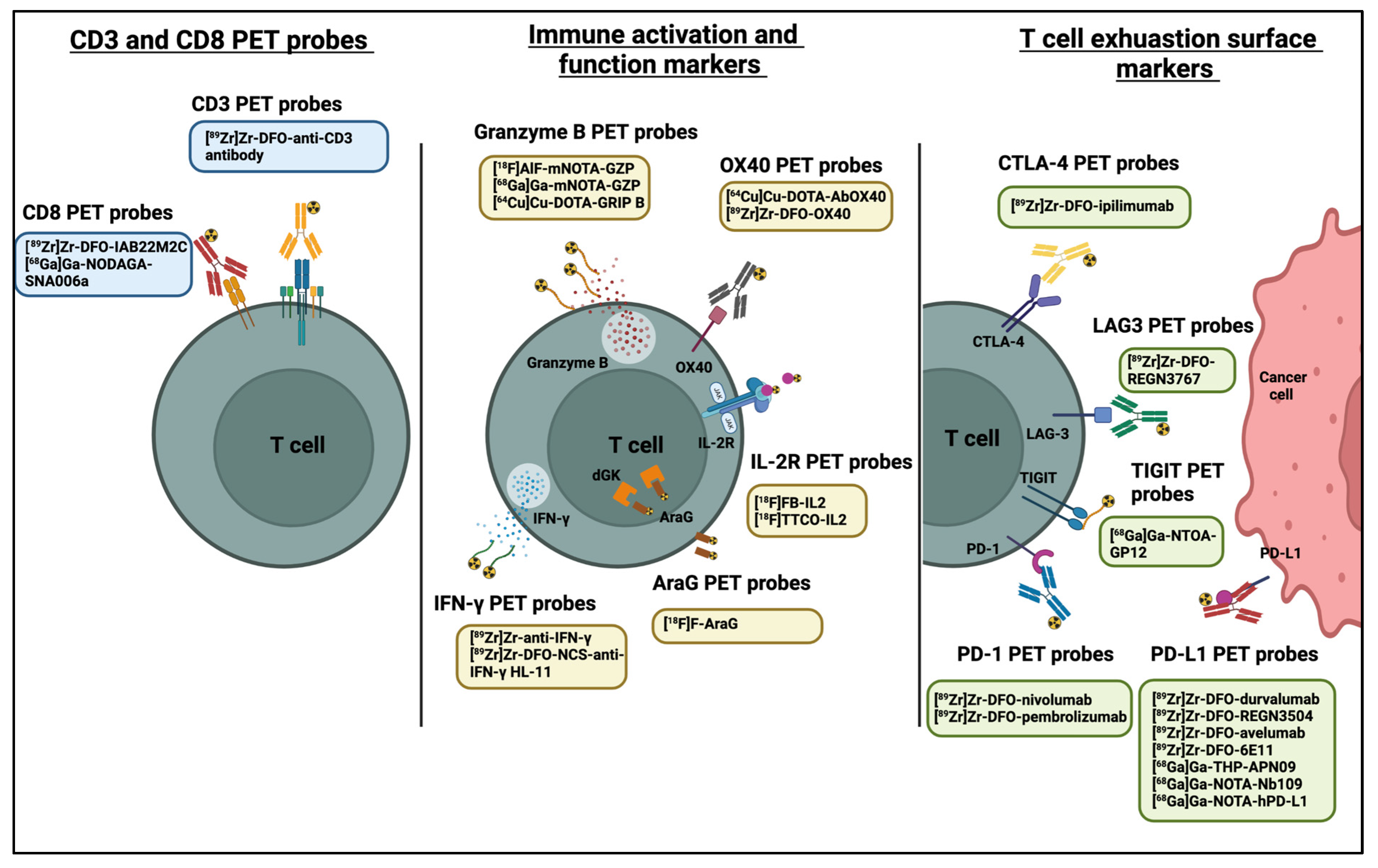
3. PET Imaging of Immune Checkpoints
4. PET Imaging of CD8+ and CD3+ T Cells
5. PET Imaging of Immune Cell Activation
5.1. OX40
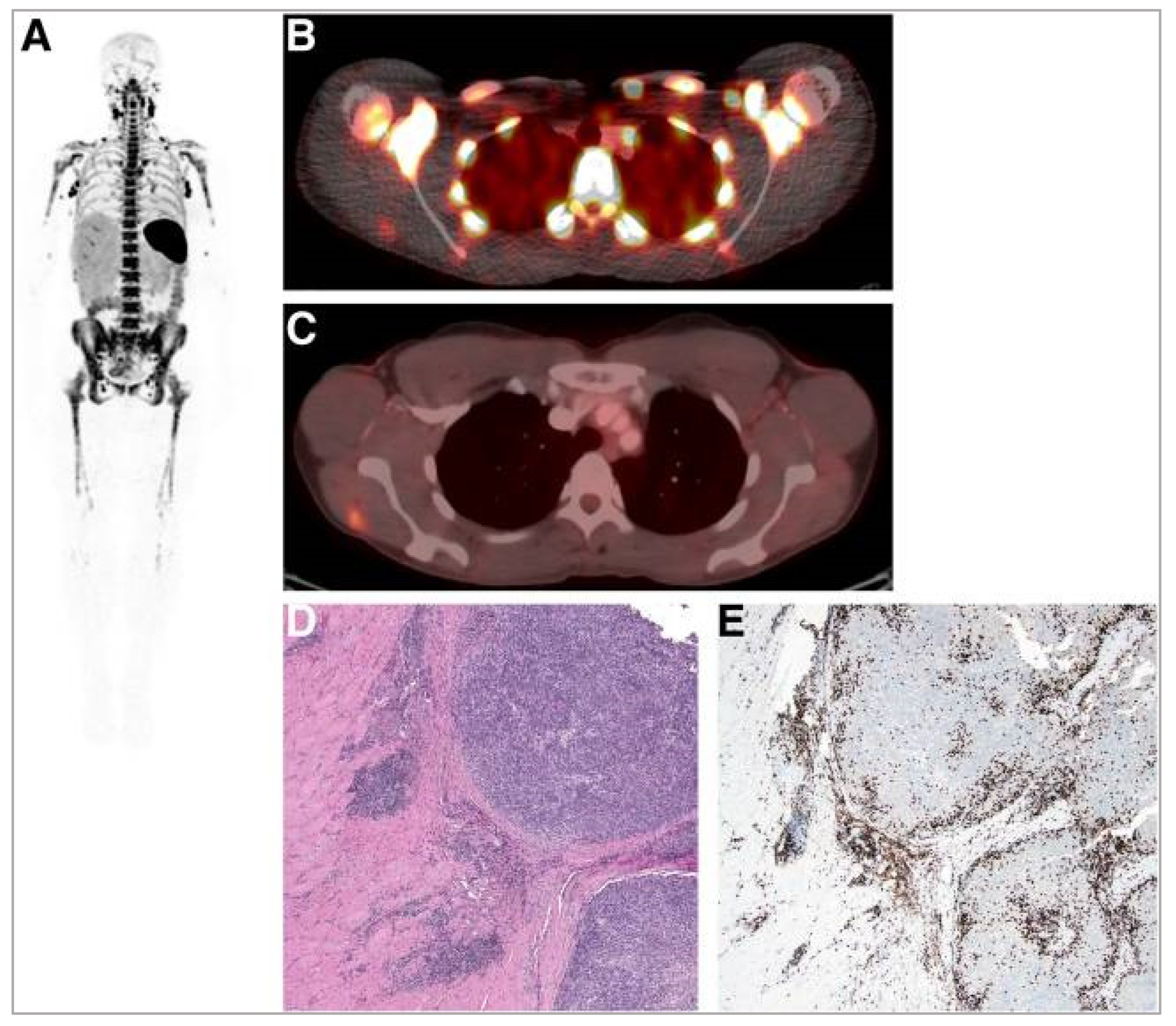
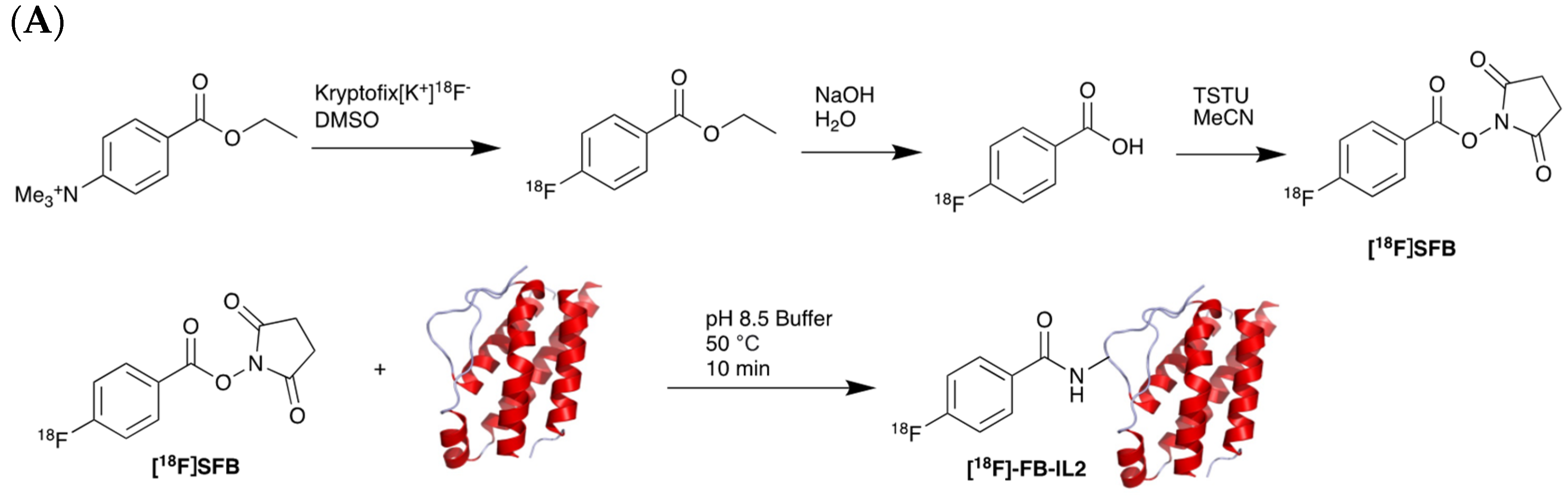
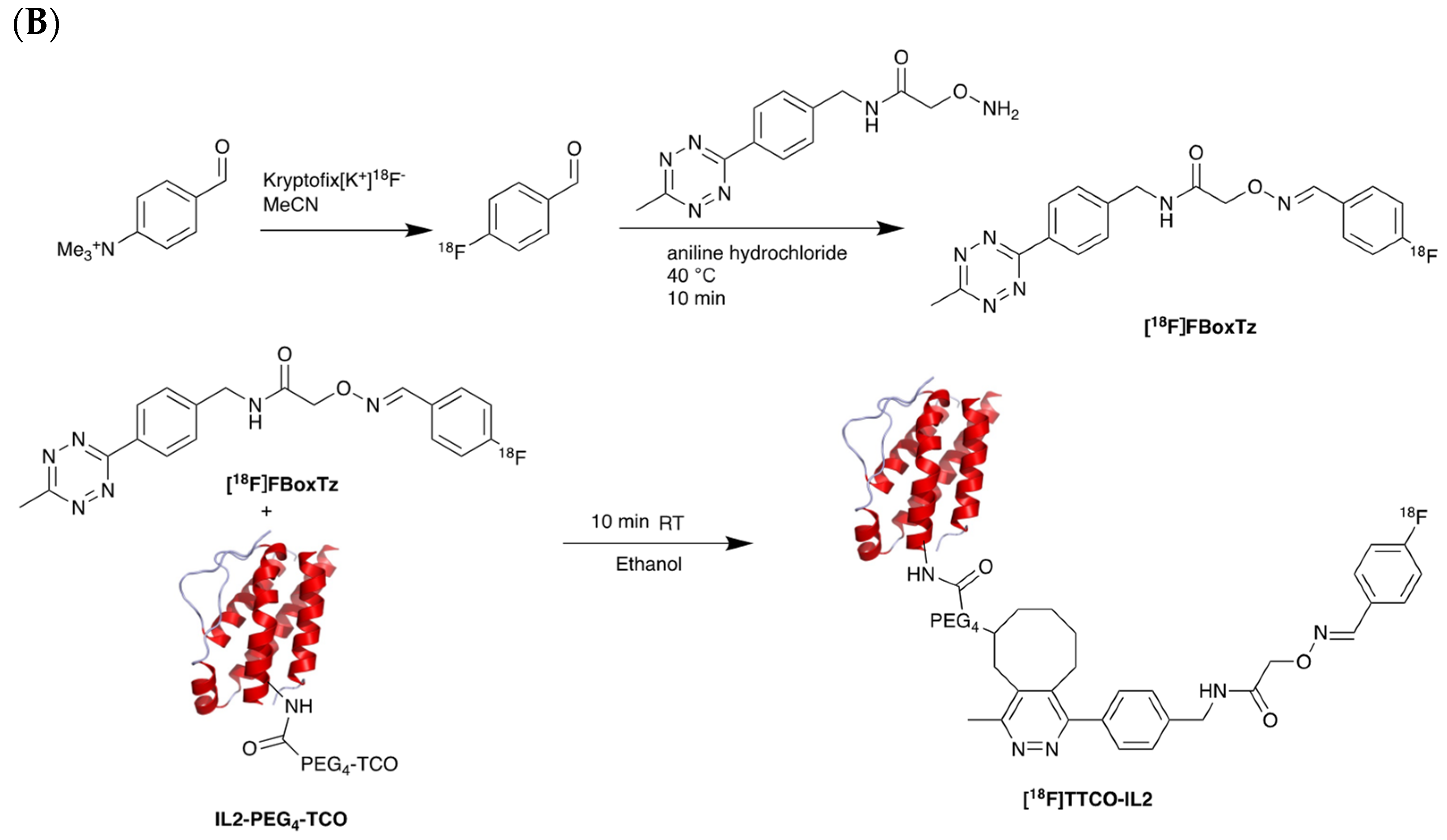
5.2. Interleukin-2 (IL-2) Receptor
5.3. Granzyme B
5.4. IFN-γ
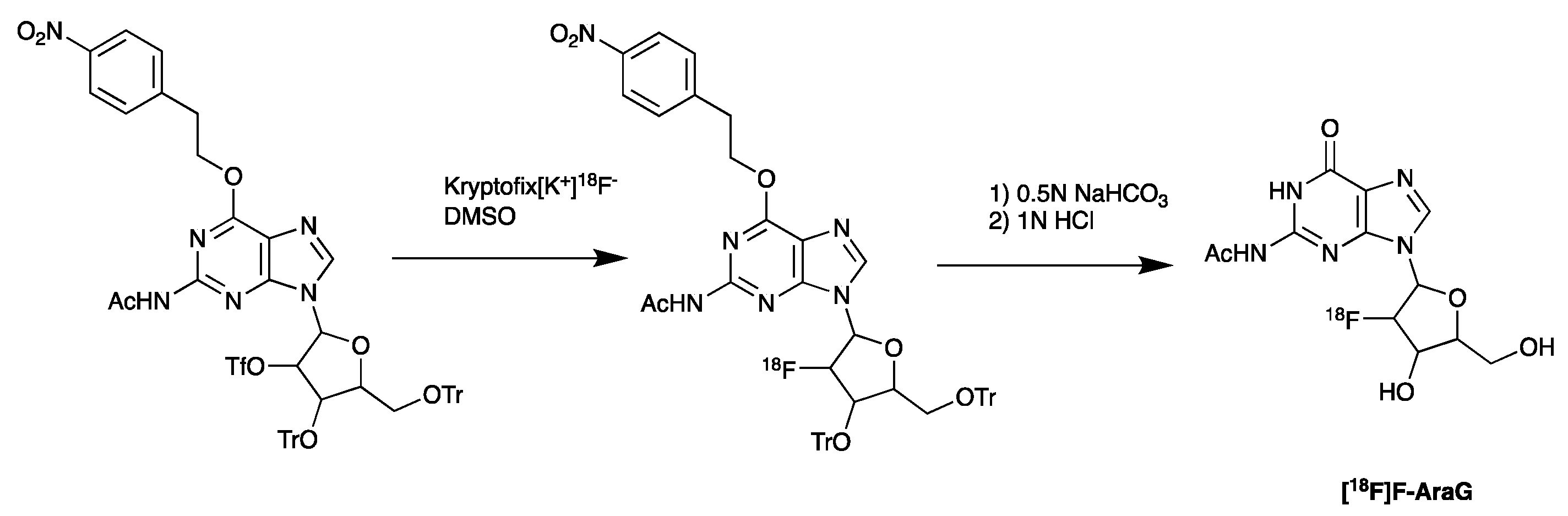
5.5. PET Imaging of Metabolic Targets Associated with Activated Immune Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Name | Format | Radioisotope | Active Clinical Trial | Trial Number | Highlights |
|---|---|---|---|---|---|---|
| PD-1 | [89Zr]Zr-DFO-nivolumab | mAb | 89Zr | Phase 1 NSCLC | EudraCT: 2015-004760-11 |
|
| Phase 2 Melanoma | NCT05289193 |
| ||||
| [89Zr]Zr-DFO-pembrolizumab | mAb | 89Zr | Phase 2 NSCLC | NCT03065764 |
| |
| PD-L1 | [89Zr]Zr-DFO-durvalumab | mAb | 89Zr | Phase 2 HNSCC | NCT03829007 |
|
| Phase 2 NSCLC | NCT03853187 |
| ||||
| [89Zr]Zr-DFO-REGN3504 | mAb | 89Zr | Phase 1 Patients with advanced PD-L1 positive malignancies | NCT03746704 |
| |
| [89Zr]Zr-DFO-avelumab | mAb | 89Zr | Phase 1 NSCLC | NCT03514719 |
| |
| [68Ga]Ga-NOTA-(hPD-L1) | nanobody | 68Ga | Pre-clinical | - |
| |
| [68Ga]Ga-NOTA-Nb109 | nanobody | 68Ga | Pre-clinical | - |
| |
| [89Zr]Zr-DFO-6E11 | mAb | 89Zr | Pre-clinical | - |
| |
| [68Ga]Ga-THP-APN09 PET | nanobody | 68Ga | Phase 1 Lung cancer, melanoma, and other solid tumours | NCT05156515 |
| |
| CTLA-4 | [89Zr]Zr-DFO-ipilimumab | mAb | 89Zr | Phase 2 Metastatic melanoma | NCT03313323 |
|
| LAG-3 | [89Zr]Zr-DFO-REGN3767 | mAb | 89Zr | Phase 1 Relapsed/Refractory DLBCL | NCT04566978 |
|
| TIGIT | [68Ga]Ga-NOTA-GP12 | Peptide antagonist | 68Ga | Pre-clinical | - |
|
| Tested in two patients with advanced NSCLC |
| |||||
| CD3 | [89Zr]Zr-DFO-anti-CD3 | mAb | 89Zr | Pre-clinical | - |
|
| CD4 | [64Cu]Cu-NOTA-IAB41 | Minibody | 64Cu | Pre-clinical | - |
|
| [89Zr]Zr-malDFO-GK1.5 cDb | Cys-Diabody | 89Zr | Pre-clinical | - |
| |
| 89Zr-labelled anti-CD4 scFv | ScFv | 89Zr | Pre-clinical | - |
| |
| CD8 | [89Zr]Zr-DFO-IAB22M2C | Minibody | 89Zr | Phase 1 Melanoma, lung, and hepatocellular carcinoma | NCT03107663 |
|
| Phase 2 Advanced and metastatic solid malignancies | NCT03802123 |
| ||||
| [68Ga]Ga-NODAGA-SNA006a | Nanobody | 68Ga | Phase 1 |
| ||
| Phase 2 |
| |||||
| OX40 | [64Cu]Cu-DOTA-AbOX40 | mAb | 64Cu | Pre-clinical | - | |
| [89Zr]Zr-DFO-OX40 | mAb | 89Zr | ||||
| IL-2R | [18F]FB-IL2 | Small protein (cytokine) | 18F | Phase 1 | NCT02922283 |
|
| [18F]FBox-TTCO-IL2 | Pre-clinical | - |
| |||
| Granzyme B | [18F]AlF-mNOTA-GZP | Peptide | 18F | Pre-clinical | - |
|
| [68Ga]Ga-mNOTA-GZP | Peptide | 68Ga | Pre-clinical | |||
| [64Cu]Cu-DOTA-GRIP B | Peptide | 64Cu | Pre-clinical |
| ||
| IFN-γ | [89Zr]Zr-DFO-anti-IFN-γ | mAb | 89Zr | Pre-clinical | - |
|
| [89Zr]Zr-DFO-NCS-anti-IFN-γ HL-11 | Diabody | 89Zr | Pre-clinical | - |
| |
| AraG | [18F]F-AraG | Small molecule (Nucleoside analog) | 18F | Early phase 1 In healthy volunteers and patients with advanced NSCLC | NCT04678440 |
|
| Phase 1 cancer patients undergoing immunotherapy and/or radiation therapy | NCT03142204 |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- U.S. Food and Drug Administration Center For Drug Evaluation and Research. Yervoy Approval Letter. 2011. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/125377Orig1s000Approv.pdf (accessed on 19 August 2022).
- U.S. Food and Drug Administration Center For Drug Evaluation and Research. Opdivo BLA Accelerated Approval Letter. 2014. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2014/125554Orig1s000ltr.pdf (accessed on 19 August 2022).
- U.S. Food and Drug Administration Center For Drug Evaluation and Research. Kymriah Approval Letter. 2017. Available online: https://www.fda.gov/media/106989/download (accessed on 19 August 2022).
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.C.; Bachmeier, C.; Kharfan-Dabaja, M.A. CAR T-cell therapy for B-cell lymphomas: Clinical trial results of available products. Ther. Adv. Hematol. 2019, 10, 2040620719841581. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, A.; Peeke, S.; Shah, N.; Mustafa, J.; Khatun, F.; Lombardo, A.; Abreu, M.; Elkind, R.; Fehn, K.; De Castro, A.; et al. Axicabtagene ciloleucel CD19 CAR-T cell therapy results in high rates of systemic and neurologic remissions in ten patients with refractory large B cell lymphoma including two with HIV and viral hepatitis. J. Hematol. Oncol. 2020, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Toschi, L.; Castello, A.; Grizzi, F.; Mansi, L.; Lopci, E. Clinical characteristics of patient selection and imaging predictors of outcome in solid tumors treated with checkpoint-inhibitors. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 2310–2325. [Google Scholar] [CrossRef] [PubMed]
- Pittet, M.J.; Grimm, J.; Berger, C.R.; Tamura, T.; Wojtkiewicz, G.; Nahrendorf, M.; Romero, P.; Swirski, F.K.; Weissleder, R. In vivo imaging of T cell delivery to tumors after adoptive transfer therapy. Proc. Natl. Acad. Sci. USA 2007, 104, 12457–12461. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Jiang, D.; Ehlerding, E.B.; Luo, Q.; Cai, W. Noninvasive PET Imaging of T cells. Trends Cancer 2018, 4, 359–373. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Z. Molecular imaging in tracking tumor-specific cytotoxic T lymphocytes (CTLs). Theranostics 2014, 4, 990–1001. [Google Scholar] [CrossRef]
- Matsui, K.; Wang, Z.; McCarthy, T.J.; Allen, P.M.; Reichert, D.E. Quantitation and visualization of tumor-specific T cells in the secondary lymphoid organs during and after tumor elimination by PET. Nucl. Med. Biol. 2004, 31, 1021–1031. [Google Scholar] [CrossRef]
- Botti, C.; Negri, D.R.; Seregni, E.; Ramakrishna, V.; Arienti, F.; Maffioli, L.; Lombardo, C.; Bogni, A.; Pascali, C.; Crippa, F.; et al. Comparison of three different methods for radiolabelling human activated T lymphocytes. Eur. J. Nucl. Med. 1997, 24, 497–504. [Google Scholar]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef]
- Hernandez, R.; Põder, J.; LaPorte, K.M.; Malek, T.R. Engineering IL-2 for immunotherapy of autoimmunity and cancer. Nat. Rev. Immunol. 2022, 22, 614–628. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration Center For Drug Evaluation and Research. Keytruda BLA Accelerated Approval Letter. 2014. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2014/125514Orig1s000ltr.pdf (accessed on 19 August 2022).
- U.S. Food and Drug Administration Center For Drug Evaluation and Research. Libtayo BLA Approval Letter. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761097Orig1s000Approv.pdf (accessed on 19 August 2022).
- U.S. Food and Drug Administration Center For Drug Evaluation and Research. Tecntriq BLA approval Letter. 2016. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/761041Orig1s000Approv.pdf (accessed on 19 August 2022).
- U.S. Food and Drug Administration Center For Drug Evaluation and Research. Bavencio BLA Accelerated Approval Letter. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761078Orig1s000Approv.pdf (accessed on 19 August 2022).
- U.S. Food and Drug Administration Center For Drug Evaluation and Research. Imfinzi BLA Approval Letter. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2017/761069Orig1s000ltr.pdf (accessed on 19 August 2022).
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Schoffski, P.; Tan, D.S.W.; Martín, M.; Ochoa-de-Olza, M.; Sarantopoulos, J.; Carvajal, R.D.; Kyi, C.; Esaki, T.; Prawira, A.; Akerley, W.; et al. Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) +/- anti-PD-1 spartalizumab (PDR001) in patients with advanced malignancies. J. Immunother. Cancer 2022, 10, e003776. [Google Scholar] [CrossRef]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef]
- Robert, C. LAG-3 and PD-1 blockade raises the bar for melanoma. Nat. Cancer 2021, 2, 1251–1253. [Google Scholar] [CrossRef]
- Long, G.V.; Hodi, F.S.; Lipson, E.J.; Schadendorf, D.; Ascierto, P.A.; Matamala, L.; Salman, P.; Gutiérrez, E.C.; Rutkowski, P.; Gogas, H.; et al. Relatlimab and nivolumab versus nivolumab in previously untreated metastatic or unresectable melanoma: Overall survival and response rates from RELATIVITY-047 (CA224-047). J. Clin. Oncol. 2022, 40, 360385. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Feasibility and Efficacy of Perioperative Nivolumab with or without Relatlimab for Patients With Potentially Resectable Hepatocellular Carcinoma (HCC), Identifier NCT04658147. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04658147 (accessed on 19 August 2022).
- U.S. National Library of Medicine. Study of Nivolumab and Relatlimab in Patients with Microsatellite Stable (MSS) Advanced Colorectal Cancer, Identifier NCT03642067. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03642067 (accessed on 19 August 2022).
- U.S. National Library of Medicine. Neoadjuvant Nivolumab Combination Treatment in Resectable Non-small Cell Lung Cancer Patients (NEOpredict), Identifier NCT04205552. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT04205552 (accessed on 19 August 2022).
- Guillerey, C.; Harjunpää, H.; Carrié, N.; Kassem, S.; Teo, T.; Miles, K.; Krumeich, S.; Weulersse, M.; Cuisinier, M.; Stannard, K.; et al. TIGIT immune checkpoint blockade restores CD8+ T-cell immunity against multiple myeloma. Blood 2018, 132, 1689–1694. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Immuno-Oncology Drugs Elotuzumab, Anti-LAG-3 and Anti-TIGIT, Identifier NCT04150965. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT04150965 (accessed on 19 August 2022).
- U.S. National Library of Medicine. AdvanTIG-203: Anti-PD-1 Monoclonal Antibody Tislelizumab (BGB-A317) Combined With or Without Anti-TIGIT Monoclonal Antibody Ociperlimab (BGB-A1217) in Participants With Recurrent or Metastatic Esophageal Squamous Cell Carcinoma, Identifier NCT04732494. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04732494 (accessed on 19 August 2022).
- U.S. National Library of Medicine. GP Chemotherapy in Combination With Anti-PD-1 and Anti-TIGIT in Unresectable Advanced BTC, Identifier NCT05023109. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT05023109 (accessed on 19 August 2022).
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration Center For Drug Evaluation and Research. Abecma BLA Approval Letter. 2021. Available online: https://www.fda.gov/media/147062/download (accessed on 19 August 2022).
- U.S. Food and Drug Administration Center For Drug Evaluation and Research. Breyanzi BLA Approval Letter. 2021. Available online: https://www.fda.gov/media/145712/download (accessed on 19 August 2022).
- U.S. Food and Drug Administration Center For Drug Evaluation and Research. Tecartus BLA Apporval Letter. 2020. Available online: https://www.fda.gov/media/140415/download (accessed on 19 August 2022).
- U.S. Food and Drug Administration Center For Drug Evaluation and Research. Yescarta BLA Approval Letter. 2017. Available online: https://www.fda.gov/media/108458/download (accessed on 19 August 2022).
- U.S. Food and Drug Administration Center For Drug Evaluation and Research. Carvykti BLA Approval Letter. 2022. Available online: https://www.fda.gov/media/156572/download (accessed on 19 August 2022).
- Xu, X.; Sun, Q.; Liang, X.; Chen, Z.; Zhang, X.; Zhou, X.; Li, M.; Tu, H.; Liu, Y.; Tu, S.; et al. Mechanisms of Relapse After CD19 CAR T-Cell Therapy for Acute Lymphoblastic Leukemia and Its Prevention and Treatment Strategies. Front. Immunol. 2019, 10, 2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, C.A.; Locke, F.L.; Miklos, D.B.; Herrera, A.F.; Westin, J.R.; Lee, J.; Rossi, J.M.; Zheng, L.; Avanzi, M.P.; Roberts, Z.J.; et al. End of Phase 1 Results from Zuma-6: Axicabtagene Ciloleucel (Axi-Cel) in Combination with Atezolizumab for the Treatment of Patients with Refractory Diffuse Large B Cell Lymphoma. Biol. Blood Marrow Transplant. 2019, 25, S173. [Google Scholar] [CrossRef]
- Maude, S.L.; E Hucks, G.; Seif, A.E.; Talekar, M.K.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Gonzalez, V.; Nazimuddin, F.; Gupta, M.; et al. The effect of pembrolizumab in combination with CD19-targeted chimeric antigen receptor (CAR) T cells in relapsed acute lymphoblastic leukemia (ALL). J. Clin. Oncol. 2017, 35, 103. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; Van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Silva-Santos, B.; Serre, K.; Norell, H. γδ T cells in cancer. Nat. Rev. Immunol. 2015, 15, 683–691. [Google Scholar] [CrossRef]
- Kabelitz, D.; Kalyan, S.; Oberg, H.H.; Wesch, D. Human Vδ2 versus non-Vδ2 γδ T cells in antitumor immunity. Oncoimmunology 2013, 2, e23304. [Google Scholar] [CrossRef]
- Das, H.; Wang, L.; Kamath, A.; Bukowski, J.F. Vγ2Vδ2 T-cell receptor-mediated recognition of aminobisphosphonates. Blood 2001, 98, 1616–1618. [Google Scholar] [CrossRef]
- Sandstrom, A.; Peigné, C.M.; Léger, A.; Crooks, J.E.; Konczak, F.; Gesnel, M.C.; Breathnach, R.; Bonneville, M.; Scotet, E.; Adams, E.J. The intracellular B30.2 domain of butyrophilin 3A1 binds phosphoantigens to mediate activation of human Vγ9Vδ2 T cells. Immunity 2014, 40, 490–500. [Google Scholar] [CrossRef]
- Rhodes, D.A.; Chen, H.-C.; Williamson, J.C.; Hill, A.; Yuan, J.; Smith, S.; Rhodes, H.; Trowsdale, J.; Lehner, P.J.; Herrmann, T.; et al. Regulation of Human γδ T Cells by BTN3A1 Protein Stability and ATP-Binding Cassette Transporters. Front. Immunol. 2018, 9, 662. [Google Scholar] [CrossRef]
- Vantourout, P.; Laing, A.; Woodward, M.J.; Zlatareva, I.; Apolonia, L.; Jones, A.W.; Snijders, A.P.; Malim, M.H.; Hayday, A.C. Heteromeric interactions regulate butyrophilin (BTN) and BTN-like molecules governing gammadelta T cell biology. Proc. Natl. Acad. Sci. USA 2018, 115, 1039–1044. [Google Scholar] [CrossRef]
- Rigau, M.; Ostrouska, S.; Fulford, T.S.; Johnson, D.N.; Woods, K.; Ruan, Z.; McWilliam, H.E.G.; Hudson, C.; Tutuka, C.; Wheatley, A.K.; et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by γδ T cells. Science 2020, 367, eaay5516. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.P.; Heuijerjans, J.; Yan, M.; Gustafsson, K.; Anderson, J. γδ T cells for cancer immunotherapy: A systematic review of clinical trials. Oncoimmunology 2014, 3, e27572. [Google Scholar] [CrossRef] [PubMed]
- Braza, M.S.; Klein, B. Anti-tumour immunotherapy with Vγ9Vδ2 T lymphocytes: From the bench to the bedside. Br. J. Haematol. 2013, 160, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Kunzmann, V.; Smetak, M.; Kimmel, B.; Weigang-Koehler, K.; Goebeler, M.; Birkmann, J.; Becker, J.; Schmidt-Wolf, I.G.H.; Einsele, H.; Wilhelm, M. Tumor-promoting versus tumor-antagonizing roles of γδ T cells in cancer immunotherapy: Results from a prospective phase I/II trial. J. Immunother. 2012, 35, 205–213. [Google Scholar] [CrossRef]
- Kobayashi, H.; Tanaka, Y. γδ T Cell Immunotherapy-A Review. Pharmaceuticals 2015, 8, 40–61. [Google Scholar] [CrossRef]
- Fournie, J.-J.; Sicard, H.; Poupot, M.; Bezombes, C.; Blanc, A.; Romagné, F.; Ysebaert, L.; Laurent, G. What lessons can be learned from γδ T cell-based cancer immunotherapy trials? Cell Mol. Immunol. 2013, 10, 35–41. [Google Scholar] [CrossRef]
- Coscia, M.; Vitale, C.; Peola, S.; Foglietta, M.; Rigoni, M.; Griggio, V.; Castella, B.; Angelini, D.; Chiaretti, S.; Riganti, C.; et al. Dysfunctional Vγ9Vδ2 T cells are negative prognosticators and markers of dysregulated mevalonate pathway activity in chronic lymphocytic leukemia cells. Blood 2012, 120, 3271–3279. [Google Scholar] [CrossRef]
- Zhao, Y.; Niu, C.; Cui, J. Gamma-delta (γδ) T cells: Friend or foe in cancer development? J. Transl. Med. 2018, 16, 3. [Google Scholar] [CrossRef]
- Oberg, H.-H.; Janitschke, L.; Sulaj, V.; Weimer, J.; Gonnermann, D.; Hedemann, N.; Arnold, N.; Kabelitz, D.; Peipp, M.; Bauerschlag, D.; et al. Bispecific antibodies enhance tumor-infiltrating T cell cytotoxicity against autologous HER-2-expressing high-grade ovarian tumors. J. Leukoc. Biol. 2020, 107, 1081–1095. [Google Scholar] [CrossRef]
- Wawrzyniecka, P.A.; Ibrahim, L.; Gritti, G.; Pule, M.A.; Maciocia, P.M. Chimeric antigen receptor T cells for gamma-delta T cell malignancies. Leukemia 2022, 36, 577–579. [Google Scholar] [CrossRef]
- Rozenbaum, M.; Meir, A.; Aharony, Y.; Itzhaki, O.; Schachter, J.; Bank, I.; Jacoby, E.; Besser, M.J. Gamma-Delta CAR-T Cells Show CAR-Directed and Independent Activity Against Leukemia. Front. Immunol. 2020, 11, 1347. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Su, M.; Liu, L.; Tang, Y.; Pan, Y.; Sun, J. Clinical Application of Cytokines in Cancer Immunotherapy. Drug Des. Dev. Ther. 2021, 15, 2269–2287. [Google Scholar] [CrossRef] [PubMed]
- Dutcher, J.P.; Schwartzentruber, D.J.; Kaufman, H.L.; Agarwala, S.S.; Tarhini, A.; Lowder, J.N.; Atkins, M.B. High dose interleukin-2 (Aldesleukin)—Expert consensus on best management practices-2014. J. Immunother. Cancer 2014, 2, 26. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef]
- Baldo, B.A. Side effects of cytokines approved for therapy. Drug Saf. 2014, 37, 921–943. [Google Scholar] [CrossRef]
- Charych, D.H.; Hoch, U.; Langowski, J.L.; Lee, S.R.; Addepalli, M.K.; Kirk, P.B.; Sheng, D.; Liu, X.; Sims, P.W.; VanderVeen, L.A.; et al. NKTR-214, an Engineered Cytokine with Biased IL2 Receptor Binding, Increased Tumor Exposure, and Marked Efficacy in Mouse Tumor Models. Clin. Cancer Res. 2016, 22, 680–690. [Google Scholar] [CrossRef]
- Bentebibel, S.-E.; Hurwitz, M.E.; Bernatchez, C.; Haymaker, C.; Hudgens, C.W.; Kluger, H.M.; Tetzlaff, M.T.; Tagliaferri, M.A.; Zalevsky, J.; Hoch, U.; et al. A First-in-Human Study and Biomarker Analysis of NKTR-214, a Novel IL2Rβγ-Biased Cytokine, in Patients with Advanced or Metastatic Solid Tumors. Cancer Discov. 2019, 9, 711–721. [Google Scholar] [CrossRef]
- Businesswire. Bristol Myers Squibb and Nektar Announce Update on Phase 3 PIVOT IO-001 Trial Evaluating Bempegaldesleukin (BEMPEG) in Combination with Opdivo (nivolumab) in Previously Untreated Unresectable or Metastatic Melanoma. Available online: https://www.businesswire.com/news/home/20220313005021/en/ (accessed on 10 August 2022).
- Falchook, G.; Gan, H.; Fu, S.; McKean, M.; Azad, A.; Sommerhalder, D.; Wang, J.; Tan, T.; Chee, C.; Barve, M.; et al. Phase 1/2 Study of Thor-707 (Sar444245), a Pegylated Recombinant Non-Alpha Il-2, as Monotherapy and in Combination with Pembrolizumab or Cetuximab in Patients (Pts) with Advanced Solid Tumors. J. Immunother. Cancer 2021, 9, A511. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration Center For Drug Evaluation and Research. Provenge Approval Letter. 2010. Available online: http://wayback.archive-it.org/7993/20170723023807/https://www.fda.gov/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/ucm210215.htm (accessed on 19 August 2022).
- Jilg, W.; Lorbeer, B.; Schmidt, M.; Wilske, B.; Zoulek, G.; Deinhardt, F. Clinical evaluation of a recombinant hepatitis B vaccine. Lancet 1984, 2, 1174–1175. [Google Scholar] [CrossRef]
- Lamm, D.L.; Morales, A.A. BCG success story: From prevention of tuberculosis to optimal bladder cancer treatment. Vaccine 2021, 39, 7308–7318. [Google Scholar] [CrossRef]
- Sankar, K.; Ye, J.C.; Li, Z.; Zheng, L.; Song, W.; Hu-Lieskovan, S. The role of biomarkers in personalized immunotherapy. Biomark. Res. 2022, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H., Jr.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Yuan, J.; Hegde, P.S.; Clynes, R.; Foukas, P.G.; Harari, A.; Kleen, T.O.; Kvistborg, P.; Maccalli, C.; Maecker, H.T.; Page, D.B.; et al. Novel technologies and emerging biomarkers for personalized cancer immunotherapy. J. Immunother. Cancer 2016, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef]
- Reckamp, K.L. Real-World Pseudoprogression: An Uncommon Phenomenon. J. Thorac. Oncol. 2018, 13, 880–882. [Google Scholar] [CrossRef]
- Nishino, M. Pseudoprogression and Measurement Variability. J. Clin. Oncol. 2016, 34, 3480–3481. [Google Scholar] [CrossRef]
- Tan, W.C.C.; Nerurkar, S.N.; Cai, H.Y.; Ng, H.H.M.; Wu, D.; Wee, Y.T.F.; Lim, J.C.T.; Yeong, J.; Lim, T.K.H. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun. 2020, 40, 135–153. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Nguyen, P.; Engle, E.L.; Kaunitz, G.J.; Cottrell, T.R.; Berry, S.; Green, B.; Soni, A.; Cuda, J.D.; Stein, J.E.; et al. Multidimensional, quantitative assessment of PD-1/PD-L1 expression in patients with Merkel cell carcinoma and association with response to pembrolizumab. J. Immunother. Cancer 2018, 6, 99. [Google Scholar] [CrossRef]
- Rahmim, A.; Zaidi, H. PET versus SPECT: Strengths, limitations and challenges. Nucl. Med. Commun. 2008, 29, 193–207. [Google Scholar] [CrossRef]
- Smith, G.; Carroll, L.; Aboagye, E.O. New frontiers in the design and synthesis of imaging probes for PET oncology: Current challenges and future directions. Mol. Imaging Biol. 2012, 14, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Badawi, R.D.; Shi, H.; Hu, P.; Chen, S.; Xu, T.; Price, P.M.; Ding, Y.; Spencer, B.A.; Nardo, L.; Liu, W.; et al. First Human Imaging Studies with the EXPLORER Total-Body PET Scanner. J. Nucl. Med. 2019, 60, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Allott, L.; Aboagye, E.O. Chemistry Considerations for the Clinical Translation of Oncology PET Radiopharmaceuticals. Mol. Pharm. 2020, 17, 2245–2259. [Google Scholar] [CrossRef]
- Kelloff, G.J.; Hoffman, J.M.; Johnson, B.; Scher, H.I.; Siegel, B.A.; Cheng, E.Y.; Cheson, B.D.; O’Shaughnessy, J.; Guyton, K.Z.; Mankoff, D.A.; et al. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin. Cancer Res. 2005, 11, 2785–2808. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.J.; Connett, J.M.; Germain, C.J.; Guo, L.W.; Rogers, B.E.; Schwarz, S.W.; Fritzberg, A.R.; Welch, M.J. Cu-64-labeled BAT-21T-NR-LU-10 Fab: An agent for PET imaging and radioimmunotherapy. J. Nucl. Med. 1996, 37, 371. [Google Scholar]
- Smith, D.L.; Breeman, W.A.; Sims-Mourtada, J. The untapped potential of Gallium 68-PET: The next wave of 68Ga-agents. Appl. Radiat. Isot. 2013, 76, 14–23. [Google Scholar] [CrossRef]
- Chames, P.; Rothbauer, U. Special Issue: Nanobody. Antibodies 2020, 9, 6. [Google Scholar] [CrossRef]
- Stumpp, M.T.; Binz, H.K.; Amstutz, P. DARPins: A new generation of protein therapeutics. Drug Discov. Today 2008, 13, 695–701. [Google Scholar] [CrossRef]
- Fu, R.; Carroll, L.; Yahioglu, G.; Aboagye, E.O.; Miller, P.W. Antibody Fragment and Affibody ImmunoPET Imaging Agents: Radiolabelling Strategies and Applications. ChemMedChem 2018, 13, 2466–2478. [Google Scholar] [CrossRef]
- Tiede, C.; Bedford, R.; Heseltine, S.; Smith, G.; Wijetunga, I.; Ross, R.; AlQallaf, D.; Roberts, A.P.; Balls, A.; Curd, A.; et al. Affimer proteins are versatile and renewable affinity reagents. eLife 2017, 6, e24903. [Google Scholar] [CrossRef]
- Laing, R.E.; Nair-Gill, E.; Witte, O.N.; Radu, C.G. Visualizing cancer and immune cell function with metabolic positron emission tomography. Curr. Opin. Genet. Dev. 2010, 20, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Treglia, G. Diagnostic Performance of 18F-FDG PET/CT in Infectious and Inflammatory Diseases according to Published Meta-Analyses. Contrast Media Mol. Imaging 2019, 2019, 3018349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamar, F.; Buscombe, J.; Chiti, A.; Christian, P.E.; Delbeke, D.; Donohoe, K.J.; Israel, O.; Martin-Comin, J.; Signore, A. EANM/SNMMI guideline for 18F-FDG use in inflammation and infection. J. Nucl. Med. 2013, 54, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Cheson, B.D.; Ansell, S.; Schwartz, L.; Gordon, L.I.; Advani, R.; Jacene, H.A.; Hoos, A.; Barrington, S.F.; Armand, P. Refinement of the Lugano Classification lymphoma response criteria in the era of immunomodulatory therapy. Blood 2016, 128, 2489–2496. [Google Scholar] [CrossRef]
- Tomita, M.; Yasui, H.; Higashikawa, K.; Nakajima, K.; Takakura, H.; Shiga, T.; Kuge, Y.; Ogawa, M. Anti PD-1 treatment increases [18F]FDG uptake by cancer cells in a mouse B16F10 melanoma model. EJNMMI Res. 2018, 8, 82. [Google Scholar] [CrossRef]
- Tomita, M.; Suzuki, M.; Kono, Y.; Nakajima, K.; Matsuda, T.; Kuge, Y.; Ogawa, M. Influence on [18F]FDG uptake by cancer cells after anti-PD-1 therapy in an enforced-immune activated mouse tumor. EJNMMI Res. 2020, 10, 24. [Google Scholar] [CrossRef]
- Kong, B.Y.; Menzies, A.; Saunders, C.A.B.; Liniker, E.; Ramanujam, S.; Guminski, A.; Kefford, R.; Long, G.; Carlino, M.S. Residual FDG-PET metabolic activity in metastatic melanoma patients with prolonged response to anti-PD-1 therapy. Pigment. Cell Melanoma Res. 2016, 29, 572–577. [Google Scholar] [CrossRef]
- Dercle, L.; Seban, R.-D.; Lazarovici, J.; Schwartz, L.H.; Houot, R.; Ammari, S.; Danu, A.; Edeline, V.; Marabelle, A.; Ribrag, V.; et al. 18F-FDG PET and CT Scans Detect New Imaging Patterns of Response and Progression in Patients with Hodgkin Lymphoma Treated by Anti-Programmed Death 1 Immune Checkpoint Inhibitor. J. Nucl. Med. 2018, 59, 15–24. [Google Scholar] [CrossRef]
- England, C.G.; Jiang, D.; Ehlerding, E.B.; Rekoske, B.T.; Ellison, P.A.; Hernandez, R.; Barnhart, T.E.; McNeel, D.G.; Huang, P.; Cai, W. 89Zr-labeled nivolumab for imaging of T-cell infiltration in a humanized murine model of lung cancer. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 110–120. [Google Scholar] [CrossRef]
- Niemeijer, A.N.; Leung, D.; Huisman, M.C.; Bahce, I.; Hoekstra, O.S.; van Dongen, G.A.M.S.; Boellaard, R.; Du, S.; Hayes, W.; Smith, R.; et al. Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer. Nat. Commun. 2018, 9, 4664. [Google Scholar] [CrossRef]
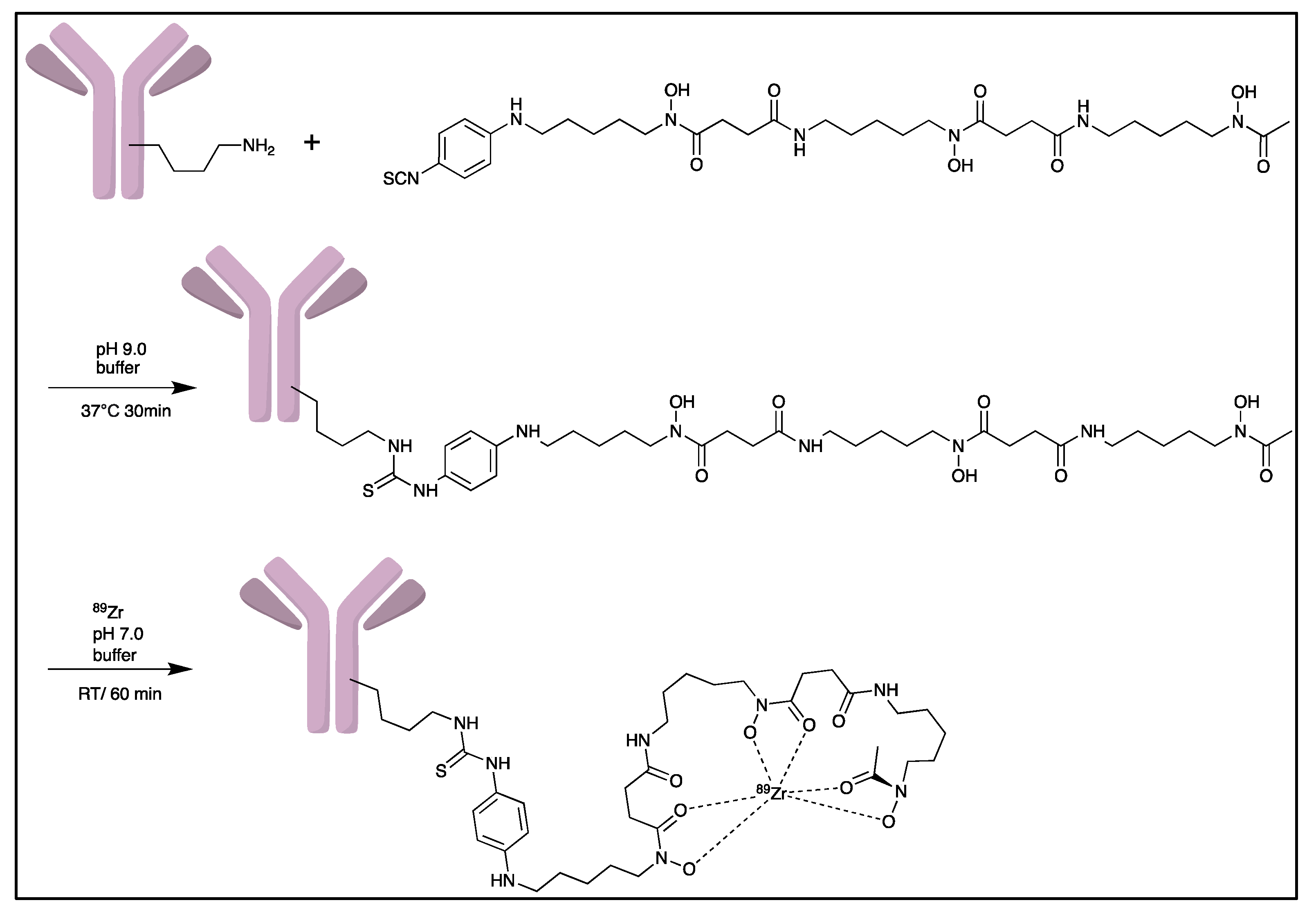
- Deri, M.A.; Zeglis, B.M.; Francesconi, L.C.; Lewis, J.S. PET imaging with 89Zr: From radiochemistry to the clinic. Nucl. Med. Biol. 2013, 40, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Pandya, D.N.; Bhatt, N.; Yuan, H.; Day, C.S.; Ehrmann, B.M.; Wright, M.; Bierbach, U.; Wadas, T.J. Zirconium tetraazamacrocycle complexes display extraordinary stability and provide a new strategy for zirconium-89-based radiopharmaceutical development. Chem. Sci. 2017, 8, 2309–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeglis, B.M.; Lewis, J.S. The bioconjugation and radiosynthesis of 89Zr-DFO-labeled antibodies. J. Vis. Exp. 2015, 96, 52521. [Google Scholar]
- Christensen, C.; Kristensen, L.K.; Alfsen, M.Z.; Nielsen, C.H.; Kjaer, A. Quantitative PET imaging of PD-L1 expression in xenograft and syngeneic tumour models using a site-specifically labelled PD-L1 antibody. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1302–1313. [Google Scholar] [CrossRef]
- Jung, K.-H.; Park, J.W.; Lee, J.H.; Moon, S.H.; Cho, Y.S.; Lee, K.-H. 89Zr-Labeled Anti-PD-L1 Antibody PET Monitors Gemcitabine Therapy-Induced Modulation of Tumor PD-L1 Expression. J. Nucl. Med. 2021, 62, 656–664. [Google Scholar] [CrossRef]
- Bensch, F.; Van der veen, E.L.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schröder, C.P.; Hiltermann, T.J.N.; Van Der Wekken, A.J.; et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 2018, 24, 1852–1858. [Google Scholar] [CrossRef]
- Smit, J.; Borm, F.J.; Niemeijer, A.-L.N.; Huisman, M.C.; Hoekstra, O.S.; Boellaard, R.; Oprea-Lager, D.E.; Vugts, D.J.; van Dongen, G.A.; Veen, B.J.d.W.v.d.; et al. PD-L1 PET/CT Imaging with Radiolabeled Durvalumab in Patients with Advanced-Stage Non-Small Cell Lung Cancer. J. Nucl. Med. 2022, 63, 686–693. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Study of ImmunoPet Imaging of PD-L1 in Tumors Using 89Zr-DFO-REGN3504 in Adult Participants With Advanced PD-L1 Positive Malignancies, Identifier NCT03746704. 2018–2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03746704 (accessed on 19 August 2022).
- U.S. National Library of Medicine. PD-L1 Imaging in Non Small Cell Lung Cancer’ (PINNACLE) (PINNACLE), Identifier NCT03514719. 2018–2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03514719 (accessed on 19 August 2022).
- Bridoux, J.; Broos, K.; Lecocq, Q.; Debie, P.; Martin, C.; Ballet, S.; Raes, G.; Neyt, S.; Vanhove, C.; Breckpot, K.; et al. Anti-human PD-L1 Nanobody for Immuno-PET Imaging: Validation of a Conjugation Strategy for Clinical Translation. Biomolecules 2020, 10, 1388. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, X.; Yang, Y.; Wang, C.; Zou, J.; Lin, J.; Qiu, L. Immuno-PET imaging of PD-L1 expression in patient-derived lung cancer xenografts with [68Ga]Ga-NOTA-Nb109. Quant. Imaging Med. Surg. 2022, 12, 3300–3313. [Google Scholar] [CrossRef]
- Liu, Q.; Jiang, L.; Li, K.; Li, H.; Lv, G.; Lin, J.; Qiu, L. Immuno-PET imaging of 68Ga-labeled nanobody Nb109 for dynamic monitoring the PD-L1 expression in cancers. Cancer Immunol. Immunother. 2021, 70, 1721–1733. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. PD-L1 Targeting Nanobody Probe for PET Imaging of Solid Tumor, Identifier NCT05156515. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT05156515 (accessed on 19 August 2022).
- Aslani, A.; Snowdon, G.M.; Bailey, D.L.; Schembri, G.P.; Bailey, E.A.; Roach, P.J. Gallium-68 DOTATATE Production with Automated PET Radiopharmaceutical Synthesis System: A Three Year Experience. Asia Ocean J. Nucl. Med. Biol. 2014, 2, 75–86. [Google Scholar] [PubMed]
- Mueller, D.; Klette, I.; Baum, R.P.; Gottschaldt, M.; Schultz, M.K.; Breeman, W.A.P. Simplified NaCl based 68Ga concentration and labeling procedure for rapid synthesis of 68Ga radiopharmaceuticals in high radiochemical purity. Bioconjug. Chem. 2012, 23, 1712–1717. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, M.; Chen, B.; Liu, H.; Fang, J.; Xiang, S.; Hu, S.; Zhang, X. Preclinical and exploratory human studies of novel 68Ga-labeled D-peptide antagonist for PET imaging of TIGIT expression in cancers. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 2584–2594. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, C.; Yang, Y.; Sun, Y.; Wei, W.; Wang, C.; Wan, L.; Zhu, C.; Li, L.; Huang, G.; et al. ImmunoPET imaging of human CD8+ T cells with novel 68Ga-labeled nanobody companion diagnostic agents. J. Nanobiotechnol. 2021, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Goggi, J.L.; Tan, Y.X.; Hartimath, S.V.; Jieu, B.; Hwang, Y.Y.; Jiang, L.; Boominathan, R.; Cheng, P.; Yuen, T.Y.; Chin, H.X.; et al. Granzyme B PET Imaging of Immune Checkpoint Inhibitor Combinations in Colon Cancer Phenotypes. Mol. Imaging Biol. 2020, 22, 1392–1402. [Google Scholar] [CrossRef] [PubMed]
- Miedema, I.H.; Zwezerijnen, G.J.; van Dongen, G.A.; Vugts, D.J.; Huisman, M.C.; Hoekstra, O.S.; de Gruijl, T.D.; Verheul, H.M.; Menke, C.W.; Eertwegh, A.J.V.D. Abstract 1136: Tumor uptake and biodistribution of 89Zirconium-labeled ipilimumab in patients with metastatic melanoma during ipilimumab treatment. Cancer Res. 2019, 79, 1136. [Google Scholar] [CrossRef]
- Higashikawa, K.; Yagi, K.; Watanabe, K.; Kamino, S.; Ueda, M.; Hiromura, M.; Enomoto, S. 64Cu-DOTA-anti-CTLA-4 mAb enabled PET visualization of CTLA-4 on the T-cell infiltrating tumor tissues. PLoS ONE 2014, 9, e109866. [Google Scholar] [CrossRef]
- Ehlerding, E.B.; England, C.G.; Majewski, R.L.; Valdovinos, H.F.; Jiang, D.; Liu, G.; McNeel, D.G.; Nickles, R.J.; Cai, W. ImmunoPET Imaging of CTLA-4 Expression in Mouse Models of Non-small Cell Lung Cancer. Mol. Pharm. 2017, 14, 1782–1789. [Google Scholar] [CrossRef]
- Zhao, N.; Bardine, C.; Lourenço, A.L.; Wang, Y.-H.; Huang, Y.; Cleary, S.J.; Wilson, D.M.; Oh, D.Y.; Fong, L.; Looney, M.R.; et al. In Vivo Measurement of Granzyme Proteolysis from Activated Immune Cells with PET. ACS Cent. Sci. 2021, 7, 1638–1649. [Google Scholar] [CrossRef]
- Alam, I.S.; Simonetta, F.; Scheller, L.; Mayer, A.T.; Murty, S.; Vermesh, O.; Nobashi, T.W.; Lohmeyer, J.K.; Hirai, T.; Baker, J.; et al. Visualization of Activated T Cells by OX40-ImmunoPET as a Strategy for Diagnosis of Acute Graft-versus-Host Disease. Cancer Res. 2020, 80, 4780–4790. [Google Scholar] [CrossRef]
- Zhang, Y.; Hong, H.; Engle, J.W.; Bean, J.; Yang, Y.; Leigh, B.R.; Barnhart, T.E.; Cai, W. Positron emission tomography imaging of CD105 expression with a 64Cu-labeled monoclonal antibody: NOTA is superior to DOTA. PLoS ONE 2011, 6, e28005. [Google Scholar] [CrossRef] [PubMed]
- Dearling, J.L.; Voss, S.D.; Dunning, P.; Snay, E.; Fahey, F.; Smith, S.V.; Huston, J.S.; Meares, C.F.; Treves, S.T.; Packard, A.B. Imaging cancer using PET--the effect of the bifunctional chelator on the biodistribution of a 64Cu-labeled antibody. Nucl. Med. Biol. 2011, 38, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.P.; Tavare, R.; Giurleo, J.T.; Makonnen, S.; Hickey, C.; Danton, M.A.; Arnold, T.C.; Ma, D.; Dai, J.; Pei, J.; et al. Abstract 3033: Immuno-PET detection of LAG-3 expressing intratumoral lymphocytes using the zirconium-89 radiolabeled fully human anti-LAG-3 antibody REGN3767. Cancer Res. 2018, 78, 3033. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. 89Zr-DFO-REGN3767 in PET Scans in People With Diffuse Large B Cell Lymphoma (DLBCL), Identifier NCT04566978. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04566978 (accessed on 19 August 2022).
- Larimer, B.M.; Wehrenberg-Klee, E.; Caraballo, A.; Mahmood, U. Quantitative CD3 PET Imaging Predicts Tumor Growth Response to Anti-CTLA-4 Therapy. J. Nucl. Med. 2016, 57, 1607–1611. [Google Scholar] [CrossRef]
- Beckford Vera, D.R.; Smith, C.C.; Bixby, L.M.; Glatt, D.M.; Dunn, S.S.; Saito, R.; Kim, W.Y.; Serody, J.S.; Vincent, B.G.; Parrott, M.C. Immuno-PET imaging of tumor-infiltrating lymphocytes using zirconium-89 radiolabeled anti-CD3 antibody in immune-competent mice bearing syngeneic tumors. PLoS ONE 2018, 13, e0193832. [Google Scholar] [CrossRef]
- Griessinger, C.M.; Olafsen, T.; Mascioni, A.; Jiang, Z.K.; Zamilpa, C.; Jia, F.; Torgov, M.; Romero, J.M.; Marchioni, F.; Satpayev, D.; et al. The PET-Tracer 89Zr-Df-IAB22M2C Enables Monitoring of Intratumoral CD8 T-cell Infiltrates in Tumor-Bearing Humanized Mice after T-cell Bispecific Antibody Treatment. Cancer Res. 2020, 80, 2903–2913. [Google Scholar] [CrossRef]
- Olafsen, T.; Torgov, M.; Zhang, G.G.; Romero, J.; Zampila, C.; Marchioni, F.; Jiang, K.; Gudas, J.; Satpayev, D. Pet imaging of cytotoxic human T cells using an 89Zr-labeled anti-CD8 minibody. J. ImmunoTherapy Cancer 2015, 3, P388. [Google Scholar] [CrossRef]
- Pandit-Taskar, N.; Postow, M.A.; Hellmann, M.D.; Harding, J.J.; Barker, C.A.; O’Donoghue, J.A.; Ziolkowska, M.; Ruan, S.; Lyashchenko, S.K.; Tsai, F.; et al. First-in-Humans Imaging with 89Zr-Df-IAB22M2C Anti-CD8 Minibody in Patients with Solid Malignancies: Preliminary Pharmacokinetics, Biodistribution, and Lesion Targeting. J. Nucl. Med. 2020, 61, 512–519. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. CD8+ PET Companion Trial, Identifier NCT05279027. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05279027 (accessed on 19 August 2022).
- Croft, M.; So, T.; Duan, W.; Soroosh, P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol. Rev. 2009, 229, 173–191. [Google Scholar] [CrossRef]
- Alam, I.S.; Mayer, A.T.; Sagiv-Barfi, I.; Wang, K.; Vermesh, O.; Czerwinski, D.K.; Johnson, E.; James, M.L.; Levy, R.; Gambhir, S.S. Imaging activated T cells predicts response to cancer vaccines. J. Clin. Investig. 2018, 128, 2569–2580. [Google Scholar] [CrossRef]
- Nobashi, T.; Mayer, A.; Xiao, Z.; Chan, C.; Chaney, A.; Gambhir, S. Imaging activated immune response following therapeutic vaccination in an orthotopic glioma model with 89Zr-DFO-OX40 mAb PET. J. Nucl. Med. 2020, 61, 2. [Google Scholar]
- Malek, T.R.; Castro, I. Interleukin-2 receptor signaling: At the interface between tolerance and immunity. Immunity 2010, 33, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Smigiel, K.S.; Richards, E.; Srivastava, S.; Thomas, K.R.; Dudda, J.C.; Klonowski, K.D.; Campbell, D.J. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J. Exp. Med. 2014, 211, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Di Gialleonardo, V.; Signore, A.; Glaudemans, A.W.; Dierckx, R.A.; De Vries, E.F. N-(4-18F-fluorobenzoyl)interleukin-2 for PET of human-activated T lymphocytes. J. Nucl. Med. 2012, 53, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Allott, L.; Amgheib, A.; Barnes, C.; Braga, M.; Brickute, D.; Wang, N.; Fu, R.; Ghaem-Maghami, S.; Aboagye, E.O. Radiolabelling an 18F biologic via facile IEDDA “click” chemistry on the GE FASTLab platform. React. Chem. Eng. 2021, 6, 1070–1078. [Google Scholar] [CrossRef]
- Barnes, C.; Nair, M.; Aboagye, E.O.; Archibald, S.J.; Allott, L. A practical guide to automating fluorine-18 PET radiochemistry using commercially available cassette-based platforms. React. Chem. Eng. 2022. [Google Scholar] [CrossRef]
- Di Gialleonardo, V.; Signore, A.; Willemsen, A.T.M.; Sijbesma, J.W.A.; Dierckx, R.A.J.O.; de Vries, E. Pharmacokinetic modelling of N-(4-[(18)F]fluorobenzoyl)interleukin-2 binding to activated lymphocytes in an xenograft model of inflammation. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 1551–1560. [Google Scholar] [CrossRef]
- Hartimath, S.V.; Draghiciu, O.; Van De Wall, S.; Manuelli, V.; Dierckx, R.A.J.O.; Nijman, H.W.; Daemen, T.; De Vries, E.F.J. Noninvasive monitoring of cancer therapy induced activated T cells using [18F]FB-IL-2 PET imaging. Oncoimmunology 2017, 6, e1248014. [Google Scholar] [CrossRef]
- van de Donk, P.P.; Wind, T.T.; Hooiveld-Noeken, J.S.; van der Veen, E.L.; Glaudemans, A.W.J.M.; Diepstra, A.; Jalving, M.; de Vries, E.G.E.; de Vries, E.F.J.; Hospers, G.A.P. Interleukin-2 PET imaging in patients with metastatic melanoma before and during immune checkpoint inhibitor therapy. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 4369–4376. [Google Scholar] [CrossRef]
- Larimer, B.M.; Wehrenberg-Klee, E.; Dubois, F.; Mehta, A.; Kalomeris, T.; Flaherty, K.; Boland, G.; Mahmood, U. Granzyme B PET Imaging as a Predictive Biomarker of Immunotherapy Response. Cancer Res. 2017, 77, 2318–2327. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Rano, T.A.; Peterson, E.P.; Rasper, D.M.; Timkey, T.; Garcia-Calvo, M.; Houtzager, V.M.; Nordstrom, P.A.; Roy, S.; Vaillancourt, J.P.; et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 1997, 272, 17907–17911. [Google Scholar] [CrossRef] [PubMed]
- Archibald, S.J.; Allott, L. The aluminium-[18F]fluoride revolution: Simple radiochemistry with a big impact for radiolabelled biomolecules. EJNMMI Radiopharm Chem. 2021, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Abed, N.S.; Chace, J.H.; Fleming, A.L.; Cowdery, J.S. Interferon-γ regulation of B lymphocyte differentiation: Activation of B cells is a prerequisite for IFN-γ-mediated inhibition of B cell differentiation. Cell. Immunol. 1994, 153, 356–366. [Google Scholar] [CrossRef]
- Kawano, Y.; Noma, T.; Yata, J. Regulation of human IgG subclass production by cytokines. IFN-gamma and IL-6 act antagonistically in the induction of human IgG1 but additively in the induction of IgG2. J. Immunol. 1994, 153, 4948–4958. [Google Scholar] [PubMed]
- Finkelman, F.D.; Katona, I.M.; Mosmann, T.R.; Coffman, R.L. IFN-gamma regulates the isotypes of Ig secreted during in vivo humoral immune responses. J. Immunol. 1988, 140, 1022–1027. [Google Scholar]
- Zaidi, M.R.; Merlino, G. The two faces of interferon-γ in cancer. Clin. Cancer Res. 2011, 17, 6118–6124. [Google Scholar] [CrossRef]
- Gibson, H.M.; McKnight, B.N.; Malysa, A.; Dyson, G.; Wiesend, W.N.; McCarthy, C.E.; Reyes, J.; Wei, W.-Z.; Viola-Villegas, N.T. IFNγ PET Imaging as a Predictive Tool for Monitoring Response to Tumor Immunotherapy. Cancer Res. 2018, 78, 5706–5717. [Google Scholar] [CrossRef]
- Rezazadeh, F.; Ramos, N.; Saliganan, A.D.; Barr, S.; Peraino, N.; Schomburg, F.; Rancour, D.; Viola, N.T. Evaluation and selection of a lead diabody for interferon-gamma PET imaging. Nucl. Med. Biol. 2022. [Google Scholar] [CrossRef]
- Radu, C.G.; Shu, C.J.; Nair-Gill, E.; Shelly, S.M.; Barrio, J.R.; Satyamurthy, N.; Phelps, M.E.; Witte, O.N. Molecular imaging of lymphoid organs and immune activation by positron emission tomography with a new [18F]-labeled 2′-deoxycytidine analog. Nat. Med. 2008, 14, 783–788. [Google Scholar] [CrossRef]
- Bitter, E.E.; Townsend, M.H.; Erickson, R.; Allen, C.; O’Neill, K.L. Thymidine kinase 1 through the ages: A comprehensive review. Cell Biosci. 2020, 10, 138. [Google Scholar] [CrossRef] [PubMed]
- Namavari, M.; Chang, Y.-F.; Kusler, B.; Yaghoubi, S.; Mitchell, B.S.; Gambhir, S.S. Synthesis of 2′-deoxy-2′-[18F]fluoro-9-beta-D-arabinofuranosylguanine: A novel agent for imaging T-cell activation with PET. Mol. Imaging Biol. 2011, 13, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Colevas, A.D.; Bedi, N.; Chang, S.; Nieves, U.Y.M.; Chatterjee, S.; Davidzon, G.A.; Srinivas, S.; Le, Q.-T.; Gambhir, A.; Sunwoo, J.B. A study to evaluate immunological response to PD-1 inhibition in squamous cell carcinoma of the head and neck (SCCHN) using novel PET imaging with [18F] F-AraG. J. Clin. Oncol. 2018, 36, 6050. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. [18F]F-AraG/Total Body PET Imaging and Healthy Subjects and Lung Cancer Patients, Identifier NCT04678440. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT004678440 (accessed on 19 August 2022).
- U.S. National Library of Medicine. [18F]F-AraG PET Imaging to Visualize Tumor Infiltrating T-cell Activation in Non-small Cell Lung Cancer. (ATTAIN), Identifier NCT05157659. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT05157659 (accessed on 19 August 2022).
- Niemeijer, A.-L.N.; Oprea-Lager, D.E.; Huisman, M.C.; Hoekstra, O.S.; Boellaard, R.; de Wit-van der Veen, B.J.; Bahce, I.; Vugts, D.J.; van Dongen, G.A.M.S.; Thunnissen, E.; et al. Study of 89Zr-Pembrolizumab PET/CT in Patients With Advanced-Stage Non-Small Cell Lung Cancer. J. Nucl. Med. 2022, 63, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Verhoeff, S.R.; van de Donk, P.P.; Aarntzen, E.H.; Oosting, S.F.; Brouwers, A.H.; Miedema, I.H.; Voortman, J.; Oordt, W.C.M.-V.d.H.v.; Boellaard, R.; Vriens, D.; et al. 89Zr-DFO-durvalumab PET/CT prior to durvalumab treatment in patients with recurrent or metastatic head and neck cancer. J. Nucl. Med. 2022, 63, 1523–1530. [Google Scholar] [CrossRef]
- Nagle, V.L.; Hertz, C.A.J.; Henry, K.E.; Graham, M.S.; Campos, C.; Pillarsetty, N.; Schietinger, A.; Mellinghoff, I.K.; Lewis, J.S. Noninvasive Imaging of CD4+ T Cells in Humanized Mice. Mol. Cancer Ther. 2022, 21, 658–666. [Google Scholar] [CrossRef]
- Freise, A.C.; Zettlitz, K.A.; Salazar, F.B.; Lu, X.; Tavaré, R.; Wu, A.M. ImmunoPET Imaging of Murine CD4+ T Cells Using Anti-CD4 Cys-Diabody: Effects of Protein Dose on T Cell Function and Imaging. Mol. Imaging Biol. 2017, 19, 599–609. [Google Scholar] [CrossRef]
- Islam, A.; Pishesha, N.; Harmand, T.J.; Heston, H.; Woodham, A.W.; Cheloha, R.W.; Bousbaine, D.; Rashidian, M.; Ploegh, H.L. Converting an Anti-Mouse CD4 Monoclonal Antibody into an scFv Positron Emission Tomography Imaging Agent for Longitudinal Monitoring of CD4+ T Cells. J. Immunol. 2021, 207, 1468–1477. [Google Scholar] [CrossRef]









| Radionuclide | Half-Life | Decay Mode (β+ Mode %) | Position Energy (MeV) | Production Method |
|---|---|---|---|---|
| 11C | 20.4 min | 99 | 0.97 | 14N(p, α)11C |
| 18F | 109.7 min | 97 | 0.65 | 18O(p, n)18F |
| 68Ga | 67.7 min | 89 | 1.9 | 68Ge/68Ga(generator) |
| 44Sc | 3.97 h | 94 | 1.47 | 44Ca(p, n)44Sc or 44Ti/44Sc (generator) |
| 64Cu | 12.7 h | 18 | 0.65 | 64Ni(p, n)64Cu |
| 89Zr | 78.4 h | 23 | 0.91 | 89Y(p, n)89Zr |
| 124I | 100.2 h | 23 | 1.54 | 124Te(p, n)124I |
| Chelator | Complex | Labelling Conditions | |
|---|---|---|---|
| 68Ga | |||
| NOTA |  |  | RT/pH 4.0/30 min |
| DOTA |  |  | 95 °C/pH 4.0/6.6 min |
| THP |  |  | RT |
| 64Cu | |||
| NOTA |  |  | 40 °C/pH 6.5/30 min |
| DOTA |  |  | 37 °C/pH 5.5/60 min or 50 °C/pH 7.0/30 min |
| 89Zr | |||
| DFO |  |  | RT/pH 6.8–7.5/60 min |
| Targeting Motif | Probes | Highlights |
|---|---|---|
| Small molecule (MW < 1000 Da) | [18F]F-AraG |
|
| Peptide (MW 1000~3000 Da) | [68Ga]Ga-mNOTA-GZP [18F]AlF-mNOTA-GZP [68Ga]Ga-NOTA-GP12 [64Cu]Cu-DOTA-GRIP B |
|
| Nanobody (MW ~15 kDa) | [68Ga]Ga-NOTA-(hPD-L1) [68Ga]Ga-NOTA-Nb109 [68Ga]Ga-NODAGA-SNA006a |
|
| Diabody (MW~55 kDa) | [89Zr]Zr-DFO-NCS-anti-IFN-γ HL-11 |
|
| Minibody (MW~80 kDa) | [89Zr]Zr-Df-IAB22M2C |
|
| mAb (MW~150 kDa) | [89Zr]Zr-DFO-nivolumab [89Zr]Zr-DFO-pembrolizumab [89Zr]Zr-DFO-durvalumab [89Zr]Zr-DFO-REGN3504 [89Zr]Zr-DFO-avelumab [89Zr]Zr-DFO-6E11 [89Zr]Zr-DFO-ipilimumab [89Zr]Zr-DFO-REGN3767 [64Cu]Cu-DOTA-AbOX40 [89Zr]Zr-DFO-OX40 [89Zr]Zr-DFO-anti-IFN-γ |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amgheib, A.; Fu, R.; Aboagye, E.O. Positron Emission Tomography Probes for Imaging Cytotoxic Immune Cells. Pharmaceutics 2022, 14, 2040. https://doi.org/10.3390/pharmaceutics14102040
Amgheib A, Fu R, Aboagye EO. Positron Emission Tomography Probes for Imaging Cytotoxic Immune Cells. Pharmaceutics. 2022; 14(10):2040. https://doi.org/10.3390/pharmaceutics14102040
Chicago/Turabian StyleAmgheib, Ala, Ruisi Fu, and Eric O. Aboagye. 2022. "Positron Emission Tomography Probes for Imaging Cytotoxic Immune Cells" Pharmaceutics 14, no. 10: 2040. https://doi.org/10.3390/pharmaceutics14102040
APA StyleAmgheib, A., Fu, R., & Aboagye, E. O. (2022). Positron Emission Tomography Probes for Imaging Cytotoxic Immune Cells. Pharmaceutics, 14(10), 2040. https://doi.org/10.3390/pharmaceutics14102040