
Findings on In Vitro Transporter-Mediated Drug Interactions and Their Follow-Up Actions for Labeling: Analysis of Drugs Approved by US FDA between 2017 and 2021


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Application of the Basic Method for In Vitro Transporter Inhibitors
2.3. Analysis of Follow-Up Actions on Possible Clinical Inhibitors or Substrates of Transporters
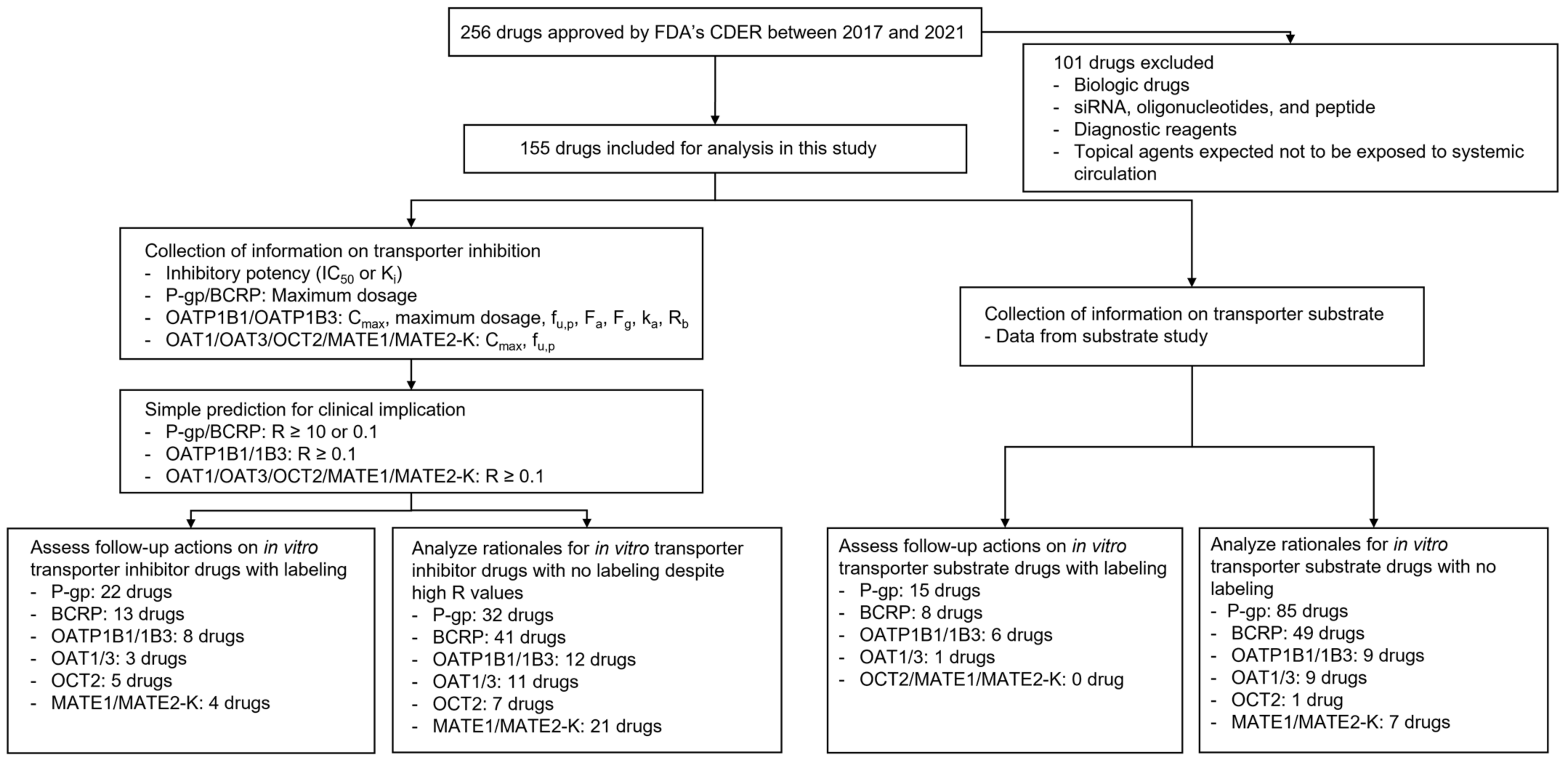
3. Results
3.1. General Findings
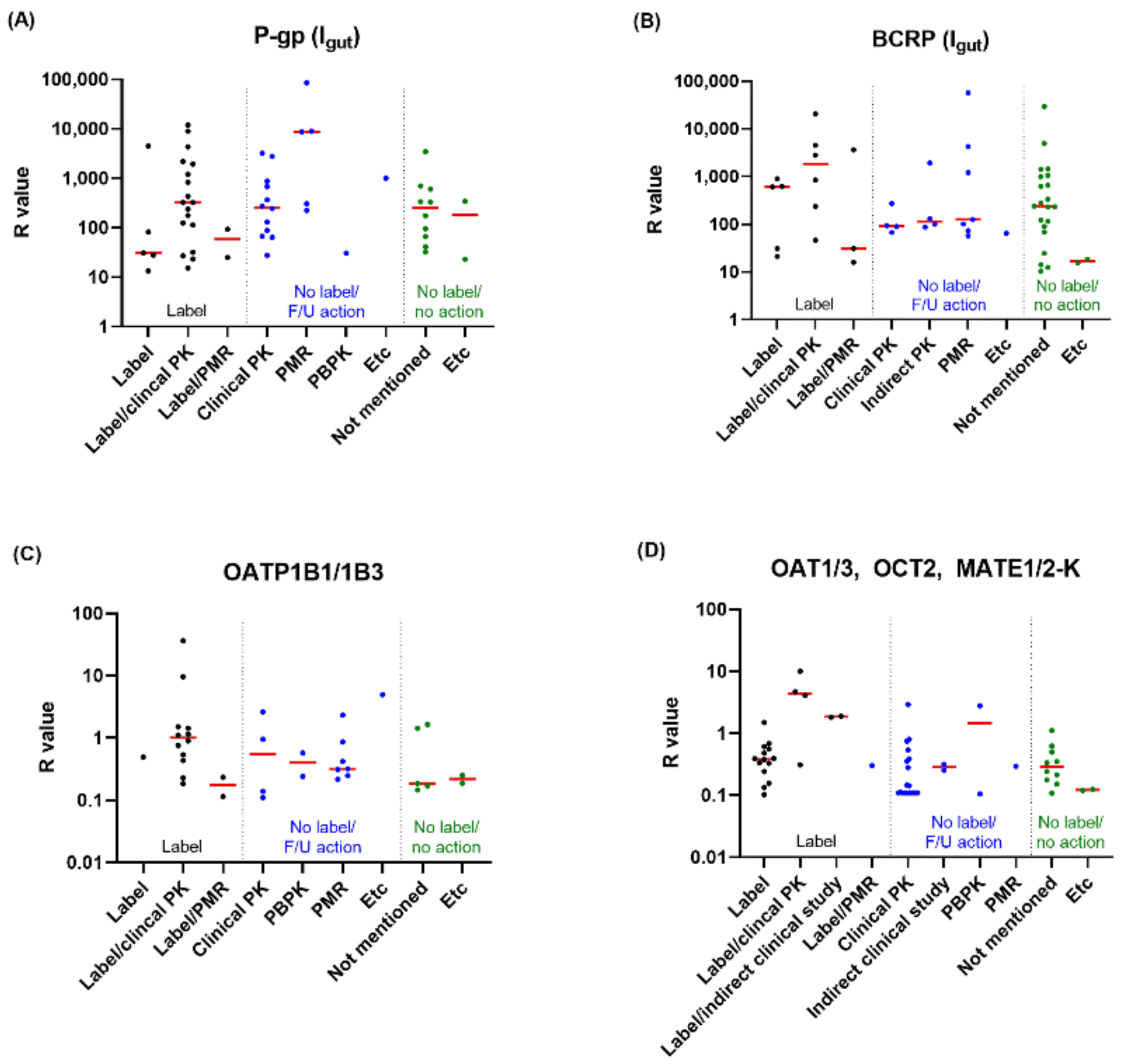
3.2. In Vitro Inhibitors and Their Follow-Up Actions
3.3. In Vitro Substrates and Their Follow-Up Actions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bjerrum, L.; Lopez-Valcarcel, B.G.; Petersen, G. Risk factors for potential drug interactions in general practice. Eur. J. Gen. Pract. 2008, 14, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Bénard-Laribière, A.; Miremont-Salamé, G.; Pérault-Pochat, M.; Noize, P.; Haramburu, F. Incidence of hospital admissions due to adverse drug reactions in France: The EMIR study. Fundam. Clin. Pharmacol. 2015, 29, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Curtis, K.; Zaidi, S.T.R.; Van, C.; Thomson, A.; Castelino, R. Prevalence, characteristics, and reporting of adverse drug reactions in an Australian hospital: A retrospective review of hospital admissions due to adverse drug reactions. Expert Opin. Drug Saf. 2021, 20, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Petzinger, E.; Geyer, J. Drug transporters in pharmacokinetics. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2006, 372, 465–475. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Lin, J.H. Drug-drug interaction mediated by inhibition and induction of P-glycoprotein. Adv. Drug Deliv. Rev. 2003, 55, 53–81. [Google Scholar] [CrossRef]
- Lee, C.A.; O’Connor, M.A.; Ritchie, T.K.; Galetin, A.; Cook, J.A.; Ragueneau-Majlessi, I.; Ellens, H.; Feng, B.; Taub, M.E.; Paine, M.F.; et al. Breast cancer resistance protein (ABCG2) in clinical pharmacokinetics and drug interactions: Practical recommendations for clinical victim and perpetrator drug-drug interaction study design. Drug Metab. Dispos. 2015, 43, 490–509. [Google Scholar] [CrossRef]
- Staud, F.; Ceckova, M.; Micuda, S.; Pavek, P. Expression and function of p-glycoprotein in normal tissues: Effect on pharmacokinetics. Methods Mol. Biol. 2010, 596, 199–222. [Google Scholar] [CrossRef]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport—An update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef]
- Obaidat, A.; Roth, M.; Hagenbuch, B. The expression and function of organic anion transporting polypeptides in normal tissues and in cancer. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 135–151. [Google Scholar] [CrossRef] [Green Version]
- Roth, M.; Obaidat, A.; Hagenbuch, B. OATPs, OATs and OCTs: The organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br. J. Pharmacol. 2012, 165, 1260–1287. [Google Scholar] [CrossRef] [PubMed]
- Damme, K.; Nies, A.T.; Schaeffeler, E.; Schwab, M. Mammalian MATE (SLC47A) transport proteins: Impact on efflux of endogenous substrates and xenobiotics. Drug Metab. Rev. 2011, 43, 499–523. [Google Scholar] [CrossRef] [PubMed]
- Okubo, H.; Atsukawa, M.; Okubo, T.; Ando, H.; Nakadera, E.; Ikejima, K.; Nagahara, A. Gadoxetic acid-enhanced magnetic resonance imaging predicts hyperbilirubinemia induced by glecaprevir during hepatitis C virus treatment. Sci. Rep. 2022, 12, 7847. [Google Scholar] [CrossRef] [PubMed]
- Zechner, J.; Britza, S.M.; Farrington, R.; Byard, R.W.; Musgrave, I.F. Flavonoid-statin interactions causing myopathy and the possible significance of OATP transport, CYP450 metabolism and mevalonate synthesis. Life Sci. 2021, 291, 119975. [Google Scholar] [CrossRef] [PubMed]
- Wang, D. Current Research Method in Transporter Study. In Drug Transporters in Drug Disposition, Effects and Toxicity; Springer: Berlin/Heidelberg, Germany, 2019; pp. 203–240. [Google Scholar]
- Jani, M.; Krajcsi, P. In vitro methods in drug transporter interaction assessment. Drug Discov. Today Technol. 2014, 12, e105–e112. [Google Scholar] [CrossRef]
- Kimoto, E.; Costales, C.; West, M.A.; Bi, Y.; Vourvahis, M.; David Rodrigues, A.; Varma, M.V.S. Biomarker-Informed Model-Based Risk Assessment of Organic Anion Transporting Polypeptide 1B Mediated Drug-Drug Interactions. Clin. Pharmacol. Ther. 2022, 111, 404–415. [Google Scholar] [CrossRef]
- Varma, M.V.S.; El-Kattan, A.; Lai, Y. Transporters-mediated drug disposition—Physiochemistry and in silico approaches. In Drug Transporters: Molecular Characterization and Role in Drug Disposition; John Wiley and Sons: Hoboken, NJ, USA, 2022; pp. 433–452. [Google Scholar]
- Guidance for Industry. In Vitro Drug Interaction Studies-Cytochrome P450 Enzyme-and Transporter-Mediated Drug Interactions. Available online: https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm (accessed on 10 January 2022).
- Guidance for Industry. Clinical Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions. Available online: https://www.fda.gov/media/134581/download (accessed on 27 July 2022).
- Tachibana, T.; Kato, M.; Watanabe, T.; Mitsui, T.; Sugiyama, Y. Method for predicting the risk of drug-drug interactions involving inhibition of intestinal CYP3A4 and P-glycoprotein. Xenobiotica 2009, 39, 430–443. [Google Scholar] [CrossRef]
- Yoshida, K.; Maeda, K.; Sugiyama, Y. Transporter-mediated drug—Drug interactions involving OATP substrates: Predictions based on in vitro inhibition studies. Clin. Pharmacol. Ther. 2012, 91, 1053–1064. [Google Scholar] [CrossRef]
- Tweedie, D.; Polli, J.W.; Berglund, E.G.; Huang, S.M.; Zhang, L.; Poirier, A.; Chu, X.; Feng, B. Transporter studies in drug development: Experience to date and follow-up on decision trees from the International Transporter Consortium. Clin. Pharmacol. Ther. 2013, 94, 113–125. [Google Scholar] [CrossRef]
- Mehvar, R. Clearance Concepts: Fundamentals and Application to Pharmacokinetic Behavior of Drugs. J. Pharm. Pharm. Sci. 2018, 21, 88s–102s. [Google Scholar] [CrossRef]
- Dong, Z.; Yang, X.; Arya, V.; Zhang, L. Comparing Various In Vitro Prediction Criteria to Assess the Potential of a New Molecular Entity (NME) to Inhibit Organic Anion Transporter 1 and 3 (OAT1 and OAT3) In Vivo. Clin. Pharmacol. Ther. 2016, 99, S94. [Google Scholar]
- Srivalli, K.M.R.; Lakshmi, P.K. Overview of P-glycoprotein inhibitors: A rational outlook. Braz. J. Pharm. Sci. 2012, 48, 353–367. [Google Scholar] [CrossRef]
- Robert, J.; Jarry, C. Multidrug resistance reversal agents. J. Med. Chem. 2003, 46, 4805–4817. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.B.; Ling, V. Effect of quercetin on Hoechst 33342 transport by purified and reconstituted P-glycoprotein. Biochem. Pharmacol. 1997, 53, 587–596. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Year | No. of drugs approved by FDA’s CDER | No. of drugs included in further analysis |
|---|---|---|
| 2017 | 46 | 28 |
| 2018 | 59 | 40 |
| 2019 | 48 | 28 |
| 2020 | 53 | 30 |
| 2021 | 50 | 29 |
| Total | 256 | 155 |
| The number of drugs with available information (%) a | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Year | P-gp | BCRP | OATP1B1 | OATP1B3 | OAT1 | OAT3 | OCT2 | MATE1 | MATE2-K |
| 2017 | 28 (100%) | 25 (89.3%) | 24 (85.7%) | 24 (85.7%) | 24 (85.7%) | 22 (78.6%) | 22 (78.6%) | 7 (25%) | 7 (25%) |
| 2018 | 35 (87.5%) | 34 (85%) | 33 (82.5%) | 32 (80%) | 34 (85%) | 34 (85%) | 35 (87.5%) | 24 (60%) | 22 (55%) |
| 2019 | 27 (96.4%) | 26 (92.9%) | 28 (100%) | 28 (100%) | 28 (100%) | 28 (100%) | 28 (100%) | 24 (85.7%) | 24 (85.7%) |
| 2020 | 27 (90%) | 26 (86.7%) | 27 (90%) | 26 (86.7%) | 25 (83.3%) | 25 (83.3%) | 27 (90%) | 28 (93.3%) | 26 (86.7%) |
| 2021 | 27 (93.1%) | 27 (93.1%) | 25 (86.2%) | 25 (86.2%) | 24 (82.8%) | 24 (82.8%) | 25 (86.2%) | 25 (86.2%) | 25 (86.2%) |
| Total | 144 (92.9%) | 138 (89%) | 137 (88.4%) | 135 (87.1%) | 135 (87.1%) | 133 (85.8%) | 137 (88.4%) | 108 (69.7%) | 104 (67.1%) |
| The number of inhibitor drugs with R value above the cut-off (%) b | |||||||||
| Year | P-gp | BCRP | OATP1B1 | OATP1B3 | OAT1 | OAT3 | OCT2 | MATE1 | MATE2-K |
| 2017 | 10 (35.7%) | 13 (52%) | 5 (20.8%) | 4 (16.7%) | 0 (0%) | 2 (9.1%) | 2 (9.1%) | 3 (42.9%) | 2 (28.6%) |
| 2018 | 15 (42.9%) | 12 (35.3%) | 5 (15.2%) | 4 (12.5%) | 1 (2.9%) | 4 (11.8%) | 4 (11.4%) | 2 (8.3%) | 2 (9.1%) |
| 2019 | 11 (40.7%) | 8 (30.8%) | 2 (7.1%) | 2 (7.1%) | 2 (7.1%) | 4 (14.3%) | 2 (7.1%) | 2 (8.3%) | 2 (8.3%) |
| 2020 | 9 (33.3%) | 10 (38.5%) | 3 (11.1%) | 3 (11.5%) | 0 (0%) | 1 (4%) | 2 (7.4%) | 8 (28.6%) | 5 (19.2%) |
| 2021 | 9 (33.3%) | 11 (40.7%) | 3 (12%) | 4 (16%) | 2 (8.3%) | 3 (12.5%) | 2 (8%) | 4 (16%) | 5 (20%) |
| Total | 54 (37.5%) | 54 (39.1%) | 18 (13.1%) | 17 (12.6%) | 5 (3.7%) | 14 (10.5%) | 12 (8.8%) | 19 (17.6%) | 16 (15.4%) |
| Category | The number of drugs (%) | |||||
|---|---|---|---|---|---|---|
| P-gp | BCRP | OATP1B1/1B3 | OAT1/3 | OCT2 | MATE1/2-K | |
| Label | 22 (40.7%) | 13 (24.1%) | 8 (40%) | 3 (21.4%) | 5 (41.7%) | 4 (16%) |
| Label (no other study/no PMR) | 3 (5.6%) | 4 (7.4%) | 1 (5%) | 2 (14.3%) | 2 (16.7%) | 2 (8%) |
| Label/clinical PK | 17 (31.5%) | 6 (11.1%) | 6 (30%) | 0 (0%) | 2 (16.7%) | 1 (4%) |
| Label/PBPK | 0 (0%) | 0 (0%) | 0 (0%) | 1 (7.1) | 0 (0%) | 0 (0%) |
| Label/PMR (clinical PK) | 2 (3.7%) | 3 (5.6%) | 1 (5%) | 0 (0%) | 1 (8.3%) | 0 (0%) |
| Label/indirect clinical study | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (4%) |
| No label | 32 (59.3%) | 41 (75.9%) | 12 (60%) | 11 (78.6%) | 7 (58.3%) | 21 (84%) |
| Clinical PK | 13 (24.1%) | 5 (9.3%) | 3 (15%) | 2 (14.3%) | 2 (16.7%) | 5 (20%) |
| PBPK | 2 (3.7%) | 0 (0%) | 1 (5%) | 1 (7.1%) | 1 (8.3%) | 0 (0%) |
| PMR | 5 (9.3%) | 7 (13%) | 3 (15%) | 0 (0%) | 1 (8.3%) | 3 (12%) |
| PMR (clinical PK) | 5 (9.3%) | 6 (11.1%) | 2 (10%) | 0 (0%) | 1 (8.3%) | 3 (12%) |
| PMR (PBPK) | 0 (0%) | 1 (1.9%) | 1 (5%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Etc | 2 (3.7%) | 7 (13%) | 1 (5%) | 1 (7.1%) | 1 (8.3%) | 5 (20%) |
| Indirect clinical study | 0 (0%) | 4 (7.4%) | 0 (0%) | 0 (0%) | 1 (8.3%) | 1 (4%) |
| Short dosing duration | 0 (0%) | 0 (0%) | 1 (5%) | 0 (0%) | 0 (0%) | 3 (12%) |
| Low solubility | 1 (1.9%) | 2 (3.7%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| No contact | 1 (1.9%) | 1 (1.9%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Static mechanistic model | 0 (0%) | 0 (0%) | 0 (0%) | 1 (7.1%) | 0 (0%) | 0 (0%) |
| No concomitant medication | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (4%) |
| Not mentioned | 10 (18.5%) | 22 (40.7%) | 4 (20%) | 7 (50%) | 2 (16.7%) | 8 (32%) |
| Total | 54 (100%) | 54 (100%) | 20 (100%) | 14 (100%) | 12 (100%) | 25 (100%) |
| The number of drugs with available information (%) a | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Year | P-gp | BCRP | OATP1B1 | OATP1B3 | OAT1 | OAT3 | OCT2 | MATE1 | MATE2-K |
| 2017 | 28 (100%) | 25 (89.3%) | 17 (60.7%) | 18 (64.3%) | 16 (57.1%) | 14 (50%) | 14 (50%) | 3 (10.7%) | 2 (7.1%) |
| 2018 | 35 (87.5%) | 31 (77.5%) | 25 (62.5%) | 25 (62.5%) | 13 (32.5%) | 14 (35%) | 14 (35%) | 7 (17.5%) | 7 (17.5%) |
| 2019 | 28 (100%) | 26 (92.9%) | 9 (32.1%) | 9 (32.1%) | 12 (42.9%) | 13 (46.4%) | 11 (39.3%) | 10 (35.7%) | 11 (39.3%) |
| 2020 | 29 (96.7%) | 25 (83.3%) | 18 (60%) | 19 (63.3%) | 9 (30%) | 9 (30%) | 11 (36.7%) | 12 (40%) | 10 (33.3%) |
| 2021 | 28 (96.6%) | 26 (89.7%) | 23 (79.3%) | 23 (79.3%) | 11 (37.9%) | 11 (37.9%) | 10 (34.5%) | 9 (31%) | 9 (31%) |
| Total | 148 (95.5%) | 133 (85.8%) | 92 (59.4%) | 94 (60.6%) | 61 (39.4%) | 61 (39.4%) | 60 (38.7%) | 41 (26.5%) | 39 (25.2%) |
| The number of in vitro transporter substrate drugs (%) b | |||||||||
| Year | P-gp | BCRP | OATP1B1 | OATP1B3 | OAT1 | OAT3 | OCT2 | MATE1 | MATE2-K |
| 2017 | 18 (64.3%) | 11 (44%) | 3 (17.6%) | 3 (16.7%) | 2 (12.5%) | 3 (21.4%) | 0 (0%) | 0 (0%) | 0 (0%) |
| 2018 | 25 (71.4%) | 14 (45.2%) | 4 (16%) | 3 (12%) | 0 (0%) | 1 (7.1%) | 0 (0%) | 0 (0%) | 1 (14.3%) |
| 2019 | 14 (50%) | 7 (26.9%) | 3 (33.3%) | 2 (22.2%) | 1 (8.3%) | 1 (7.7%) | 1 (9.1%) | 2 (20%) | 2 (18.2%) |
| 2020 | 25 (86.2%) | 16 (64%) | 1 (5.6%) | 2 (10.5%) | 0 (0%) | 1 (11.1%) | 0 (0%) | 1 (8.3%) | 1 (10%) |
| 2021 | 18 (64.3%) | 9 (34.6%) | 4 (17.4%) | 4 (17.4%) | 2 (18.2%) | 2 (18.2%) | 0 (0%) | 1 (11.1%) | 1 (11.1%) |
| Total | 100 (67.6%) | 57 (42.9%) | 15 (16.3%) | 14 (14.9%) | 5 (8.2%) | 8 (13.1%) | 1 (1.7%) | 4 (9.8%) | 5 (12.8%) |
| Category | The number of drugs (%) | |||||
|---|---|---|---|---|---|---|
| P-gp | BCRP | OATP1B1/1B3 | OAT1/3 | OCT2 | MATE1 /2-K | |
| Label | 15 (15%) | 8 (14%) | 6 (40%) | 1 (10%) | 0 (0%) | 0 (0%) |
| Label | 4 (4%) | 1 (6.7%) | 1 (6.7%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Label/clinical PK | 7 (7%) | 5 (33.3%) | 5 (33.3%) | 1 (10%) | 0 (0%) | 0 (0%) |
| Label/PMR (clinical PK) | 4 (4%) | 1 (1.8%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| No label | 85 (85%) | 49 (86%) | 9 (60%) | 9 (90%) | 1 (100%) | 7 (100%) |
| Clinical PK | 19 (19%) | 5 (8.8%) | 1 (6.7%) | 1 (10%) | 0 (0%) | 0 (0%) |
| PBPK | 1 (1%) | 2 (3.5%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| PMR | 3 (3%) | 1 (1.8%) | 1 (6.7%) | 0 (0%) | 0 (0%) | 0 (0%) |
| PMR (clinical PK) | 2 (2%) | 1 (1.8%) | 1 (6.7%) | 0 (0%) | 0 (0%) | 0 (0%) |
| PMR (in vitro study) | 1 (1%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Etc | 38 (38%) | 22 (38.6%) | 4 (26.7%) | 8 (80%) | 1 (100%) | 5 (71.4%) |
| High permeability | 14 (14%) | 7 (12.3%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Weak substrate | 7 (7%) | 3 (5.3%) | 2 (13.3%) | 2 (20%) | 1 (100%) | 2 (28.6%) |
| Not major elimination route | 1 (1%) | 1 (1.8%) | 2 (13.3%) | 2 (20%) | 0 (0%) | 1 (14.3%) |
| Saturation | 2 (2%) | 1 (1.8%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Wide safety range | 1 (1%) | 2 (3.5%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Indirect clinical study | 2 (2%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (14.3%) |
| IV dosing/no safety concern | 1 (1%) | 1 (1.8%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Low solubility | 1 (1%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Short dosing duration | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (14.3%) |
| Not mentioned | 33 (33%) | 26 (45.6%) | 3 (20%) | 4 (40%) | 0 (0%) | 2 (28.6%) |
| Total | 100 (100%) | 57 (100%) | 15 (100%) | 10 (100%) | 1 (100%) | 7 (100%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.-R.; Chang, J.-E.; Yoon, J.; Jin, H.; Chae, Y.-J. Findings on In Vitro Transporter-Mediated Drug Interactions and Their Follow-Up Actions for Labeling: Analysis of Drugs Approved by US FDA between 2017 and 2021. Pharmaceutics 2022, 14, 2078. https://doi.org/10.3390/pharmaceutics14102078
Lee K-R, Chang J-E, Yoon J, Jin H, Chae Y-J. Findings on In Vitro Transporter-Mediated Drug Interactions and Their Follow-Up Actions for Labeling: Analysis of Drugs Approved by US FDA between 2017 and 2021. Pharmaceutics. 2022; 14(10):2078. https://doi.org/10.3390/pharmaceutics14102078
Chicago/Turabian StyleLee, Kyeong-Ryoon, Ji-Eun Chang, Jongmin Yoon, Hyojeong Jin, and Yoon-Jee Chae. 2022. "Findings on In Vitro Transporter-Mediated Drug Interactions and Their Follow-Up Actions for Labeling: Analysis of Drugs Approved by US FDA between 2017 and 2021" Pharmaceutics 14, no. 10: 2078. https://doi.org/10.3390/pharmaceutics14102078
APA StyleLee, K. -R., Chang, J. -E., Yoon, J., Jin, H., & Chae, Y. -J. (2022). Findings on In Vitro Transporter-Mediated Drug Interactions and Their Follow-Up Actions for Labeling: Analysis of Drugs Approved by US FDA between 2017 and 2021. Pharmaceutics, 14(10), 2078. https://doi.org/10.3390/pharmaceutics14102078