

Bispecific Antibodies: A Novel Approach for the Treatment of Solid Tumors
 , , , ,
, , , ,  , , , , ,
, , , , , 

Abstract
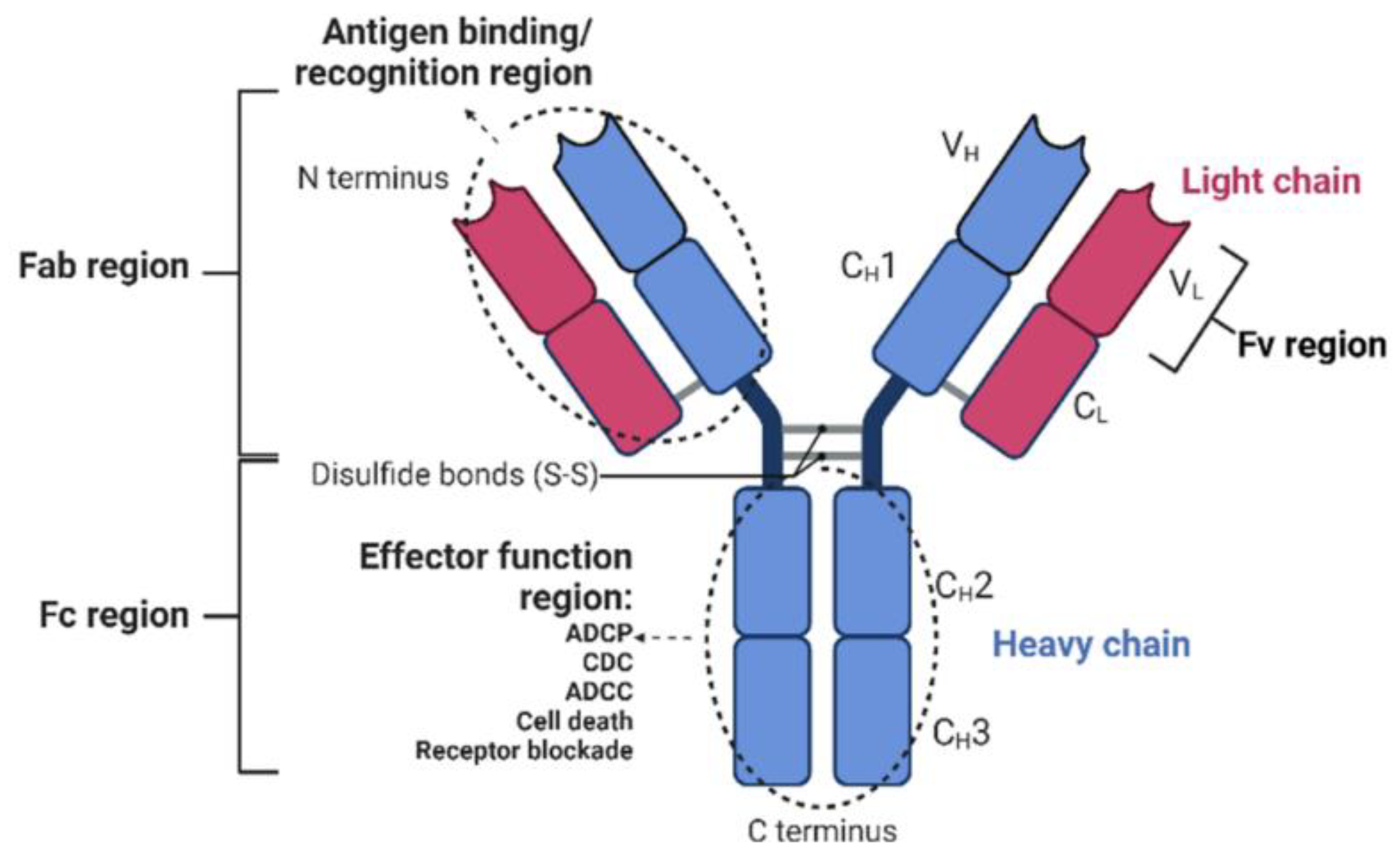
:1. Introduction: From Monoclonal Antibodies to Bispecific Antibodies
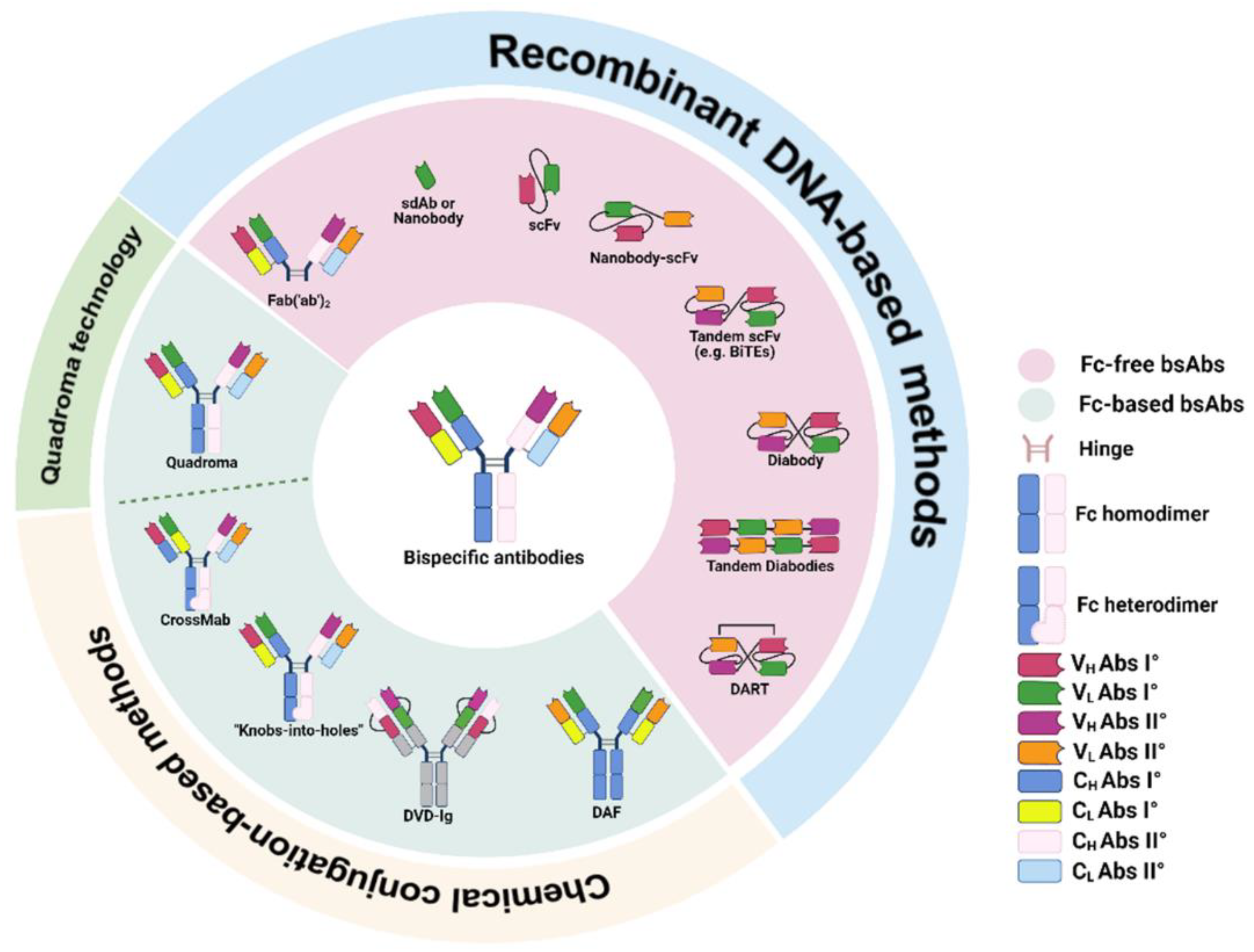
2. Classification and Production of Bispecific Antibodies
2.1. Fc-Based bsAbs
2.2. Fc-Free bsAbs
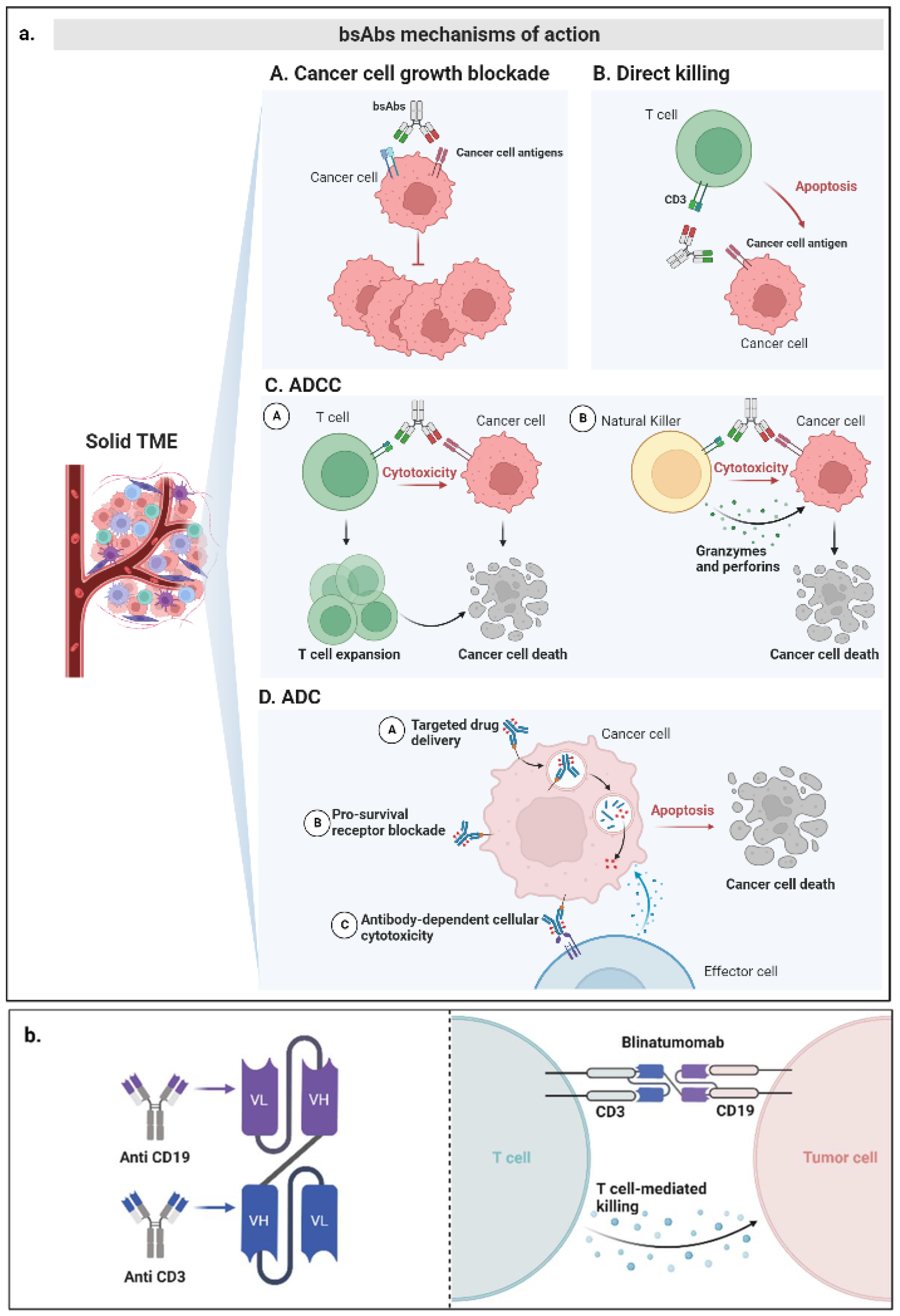
3. Mechanisms of Action of bsAbs
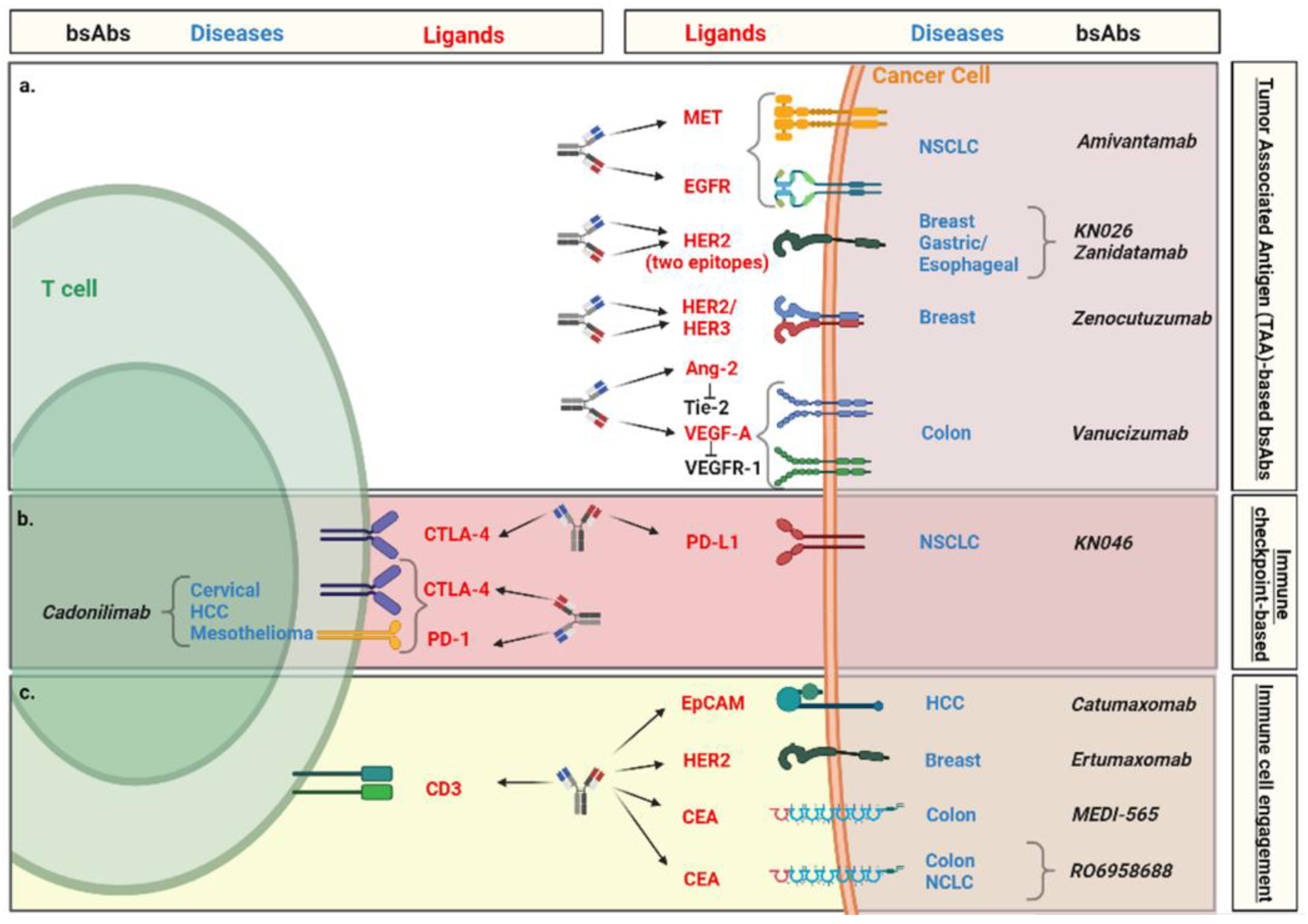
4. Clinical Evidence of Bispecific Antibodies in Solid Malignancies
4.1. Tumor-Associated Antigen (TAA)-Based bsAbs
4.1.1. Amivantamab
4.1.2. KN026
4.1.3. Zanidatamab
4.1.4. Zenocutuzumab
4.1.5. Vanucizumab
4.2. Immune Checkpoint-Based bsAbs
4.2.1. KN046
4.2.2. Cadonilimab
4.3. Immune Cell Engagement by bsAbs
4.3.1. Catumaxomab
4.3.2. Ertumaxomab
4.3.3. MEDI-565
4.3.4. RO6958688
5. Challenges of bsAbs in Solid Malignancies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shuptrine, C.W.; Surana, R.; Weiner, L.M. Monoclonal Antibodies for the Treatment of Cancer. Semin. Cancer Biol. 2012, 22, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [Green Version]
- Porter, R.R. The Hydrolysis of Rabbit Y-Globulin and Antibodies with Crystalline Papain. Biochem. J. 1959, 73, 119–127. [Google Scholar] [CrossRef]
- Chiu, M.L.; Goulet, D.R.; Teplyakov, A.; Gilliland, G.L. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies 2019, 8, 55. [Google Scholar] [CrossRef] [Green Version]
- Kellner, C.; Derer, S.; Valerius, T.; Peipp, M. Boosting ADCC and CDC Activity by Fc Engineering and Evaluation of Antibody Effector Functions. Methods 2014, 65, 105–113. [Google Scholar] [CrossRef]
- Elshiaty, M.; Schindler, H.; Christopoulos, P. Principles and Current Clinical Landscape of Multispecific Antibodies against Cancer. Int. J. Mol. Sci. 2021, 22, 5632. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef]
- Yoneda, K.; Imanishi, N.; Ichiki, Y.; Tanaka, F. Treatment of Non-Small Cell Lung Cancer with EGFR-Mutations. J. UOEH 2019, 41, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Aldea, M.; Andre, F.; Marabelle, A.; Dogan, S.; Barlesi, F.; Soria, J.-C. Overcoming Resistance to Tumor-Targeted and Immune-Targeted Therapies. Cancer Discov. 2021, 11, 874–899. [Google Scholar] [CrossRef]
- Rader, C. Bispecific Antibodies in Cancer Immunotherapy. Curr. Opin. Biotechnol. 2020, 65, 9–16. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific Antibodies: A Mechanistic Review of the Pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Wu, Y.; Yi, M.; Zhu, S.; Wang, H.; Wu, K. Recent Advances and Challenges of Bispecific Antibodies in Solid Tumors. Exp. Hematol. Oncol. 2021, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, S.S.; Khalili, S.; Baradaran, B.; Bidar, N.; Shahbazi, M.-A.; Mosafer, J.; Hashemzaei, M.; Mokhtarzadeh, A.; Hamblin, M.R. Bispecific Monoclonal Antibodies for Targeted Immunotherapy of Solid Tumors: Recent Advances and Clinical Trials. Int. J. Biol. Macromol. 2021, 167, 1030–1047. [Google Scholar] [CrossRef]
- Nisonoff, A.; Wissler, F.C.; Lipman, L.N. Properties of the Major Component of a Peptic Digest of Rabbit Antibody. Science 1960, 132, 1770–1771. [Google Scholar] [CrossRef]
- Staerz, U.D.; Kanagawa, O.; Bevan, M.J. Hybrid Antibodies Can Target Sites for Attack by T Cells. Nature 1985, 314, 628–631. [Google Scholar] [CrossRef]
- Riethmüller, G. Symmetry Breaking: Bispecific Antibodies, the Beginnings, and 50 Years On. Cancer Immun. 2012, 12, 12. [Google Scholar] [PubMed]
- Chames, P.; Baty, D. Bispecific Antibodies for Cancer Therapy: The Light at the End of the Tunnel? MAbs 2009, 1, 539–547. [Google Scholar] [CrossRef]
- Shin, H.G.; Yang, H.R.; Yoon, A.; Lee, S. Bispecific Antibody-Based Immune-Cell Engagers and Their Emerging Therapeutic Targets in Cancer Immunotherapy. Int. J. Mol. Sci. 2022, 23, 5686. [Google Scholar] [CrossRef]
- Yu, S.; Li, A.; Liu, Q.; Yuan, X.; Xu, H.; Jiao, D.; Pestell, R.G.; Han, X.; Wu, K. Recent Advances of Bispecific Antibodies in Solid Tumors. J. Hematol. Oncol. 2017, 10, 155. [Google Scholar] [CrossRef]
- Viardot, A.; Bargou, R. Bispecific Antibodies in Haematological Malignancies. Cancer Treat. Rev. 2018, 65, 87–95. [Google Scholar] [CrossRef]
- Curran, E.; Stock, W. Taking a “BiTE out of ALL”: Blinatumomab Approval for MRD-Positive ALL. Blood 2019, 133, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Goebeler, M.-E.; Bargou, R.C. T Cell-Engaging Therapies—BiTEs and Beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434. [Google Scholar] [CrossRef] [PubMed]
- Antonarelli, G.; Giugliano, F.; Corti, C.; Repetto, M.; Tarantino, P.; Curigliano, G. Research and Clinical Landscape of Bispecific Antibodies for the Treatment of Solid Malignancies. Pharmaceuticals 2021, 14, 884. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, K.; Lei, Q.; Ma, P.; Yuan, A.Q.; Zhao, Y.; Jiang, Y.; Fang, H.; Xing, S.; Fang, Y.; et al. The State of the Art of Bispecific Antibodies for Treating Human Malignancies. EMBO Mol. Med. 2021, 13, e14291. [Google Scholar] [CrossRef]
- Löffler, A.; Kufer, P.; Lutterbüse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmüller, G.; Dörken, B.; Bargou, R.C. A Recombinant Bispecific Single-Chain Antibody, CD19 x CD3, Induces Rapid and High Lymphoma-Directed Cytotoxicity by Unstimulated T Lymphocytes. Blood 2000, 95, 2098–2103. [Google Scholar] [CrossRef] [PubMed]
- Holliger, P.; Brissinck, J.; Williams, R.L.; Thielemans, K.; Winter, G. Specific Killing of Lymphoma Cells by Cytotoxic T-Cells Mediated by a Bispecific Diabody. Protein Eng. Des. Sel. 1996, 9, 299–305. [Google Scholar] [CrossRef] [Green Version]
- Kontermann, R.E. Strategies for Extended Serum Half-Life of Protein Therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef]
- Carter, P.J. Potent Antibody Therapeutics by Design. Nat. Rev. Immunol. 2006, 6, 343–357. [Google Scholar] [CrossRef]
- Menard, S.; Canevari, S.; Colnaghi, M.I. Hybrid Antibodies in Cancer Diagnosis and Therapy. Int. J. Biol. Markers 1989, 4, 131–134. [Google Scholar] [CrossRef]
- Morrison, S.L. Two Heads Are Better than One. Nat. Biotechnol. 2007, 25, 1233–1234. [Google Scholar] [CrossRef]
- Lindhofer, H.; Mocikat, R.; Steipe, B.; Thierfelder, S. Preferential Species-Restricted Heavy/Light Chain Pairing in Rat/Mouse Quadromas. Implications for a Single-Step Purification of Bispecific Antibodies. J. Immunol. 1995, 155, 219–225. [Google Scholar]
- Li, H.; Saw, P.E.; Song, E. Challenges and Strategies for Next-Generation Bispecific Antibody-Based Antitumor Therapeutics. Cell. Mol. Immunol. 2020, 17, 451–461. [Google Scholar] [CrossRef]
- Jakob, C.G.; Edalji, R.; Judge, R.A.; DiGiammarino, E.; Li, Y.; Gu, J.; Ghayur, T. Structure Reveals Function of the Dual Variable Domain Immunoglobulin (DVD-IgTM) Molecule. MAbs 2013, 5, 358–363. [Google Scholar] [CrossRef] [Green Version]
- Eigenbrot, C.; Fuh, G. Two-in-One Antibodies with Dual Action Fabs. Curr. Opin. Chem. Biol. 2013, 17, 400–405. [Google Scholar] [CrossRef]
- Ridgway, J.B.B.; Presta, L.G.; Carter, P. ‘Knobs-into-Holes’ Engineering of Antibody CH3 Domains for Heavy Chain Heterodimerization. Protein Eng. Des. Sel. 1996, 9, 617–621. [Google Scholar] [CrossRef] [Green Version]
- Holliger, P.; Hudson, P.J. Engineered Antibody Fragments and the Rise of Single Domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Shatz, W.; Ng, D.; Dutina, G.; Wong, A.W.; Dunshee, D.R.; Sonoda, J.; Shen, A.; Scheer, J.M. An Efficient Route to Bispecific Antibody Production Using Single-Reactor Mammalian Co-Culture. MAbs 2016, 8, 1487–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouet, R.; Christ, D. Bispecific Antibodies with Native Chain Structure. Nat. Biotechnol. 2014, 32, 136–137. [Google Scholar] [CrossRef] [PubMed]
- Dhimolea, E.; Reichert, J.M. World Bispecific Antibody Summit, September 27-28, 2011, Boston, MA. MAbs 2012, 4, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onitsuka, M.; Kawaguchi, A.; Asano, R.; Kumagai, I.; Honda, K.; Ohtake, H.; Omasa, T. Glycosylation Analysis of an Aggregated Antibody Produced by Chinese Hamster Ovary Cells in Bioreactor Culture. J. Biosci. Bioeng. 2014, 117, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, W.; Regula, J.T.; Bähner, M.; Schanzer, J.; Croasdale, R.; Dürr, H.; Gassner, C.; Georges, G.; Kettenberger, H.; Imhof-Jung, S.; et al. Immunoglobulin Domain Crossover as a Generic Approach for the Production of Bispecific IgG Antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 11187–11192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenn, S.; Schiller, C.B.; Griese, J.J.; Duerr, H.; Imhof-Jung, S.; Gassner, C.; Moelleken, J.; Regula, J.T.; Schaefer, W.; Thomas, M.; et al. Crystal Structure of an Anti-Ang2 CrossFab Demonstrates Complete Structural and Functional Integrity of the Variable Domain. PLoS ONE 2013, 8, e61953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahamadi-Fesharaki, R.; Fateh, A.; Vaziri, F.; Solgi, G.; Siadat, S.D.; Mahboudi, F.; Rahimi-Jamnani, F. Single-Chain Variable Fragment-Based Bispecific Antibodies: Hitting Two Targets with One Sophisticated Arrow. Mol. Ther. Oncolytics 2019, 14, 38–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, U.; Kontermann, R.E. The Making of Bispecific Antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [Green Version]
- Revets, H.; De Baetselier, P.; Muyldermans, S. Nanobodies as Novel Agents for Cancer Therapy. Expert Opin. Biol. Ther. 2005, 5, 111–124. [Google Scholar] [CrossRef]
- Johnson, S.; Burke, S.; Huang, L.; Gorlatov, S.; Li, H.; Wang, W.; Zhang, W.; Tuaillon, N.; Rainey, J.; Barat, B.; et al. Effector Cell Recruitment with Novel Fv-Based Dual-Affinity Re-Targeting Protein Leads to Potent Tumor Cytolysis and in Vivo B-Cell Depletion. J. Mol. Biol. 2010, 399, 436–449. [Google Scholar] [CrossRef]
- Li, Z.; Krippendorff, B.-F.; Sharma, S.; Walz, A.C.; Lavé, T.; Shah, D.K. Influence of Molecular Size on Tissue Distribution of Antibody Fragments. MAbs 2016, 8, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Penny, H.L.; Kroenke, M.A.; Bautista, B.; Hainline, K.; Chea, L.S.; Parnes, J.; Mytych, D.T. Immunogenicity Assessment of Bispecific Antibody-Based Immunotherapy in Oncology. J. Immunother. Cancer 2022, 10, e004225. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, M.; Ren, F.; Meng, X.; Yu, J. The Landscape of Bispecific T Cell Engager in Cancer Treatment. Biomark. Res. 2021, 9, 38. [Google Scholar] [CrossRef]
- Suryadevara, C.M.; Gedeon, P.C.; Sanchez-Perez, L.; Verla, T.; Alvarez-Breckenridge, C.; Choi, B.D.; Fecci, P.E.; Sampson, J.H. Are BiTEs the “Missing Link” in Cancer Therapy? Oncoimmunology 2015, 4, e1008339. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Gökbuget, N.; Zugmaier, G.; Klappers, P.; Stelljes, M.; Neumann, S.; Viardot, A.; Marks, R.; Diedrich, H.; Faul, C.; et al. Phase II Trial of the Anti-CD19 Bispecific T Cell-Engager Blinatumomab Shows Hematologic and Molecular Remissions in Patients with Relapsed or Refractory B-Precursor Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2014, 32, 4134–4140. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.; Hofmeister, R.; Kufer, P.; Schlereth, B.; Baeuerle, P.A. BiTEs: Bispecific Antibody Constructs with Unique Anti-Tumor Activity. Drug Discov. Today 2005, 10, 1237–1244. [Google Scholar] [CrossRef]
- Igawa, T.; Tsunoda, H.; Kikuchi, Y.; Yoshida, M.; Tanaka, M.; Koga, A.; Sekimori, Y.; Orita, T.; Aso, Y.; Hattori, K.; et al. VH/VL Interface Engineering to Promote Selective Expression and Inhibit Conformational Isomerization of Thrombopoietin Receptor Agonist Single-Chain Diabody. Protein Eng. Des. Sel. 2010, 23, 667–677. [Google Scholar] [CrossRef] [Green Version]
- Kwon, N.-Y.; Kim, Y.; Lee, J.-O. Structural Diversity and Flexibility of Diabodies. Methods 2019, 154, 136–142. [Google Scholar] [CrossRef]
- Wu, C. Diabodies: Molecular Engineering and Therapeutic Applications. Drug News Perspect 2009, 22, 453–458. [Google Scholar]
- Lameris, R.; de Bruin, R.C.G.; Schneiders, F.L.; van Bergen en Henegouwen, P.M.P.; Verheul, H.M.W.; de Gruijl, T.D.; van der Vliet, H.J. Bispecific Antibody Platforms for Cancer Immunotherapy. Crit. Rev. Oncol. Hematol. 2014, 92, 153–165. [Google Scholar] [CrossRef]
- Fucà, G.; Spagnoletti, A.; Ambrosini, M.; de Braud, F.; Di Nicola, M. Immune Cell Engagers in Solid Tumors: Promises and Challenges of the next Generation Immunotherapy. ESMO Open 2021, 6, 100046. [Google Scholar] [CrossRef]
- Nguyen, H.H.; Kim, T.; Song, S.Y.; Park, S.; Cho, H.H.; Jung, S.-H.; Ahn, J.-S.; Kim, H.-J.; Lee, J.-J.; Kim, H.-O.; et al. Naïve CD8(+) T Cell Derived Tumor-Specific Cytotoxic Effectors as a Potential Remedy for Overcoming TGF-β Immunosuppression in the Tumor Microenvironment. Sci. Rep. 2016, 6, 28208. [Google Scholar] [CrossRef]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-Cell Engagers for Cancer Immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Hamblett, K.; Barnscher, S.; Davies, R.; Hammond, P.; Hernandez, A.; Wickman, G.; Fung, V.; Ding, T.; Garnett, G.; Galey, A.; et al. Abstract P6-17-13: ZW49, a HER2 Targeted Biparatopic Antibody Drug Conjugate for the Treatment of HER2 Expressing Cancers. Cancer Res. 2019, 79, P6-17-13. [Google Scholar] [CrossRef]
- de Goeij, B.E.; Vink, T.; Napel, H.T.; Breij, E.C.; Satijn, D.; Wubbolts, R.; Miao, D.; Parren, P.W. Efficient Payload Delivery by a Bispecific Antibody-Drug Conjugate Targeting HER2 and CD63. Mol. Cancer Ther. 2016, 15, 2688–2697. [Google Scholar] [CrossRef] [Green Version]
- Rossi, E.A.; Rossi, D.L.; Stein, R.; Goldenberg, D.M.; Chang, C.-H. A Bispecific Antibody-IFNα2b Immunocytokine Targeting CD20 and HLA-DR Is Highly Toxic to Human Lymphoma and Multiple Myeloma Cells. Cancer Res. 2010, 70, 7600–7609. [Google Scholar] [CrossRef] [Green Version]
- Neijssen, J.; Cardoso, R.M.F.; Chevalier, K.M.; Wiegman, L.; Valerius, T.; Anderson, G.M.; Moores, S.L.; Schuurman, J.; Parren, P.W.H.I.; Strohl, W.R.; et al. Discovery of Amivantamab (JNJ-61186372), a Bispecific Antibody Targeting EGFR and MET. J. Biol. Chem. 2021, 296, 100641. [Google Scholar] [CrossRef]
- Yun, J.; Lee, S.-H.; Kim, S.-Y.; Jeong, S.-Y.; Kim, J.-H.; Pyo, K.-H.; Park, C.-W.; Heo, S.G.; Yun, M.R.; Lim, S.; et al. Antitumor Activity of Amivantamab (JNJ-61186372), an EGFR–MET Bispecific Antibody, in Diverse Models of EGFR Exon 20 Insertion–Driven NSCLC. Cancer Discov. 2020, 10, 1194–1209. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Lipfert, L.; Chevalier, K.; Bushey, B.S.; Henley, B.; Lenhart, R.; Sendecki, J.; Beqiri, M.; Millar, H.J.; Packman, K.; et al. Amivantamab (JNJ-61186372), an Fc Enhanced EGFR/CMet Bispecific Antibody, Induces Receptor Downmodulation and Antitumor Activity by Monocyte/Macrophage Trogocytosis. Mol. Cancer Ther. 2020, 19, 2044–2056. [Google Scholar] [CrossRef]
- Park, K.; Haura, E.B.; Leighl, N.B.; Mitchell, P.; Shu, C.A.; Girard, N.; Viteri, S.; Han, J.-Y.; Kim, S.-W.; Lee, C.K.; et al. Amivantamab in EGFR Exon 20 Insertion-Mutated Non-Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS Phase I Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 3391–3402. [Google Scholar] [CrossRef]
- Bauml, J.; Cho, B.C.; Park, K.; Lee, K.H.; Cho, E.K.; Kim, D.-W.; Kim, S.-W.; Haura, E.B.; Sabari, J.K.; Sanborn, R.E.; et al. Amivantamab in Combination with Lazertinib for the Treatment of Osimertinib-Relapsed, Chemotherapy-Naïve EGFR Mutant (EGFRm) Non-Small Cell Lung Cancer (NSCLC) and Potential Biomarkers for Response. J. Clin. Oncol. 2021, 39, 9006. [Google Scholar] [CrossRef]
- Wei, H.; Cai, H.; Jin, Y.; Wang, P.; Zhang, Q.; Lin, Y.; Wang, W.; Cheng, J.; Zeng, N.; Xu, T.; et al. Structural Basis of a Novel Heterodimeric Fc for Bispecific Antibody Production. Oncotarget 2017, 8, 51037–51049. [Google Scholar] [CrossRef] [Green Version]
- Ji, D.; Zhang, J.; Shen, W.; Du, Y.; Xu, J.; Yang, J.; Luo, X.; Kong, P.; Yang, F.; Hu, X.-C. Preliminary Safety, Efficacy and Pharmacokinetics (PK) Results of KN026, a HER2 Bispecific Antibody in Patients (Pts) with HER2-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2020, 38, 1041. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Wu, J.; Xu, N.; Ying, J.; Xiang, X.; Zhang, Y.; Wang, J.; Zhao, R.; Ye, F.; et al. The Preliminary Efficacy of KN026 (Anti-HER2 BsAb) in Advanced Gastric and Gastroesophageal Junction Cancer Patients with HER2 Expression. J. Clin. Oncol. 2021, 39, e16005. [Google Scholar] [CrossRef]
- Gong, J.; Dong, Z.; Liu, D.; Xu, J.; Yang, J.; Yang, Y.; Qi, Y.; Men, J.; Kong, P.; Xu, T.; et al. 339 Preliminary Safety, Tolerability and Efficacy Results of KN026 (a HER2-Targeted Bispecific Antibody) in Combination with KN046 (an Anti-PD-L1/CTLA-4 Bispecific Antibody) in Patients (Pts) with HER2 Aberrated Solid Tumors. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Weisser, N.E.; Wickman, G.; Abraham, L.; O’Toole, J.; Harbourne, B.; Guedia, J.; Cheng, C.W.; Chan, P.; Browman, D.; Gold, M.R.; et al. Abstract 1005: The Bispecific Antibody Zanidatamab’s (ZW25’s) Unique Mechanisms of Action and Durable Anti-Tumor Activity in HER2-Expressing Cancers. Cancer Res. 2021, 81, 1005. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Beeram, M.; Mayordomo, J.I.; Hanna, D.L.; Ajani, J.A.; Murphy, M.A.B.; Murthy, R.K.; Piha-Paul, S.A.; Bauer, T.M.; Bendell, J.C.; et al. Single Agent Activity of ZW25, a HER2-Targeted Bispecific Antibody, in Heavily Pretreated HER2-Expressing Cancers. J. Clin. Oncol. 2018, 36, 2500. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Hanna, D.L.; El-Khoueiry, A.B.; Kang, Y.-K.; Oh, D.-Y.; Chaves, J.M.; Rha, S.Y.; Hamilton, E.P.; Pant, S.; Javle, M.M.; et al. Zanidatamab (ZW25) in HER2-Positive Biliary Tract Cancers (BTCs): Results from a Phase I Study. J. Clin. Oncol. 2021, 39, 299. [Google Scholar] [CrossRef]
- Lee, K.W.; Im, Y.-H.; Lee, K.S.; Cho, J.Y.; Oh, D.-Y.; Chung, H.C.C.; Chao, Y.; Bai, L.-Y.; Yen, C.J.; Kim, I.-H.; et al. Zanidatamab, an Anti-HER2 Bispecific Antibody, plus Chemotherapy with/without Tislelizumab as First-Line Treatment for Patients with Advanced HER2-Positive Breast Cancer or Gastric/Gastroesophageal Junction Adenocarcinoma: A Phase 1B/2 Trial-in-Progress. J. Clin. Oncol. 2021, 39, TPS2656. [Google Scholar] [CrossRef]
- Geuijen, C.A.W.; De Nardis, C.; Maussang, D.; Rovers, E.; Gallenne, T.; Hendriks, L.J.A.; Visser, T.; Nijhuis, R.; Logtenberg, T.; de Kruif, J.; et al. Unbiased Combinatorial Screening Identifies a Bispecific IgG1 That Potently Inhibits HER3 Signaling via HER2-Guided Ligand Blockade. Cancer Cell 2018, 33, 922–936.e10. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Yonesaka, K.; Tanizaki, J.; Nonagase, Y.; Takegawa, N.; Haratani, K.; Kawakami, H.; Hayashi, H.; Takeda, M.; Tsurutani, J.; et al. Targeting of the HER2/HER3 Signaling Axis Overcomes Ligand-Mediated Resistance to Trastuzumab in HER2-Positive Breast Cancer. Cancer Med. 2019, 8, 1258–1268. [Google Scholar] [CrossRef] [Green Version]
- Alsina, M.; Varga, A.; Amatu, A.; Schellens, J.H.M.; Witteveen, P.O.; Boni, V.; Moreno, V.; Bol, K.; Lourbakos, A.; Ferrer, M.M.; et al. Phase I/II Study of Single Agent MCLA-128, a Full Length IgG1 Bispecific Antibody Targeting the HER3 Pathway: Overall Safety at the Recommended Phase II Dose (R2PD) and Preliminary Activity in HER2+ Metastatic Gastric/Gastroesophageal Junction Cancer (GC/GEJ). Ann. Oncol. 2018, 29, viii223–viii224. [Google Scholar] [CrossRef]
- Hamilton, E.P.; Petit, T.; Pistilli, B.; Goncalves, A.; Ferreira, A.A.; Dalenc, F.; Cardoso, F.; Mita, M.M.; Dezentjé, V.O.; Manso, L.; et al. Clinical Activity of MCLA-128 (Zenocutuzumab), Trastuzumab, and Vinorelbine in HER2 Amplified Metastatic Breast Cancer (MBC) Patients (Pts) Who Had Progressed on Anti-HER2 ADCs. J. Clin. Oncol. 2020, 38, 3093. [Google Scholar] [CrossRef]
- Pistilli, B.; Wildiers, H.; Hamilton, E.P.; Ferreira, A.A.; Dalenc, F.; Vidal, M.; Gavilá, J.; Goncalves, A.; Murias, C.; Mouret-Reynier, M.-A.; et al. Clinical Activity of MCLA-128 (Zenocutuzumab) in Combination with Endocrine Therapy (ET) in ER+/HER2-Low, Non-Amplified Metastatic Breast Cancer (MBC) Patients (Pts) with ET-Resistant Disease Who Had Progressed on a CDK4/6 Inhibitor (CDK4/6i). J. Clin. Oncol. 2020, 38, 1037. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerbel, R.S. Tumor Angiogenesis. N. Engl. J. Med. 2008, 358, 2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kienast, Y.; Klein, C.; Scheuer, W.; Raemsch, R.; Lorenzon, E.; Bernicke, D.; Herting, F.; Yu, S.; The, H.H.; Martarello, L.; et al. Ang-2-VEGF-A CrossMab, a Novel Bispecific Human IgG1 Antibody Blocking VEGF-A and Ang-2 Functions Simultaneously, Mediates Potent Antitumor, Antiangiogenic, and Antimetastatic Efficacy. Clin. Cancer Res. 2013, 19, 6730–6740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendell, J.C.; Sauri, T.; Cubillo, A.; López-López, C.; Garcia Alfonso, P.; Hussein, M.A.; Limon, M.L.; Cervantes, A.; Montagut, C.; Santos, C.; et al. Final Results of the McCAVE Trial: A Double-Blind, Randomized Phase 2 Study of Vanucizumab (VAN) plus FOLFOX vs. Bevacizumab (BEV) plus FOLFOX in Patients (Pts) with Previously Untreated Metastatic Colorectal Carcinoma (MCRC). J. Clin. Oncol. 2017, 35, 3539. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Sabbatino, F.; Liguori, L.; Pepe, S.; Ferrone, S. Immune Checkpoint Inhibitors for the Treatment of Melanoma. Expert Opin. Biol. Ther. 2022, 22, 563–576. [Google Scholar] [CrossRef]
- Sabbatino, F.; Marra, A.; Liguori, L.; Scognamiglio, G.; Fusciello, C.; Botti, G.; Ferrone, S.; Pepe, S. Resistance to Anti-PD-1-Based Immunotherapy in Basal Cell Carcinoma: A Case Report and Review of the Literature. J. Immunother. Cancer 2018, 6, 126. [Google Scholar] [CrossRef]
- Jiang, C.; Zhang, L.; Xu, X.; Qi, M.; Zhang, J.; He, S.; Tian, Q.; Song, S. Engineering a Smart Agent for Enhanced Immunotherapy Effect by Simultaneously Blocking PD-L1 and CTLA-4. Adv. Sci. 2021, 8, 2102500. [Google Scholar] [CrossRef]
- Zhao, H.; Ma, Y.; Zhang, Y.; Hong, S.; Yang, Y.; Fang, W.; Xu, J.; Van, H.; Kong, P.; Yang, F.; et al. The Preliminary Efficacy and Safety Data of KN046 in Patients Failed on Prior Immune Checkpoint Inhibitors Therapy. J. Clin. Oncol. 2020, 38, 3020. [Google Scholar] [CrossRef]
- Xu, B.; Li, Q.; Zhang, Q.; Zhang, Y.; Ouyang, Q.; Zhang, Y.; Liu, Q.; Sun, T.; Xu, J.; Yang, J.; et al. Abstract 1660: Preliminary Safety Tolerability & Efficacy Results of KN046 (an Anti-PD-L1/CTLA-4 Bispecific Antibody) in Combination with Nab-Paclitaxel in Patients with Metastatic Triple-Negative Breast Cancer (MTNBC). Cancer Res. 2021, 81, 1660. [Google Scholar] [CrossRef]
- Akesobio Australia Pty Ltd. A Phase 1A/1B Multicenter, Open-Label, Dose-Escalation, and Dose-Expansion Study to Evaluate the Safety, Pharmacokinetics, and Antitumor Activity of AK104 in Subjects with Advanced Solid Tumors; Clinicaltrials.gov: Bethesda, MD, USA, 2018.
- Ji, J.; Shen, L.; Li, Z.; Xu, N.; Liu, T.; Chen, Y.; Li, C.; Gao, X.; Ji, K.; Mao, C.; et al. AK104 (PD-1/CTLA-4 Bispecific) Combined with Chemotherapy as First-Line Therapy for Advanced Gastric (G) or Gastroesophageal Junction (GEJ) Cancer: Updated Results from a Phase Ib Study. J. Clin. Oncol. 2021, 39, 232. [Google Scholar] [CrossRef]
- Bai, L.; Sun, M.; Xu, A.; Bai, Y.; Wu, J.; Shao, G.; Song, L.; Jin, X.; Song, W.; Li, B.; et al. Phase 2 Study of AK104 (PD-1/CTLA-4 Bispecific Antibody) plus Lenvatinib as First-Line Treatment of Unresectable Hepatocellular Carcinoma. J. Clin. Oncol. 2021, 39, 4101. [Google Scholar] [CrossRef]
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab: Clinical Development and Future Directions. MAbs 2010, 2, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Heiss, M.M.; Murawa, P.; Koralewski, P.; Kutarska, E.; Kolesnik, O.O.; Ivanchenko, V.V.; Dudnichenko, A.S.; Aleknaviciene, B.; Razbadauskas, A.; Gore, M.; et al. The Trifunctional Antibody Catumaxomab for the Treatment of Malignant Ascites Due to Epithelial Cancer: Results of a Prospective Randomized Phase II/III Trial. Int. J. Cancer. J. Int. Du Cancer 2010, 127, 2209. [Google Scholar] [CrossRef] [Green Version]
- Knödler, M.; Körfer, J.; Kunzmann, V.; Trojan, J.; Daum, S.; Schenk, M.; Kullmann, F.; Schroll, S.; Behringer, D.; Stahl, M.; et al. Randomised Phase II Trial to Investigate Catumaxomab (Anti-EpCAM × Anti-CD3) for Treatment of Peritoneal Carcinomatosis in Patients with Gastric Cancer. Br. J. Cancer 2018, 119, 296–302. [Google Scholar] [CrossRef]
- EMA Removab. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/removab (accessed on 19 June 2022).
- Kiewe, P.; Thiel, E. Ertumaxomab: A Trifunctional Antibody for Breast Cancer Treatment. Expert Opin. Investig. Drugs 2008, 17, 1553–1558. [Google Scholar] [CrossRef]
- Haense, N.; Atmaca, A.; Pauligk, C.; Steinmetz, K.; Marmé, F.; Haag, G.M.; Rieger, M.; Ottmann, O.G.; Ruf, P.; Lindhofer, H.; et al. A Phase I Trial of the Trifunctional Anti Her2 × Anti CD3 Antibody Ertumaxomab in Patients with Advanced Solid Tumors. BMC Cancer 2016, 16, 420. [Google Scholar] [CrossRef] [Green Version]
- Lutterbuese, R.; Raum, T.; Kischel, R.; Lutterbuese, P.; Schlereth, B.; Schaller, E.; Mangold, S.; Rau, D.; Meier, P.; Kiener, P.A.; et al. Potent Control of Tumor Growth by CEA/CD3-Bispecific Single-Chain Antibody Constructs That Are Not Competitively Inhibited by Soluble CEA. J. Immunother. 2009, 32, 341–352. [Google Scholar] [CrossRef]
- Osada, T.; Hsu, D.; Hammond, S.; Hobeika, A.; Devi, G.; Clay, T.M.; Lyerly, H.K.; Morse, M.A. Metastatic Colorectal Cancer Cells from Patients Previously Treated with Chemotherapy Are Sensitive to T-Cell Killing Mediated by CEA/CD3-Bispecific T-Cell-Engaging BiTE Antibody. Br. J. Cancer 2010, 102, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Pishvaian, M.; Morse, M.A.; McDevitt, J.; Norton, J.D.; Ren, S.; Robbie, G.J.; Ryan, P.C.; Soukharev, S.; Bao, H.; Denlinger, C.S. Phase 1 Dose Escalation Study of MEDI-565, a Bispecific T-Cell Engager That Targets Human Carcinoembryonic Antigen, in Patients With Advanced Gastrointestinal Adenocarcinomas. Clin. Color. Cancer 2016, 15, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Moek, K.L.; Fiedler, W.M.; von Einem, J.C.; Verheul, H.M.; Seufferlein, T.; de Groot, D.J.; Heinemann, V.; Kebenko, M.; Oordt, C.W.M.d.H.v.; Ettrich, T.J.; et al. Phase I Study of AMG 211/MEDI-565 Administered as Continuous Intravenous Infusion (CIV) for Relapsed/Refractory Gastrointestinal (GI) Adenocarcinoma. Ann. Oncol. 2018, 29, viii139–viii140. [Google Scholar] [CrossRef]
- Bacac, M.; Klein, C.; Umana, P. CEA TCB: A Novel Head-to-Tail 2:1 T Cell Bispecific Antibody for Treatment of CEA-Positive Solid Tumors. Oncoimmunology 2016, 5, e1203498. [Google Scholar] [CrossRef] [Green Version]
- Bacac, M.; Fauti, T.; Sam, J.; Colombetti, S.; Weinzierl, T.; Ouaret, D.; Bodmer, W.; Lehmann, S.; Hofer, T.; Hosse, R.J.; et al. A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors. Clin. Cancer Res. 2016, 22, 3286–3297. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Melero, I.; Ros, W.; Argiles, G.; Marabelle, A.; Rodriguez-Ruiz, M.E.; Albanell, J.; Calvo, E.; Moreno, V.; Cleary, J.M.; et al. Phase Ia and Ib Studies of the Novel Carcinoembryonic Antigen (CEA) T-Cell Bispecific (CEA CD3 TCB) Antibody as a Single Agent and in Combination with Atezolizumab: Preliminary Efficacy and Safety in Patients with Metastatic Colorectal Cancer (MCRC). J. Clin. Oncol. 2017, 35, 3002. [Google Scholar] [CrossRef]
- Suurs, F.V.; Hooge, M.N.L.-D.; de Vries, E.G.E.; de Groot, D.J.A. A Review of Bispecific Antibodies and Antibody Constructs in Oncology and Clinical Challenges. Pharmacol. Ther. 2019, 201, 103–119. [Google Scholar] [CrossRef]
- Leconet, W.; Liu, H.; Guo, M.; Le Lamer-Déchamps, S.; Molinier, C.; Kim, S.; Vrlinic, T.; Oster, M.; Liu, F.; Navarro, V.; et al. Anti-PSMA/CD3 Bispecific Antibody Delivery and Antitumor Activity Using a Polymeric Depot Formulation. Mol. Cancer Ther. 2018, 17, 1927–1940. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.T.; Ravetch, J.V. Functional Diversification of IgGs through Fc Glycosylation. J. Clin. Investig. 2019, 129, 3492–3498. [Google Scholar] [CrossRef] [Green Version]
- Borlak, J.; Länger, F.; Spanel, R.; Schöndorfer, G.; Dittrich, C. Immune-Mediated Liver Injury of the Cancer Therapeutic Antibody Catumaxomab Targeting EpCAM, CD3 and Fcγ Receptors. Oncotarget 2016, 7, 28059–28074. [Google Scholar] [CrossRef]
- Wang, L.; Hoseini, S.S.; Xu, H.; Ponomarev, V.; Cheung, N.-K. Silencing Fc Domains in T Cell-Engaging Bispecific Antibodies Improves T-Cell Trafficking and Antitumor Potency. Cancer Immunol. Res. 2019, 7, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Blanco, B.; Compte, M.; Lykkemark, S.; Sanz, L.; Alvarez-Vallina, L. T Cell-Redirecting Strategies to “STAb” Tumors: Beyond CARs and Bispecific Antibodies. Trends Immunol. 2019, 40, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Edeline, J.; Houot, R.; Marabelle, A.; Alcantara, M. CAR-T Cells and BiTEs in Solid Tumors: Challenges and Perspectives. J. Hematol. Oncol. 2021, 14, 65. [Google Scholar] [CrossRef] [PubMed]
- Oates, J.; Hassan, N.J.; Jakobsen, B.K. ImmTACs for Targeted Cancer Therapy: Why, What, How, and Which. Mol. Immunol. 2015, 67, 67–74. [Google Scholar] [CrossRef]
- Oates, J.; Jakobsen, B.K. ImmTACs: Novel Bi-Specific Agents for Targeted Cancer Therapy. Oncoimmunology 2013, 2, e22891. [Google Scholar] [CrossRef] [Green Version]
- Liddy, N.; Bossi, G.; Adams, K.J.; Lissina, A.; Mahon, T.M.; Hassan, N.J.; Gavarret, J.; Bianchi, F.C.; Pumphrey, N.J.; Ladell, K.; et al. Monoclonal TCR-Redirected Tumor Cell Killing. Nat. Med. 2012, 18, 980–987. [Google Scholar] [CrossRef]
- Dahan, R.; Reiter, Y. T-Cell-Receptor-like Antibodies—Generation, Function and Applications. Expert Rev. Mol. Med. 2012, 14, e6. [Google Scholar] [CrossRef]
- Middelburg, J.; Kemper, K.; Engelberts, P.; Labrijn, A.F.; Schuurman, J.; van Hall, T. Overcoming Challenges for CD3-Bispecific Antibody Therapy in Solid Tumors. Cancers 2021, 13, 287. [Google Scholar] [CrossRef]
- Ahmed, M.; Cheng, M.; Cheung, I.Y.; Cheung, N.-K.V. Human Derived Dimerization Tag Enhances Tumor Killing Potency of a T-Cell Engaging Bispecific Antibody. Oncoimmunology 2015, 4, e989776. [Google Scholar] [CrossRef] [Green Version]
- Slaga, D.; Ellerman, D.; Lombana, T.N.; Vij, R.; Li, J.; Hristopoulos, M.; Clark, R.; Johnston, J.; Shelton, A.; Mai, E.; et al. Avidity-Based Binding to HER2 Results in Selective Killing of HER2-Overexpressing Cells by Anti-HER2/CD3. Sci. Transl. Med. 2018, 10, eaat5775. [Google Scholar] [CrossRef] [Green Version]
- Asano, R.; Ikoma, K.; Sone, Y.; Kawaguchi, H.; Taki, S.; Hayashi, H.; Nakanishi, T.; Umetsu, M.; Katayose, Y.; Unno, M.; et al. Highly Enhanced Cytotoxicity of a Dimeric Bispecific Diabody, the HEx3 Tetrabody*. J. Biol. Chem. 2010, 285, 20844–20849. [Google Scholar] [CrossRef] [Green Version]
- Voynov, V.; Adam, P.J.; Nixon, A.E.; Scheer, J.M. Discovery Strategies to Maximize the Clinical Potential of T-Cell Engaging Antibodies for the Treatment of Solid Tumors. Antibodies 2020, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Dees, S.; Grewal, I.S. Overcoming the Challenges Associated with CD3+ T-Cell Redirection in Cancer. Br. J. Cancer 2021, 124, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Boustany, L.M.; Porte, S.L.L.; Wong, L.; White, C.W.; Diep, L.; Huang, Y.; Liu, S.; Richardson, J.H.; Kavanaugh, W.M.; Irving, B.A. EGFR-CD3 Bispecific ProbodyTM Therapeutic Induces Tumor Regressions and Increases Maximum Tolerated Dose > 60 Fold in Preclinical Studies. Mol. Cancer Ther. 2018, 17 (Suppl. S1), A164. [Google Scholar] [CrossRef]
- Guo, Z.S.; Lotze, M.T.; Zhu, Z.; Storkus, W.J.; Song, X.-T. Bi- and Tri-Specific T Cell Engager-Armed Oncolytic Viruses: Next-Generation Cancer Immunotherapy. Biomedicines 2020, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic Viruses as Engineering Platforms for Combination Immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef]
- Yu, F.; Wang, X.; Guo, Z.S.; Bartlett, D.L.; Gottschalk, S.M.; Song, X.-T. T-Cell Engager-Armed Oncolytic Vaccinia Virus Significantly Enhances Antitumor Therapy. Mol. Ther. 2014, 22, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Blanco, B.; Ramírez-Fernández, Á.; Alvarez-Vallina, L. Engineering Immune Cells for in Vivo Secretion of Tumor-Specific T Cell-Redirecting Bispecific Antibodies. Front. Immunol. 2020, 11, 1792. [Google Scholar] [CrossRef]
- Iwahori, K.; Kakarla, S.; Velasquez, M.P.; Yu, F.; Yi, Z.; Gerken, C.; Song, X.-T.; Gottschalk, S. Engager T Cells: A New Class of Antigen-Specific T Cells That Redirect Bystander T Cells. Mol. Ther. 2015, 23, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.M.; Pyo, K.-H.; Soo, R.A.; Cho, B.C. The Promise of Bispecific Antibodies: Clinical Applications and Challenges. Cancer Treat. Rev. 2021, 99, 102240. [Google Scholar] [CrossRef]
- Kamakura, D.; Asano, R.; Yasunaga, M. T Cell Bispecific Antibodies: An Antibody-Based Delivery System for Inducing Antitumor Immunity. Pharmaceuticals 2021, 14, 1172. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Groeneveldt, C.; Kinderman, P.; van den Wollenberg, D.J.M.; van den Oever, R.L.; Middelburg, J.; Mustafa, D.A.M.; Hoeben, R.C.; van der Burg, S.H.; van Hall, T.; van Montfoort, N. Preconditioning of the Tumor Microenvironment with Oncolytic Reovirus Converts CD3-Bispecific Antibody Treatment into Effective Immunotherapy. J. Immunother. Cancer 2020, 8, e001191. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Huang, M.; Lum, L.G. Bispecific Antibody Based Therapeutics: Strengths and Challenges. Blood Rev. 2018, 32, 339–347. [Google Scholar] [CrossRef]
- Walker, C.; Mojares, E.; Del Río Hernández, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [Green Version]
- Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Cell Surface Glycoprotein of Reactive Stromal Fibroblasts as a Potential Antibody Target in Human Epithelial Cancers. Proc. Natl. Acad. Sci. USA 1990, 87, 7235–7239. [Google Scholar] [CrossRef] [Green Version]
- de Sostoa, J.; Fajardo, C.A.; Moreno, R.; Ramos, M.D.; Farrera-Sal, M.; Alemany, R. Targeting the Tumor Stroma with an Oncolytic Adenovirus Secreting a Fibroblast Activation Protein-Targeted Bispecific T-Cell Engager. J. Immunother. Cancer 2019, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Hong, B.; Song, X.-T. A T-Cell Engager-Armed Oncolytic Vaccinia Virus to Target the Tumor Stroma. Cancer Transl. Med. 2017, 3, 122. [Google Scholar]
- Chiu, D.; Tavaré, R.; Haber, L.; Aina, O.H.; Vazzana, K.; Ram, P.; Danton, M.; Finney, J.; Jalal, S.; Krueger, P.; et al. A PSMA-Targeting CD3 Bispecific Antibody Induces Antitumor Responses That Are Enhanced by 4-1BB Costimulation. Cancer Immunol. Res. 2020, 8, 596–608. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-H.; Wang, Y.; Li, R.; Rossi, D.L.; Liu, D.; Rossi, E.A.; Cardillo, T.M.; Goldenberg, D.M. Combination Therapy with Bispecific Antibodies and PD-1 Blockade Enhances the Antitumor Potency of T Cells. Cancer Res. 2017, 77, 5384–5394. [Google Scholar] [CrossRef] [Green Version]
- Sam, J.; Colombetti, S.; Fauti, T.; Roller, A.; Biehl, M.; Fahrni, L.; Nicolini, V.; Perro, M.; Nayak, T.; Bommer, E.; et al. Combination of T-Cell Bispecific Antibodies With PD-L1 Checkpoint Inhibition Elicits Superior Anti-Tumor Activity. Front. Oncol. 2020, 10, 575737. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Sano, Y.; Komatsu, S.-I.; Kamata-Sakurai, M.; Kaneko, A.; Kinoshita, Y.; Shiraiwa, H.; Azuma, Y.; Tsunenari, T.; Kayukawa, Y.; et al. An Anti-Glypican 3/CD3 Bispecific T Cell-Redirecting Antibody for Treatment of Solid Tumors. Sci. Transl. Med. 2017, 9, eaal4291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hettich, M.; Lahoti, J.; Prasad, S.; Niedermann, G. Checkpoint Antibodies but Not T Cell-Recruiting Diabodies Effectively Synergize with TIL-Inducing γ-Irradiation. Cancer Res. 2016, 76, 4673–4683. [Google Scholar] [CrossRef]
- Junttila, T.T.; Li, J.; Johnston, J.; Hristopoulos, M.; Clark, R.; Ellerman, D.; Wang, B.-E.; Li, Y.; Mathieu, M.; Li, G.; et al. Antitumor Efficacy of a Bispecific Antibody That Targets HER2 and Activates T Cells. Cancer Res. 2014, 74, 5561–5571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osada, T.; Patel, S.P.; Hammond, S.A.; Osada, K.; Morse, M.A.; Lyerly, H.K. CEA/CD3-Bispecific T Cell-Engaging (BiTE) Antibody-Mediated T Lymphocyte Cytotoxicity Maximized by Inhibition of Both PD1 and PD-L1. Cancer Immunol. Immunother. 2015, 64, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Albaitero, A.; Xu, H.; Guo, H.; Wang, L.; Wu, Z.; Tran, H.; Chandarlapaty, S.; Scaltriti, M.; Janjigian, Y.; de Stanchina, E.; et al. Overcoming Resistance to HER2-Targeted Therapy with a Novel HER2/CD3 Bispecific Antibody. Oncoimmunology 2017, 6, e1267891. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Yuan, Q.; Yuan, X.; Wang, Y.; Mo, W.; Wang, H.; Yu, M. A Novel Tetravalent Bispecific Antibody Targeting Programmed Death 1 and Tyrosine-Protein Kinase Met for Treatment of Gastric Cancer. Investig. New Drugs 2019, 37, 876–889. [Google Scholar] [CrossRef]
- Elgundi, Z.; Reslan, M.; Cruz, E.; Sifniotis, V.; Kayser, V. The State-of-Play and Future of Antibody Therapeutics. Adv. Drug Deliv. Rev. 2017, 122, 2–19. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agents | Format | Targets | Cancer | Trial Phase | NCT Number | Status | Company |
|---|---|---|---|---|---|---|---|
| 1. Tumor Associated Antigen (TAA)-Based bsAbs | |||||||
| Amivantamab | Quadroma | c-Met/EGFR | NSCL | 1 | NCT02609776 | R | Janssen Research & Development, LLC (Raritan, NJ, USA) |
| Solid tumors | 1 | NCT04606381 | R | ||||
| NSCL | 1 | NCT04077463 | R | ||||
| UGI | 2 | NCT04945733 | R | ||||
| NSCL | 3 | NCT04538664 | R | ||||
| NSCL | 3 | NCT04487080 | ANR | ||||
| KN026 | CrossMab | HER2 | Breast/UGI | 1 | NCT03619681 | ANR | Jiangsu Alphamab Biopharmaceuticals Co., Ltd. (Suzhou, China) |
| Breast/UGI | 1 | NCT03847168 | ANR | ||||
| Breast | 2 | NCT04881929 | R | ||||
| GI | 2 | NCT03925974 | U | ||||
| Solid tumors | 2 | NCT04521179 | ANR | ||||
| Breast | 2 | NCT04778982 | R | ||||
| Breast | 2 | NCT04165993 | ANR | ||||
| Vanucizumab | CrossMab | Ang-2/VEGF-A | Solid tumors | 1 | NCT02665416 | C | Hoffmann-La Roche (Basel, Switzerland) |
| Solid tumors | 1 | NCT01688206 | C | ||||
| Solid tumors | 1 | NCT02715531 | C | ||||
| CR | 2 | NCT02141295 | T | ||||
| Zanidatamab | CrossMab | HER2 | Solid tumors | 1 | NCT02892123 | ANR | Zymeworks Inc./BeiGene, Ltd. (Vancouver, BC, Canada) |
| Breast/UGI | 1/2 | NCT04276493 | ANR | ||||
| Biliary | 2 | NCT04466891 | ANR | ||||
| Breast | 2 | NCT04224272 | R | ||||
| GI | 2 | NCT03929666 | R | ||||
| Zenocutuzumab | CrossMab | HER2/HER3 | Solid tumors | 1/2 | NCT02912949 | R | Merus N.V. (Utrecht, The Netherlands) |
| Breast | 2 | NCT03321981 | ANR | ||||
| 2. Antibody Drug Conjugates (ADC)-Based bsAbs | |||||||
| ZW49 | CrossMab | HER2 | Solid tumors | 1 | NCT03821233 | R | Zymeworks Inc. (Vancouver, BC, Canada) |
| 3. Immune Checkpoint-Based bsAbs | |||||||
| Cadonilimab | IgG-scFv | CTLA-4/PD-1 | Solid tumors | 1 | NCT03261011 | U | Akeso|Akeso Pharmaceutical, Inc. (Hong Kong, China) |
| Solid tumors | 1 | NCT04572152 | R | ||||
| NSCL | 1/2 | NCT04647344 | NYR | ||||
| Solid tumors | 1/2 | NCT03852251 | U | ||||
| Solid tumors | 1/2 | NCT04172454 | U | ||||
| HC | 1/2 | NCT04444167 | U | ||||
| NSCL | 1/2 | NCT04646330 | ANR | ||||
| NSCL | 2 | NCT04544644 | NYR | ||||
| HC | 2 | NCT04728321 | R | ||||
| GI | 2 | NCT04556253 | NYR | ||||
| Cervical | 2 | NCT04380805 | ANR | ||||
| Solid tumors | 2 | NCT04547101 | R | ||||
| NF | 2 | NCT04220307 | U | ||||
| Cervical | 2 | NCT04868708 | ANR | ||||
| KN046 | Quadroma | CTLA-4/PD-L1 | Solid tumors | 1 | NCT03529526 | U | Jiangsu Alphamab Biopharmaceuticals Co., Ltd. (Suzhou, China) |
| Solid tumors | 1 | NCT03733951 | R | ||||
| Solid tumors | 1 | NCT04040699 | R | ||||
| Breast | 1/2 | NCT03872791 | ANR | ||||
| GI | 1/2 | NCT04612712 | R | ||||
| HC | 1/2 | NCT04601610 | ANR | ||||
| Thymic | 2 | NCT04925947 | R | ||||
| Thymic | 2 | NCT04469725 | R | ||||
| UGI | 2 | NCT03925870 | U | ||||
| UGI | 2 | NCT03927495 | U | ||||
| NSCL | 2 | NCT03838848 | U | ||||
| NSCL | 2 | NCT04054531 | U | ||||
| Breast | 2 | NCT04165993 | ANR | ||||
| HC | 2 | NCT04542837 | R | ||||
| Solid tumors | 2 | NCT04521179 | ANR | ||||
| NSCL | 3 | NCT04474119 | ANR | ||||
| 4. Immune Cell Engagement by bsAbs | |||||||
| Catumaxomab | Qaudroma | CD3/EpCAM | Solid tumors | 1 | NCT01320020 | T | Neovii Biotech/ Fresenius Biotech NorthAmerica (Boston, MA, USA) |
| Bladder | 1 | NCT04819399 | R | ||||
| Bladder | 1/2 | NCT04799847 | R | ||||
| Ovarian | 2 | NCT00189345 | C | ||||
| Ovarian | 2 | NCT00377429 | C | ||||
| Ovarian | 2 | NCT01815528 | C | ||||
| Ovarian | 2 | NCT01246440 | C | ||||
| Ovarian | 2 | NCT00563836 | C | ||||
| Gastric | 2 | NCT00464893 | C | ||||
| Gastric | 2 | NCT00352833 | C | ||||
| Gastric | 2 | NCT01784900 | T | ||||
| GI | 2 | NCT01504256 | C | ||||
| MA | 2 | NCT01065246 | C | ||||
| MA | 2 | NCT00326885 | C | ||||
| MA | 2/3 | NCT00836654 | C | ||||
| MA | 3 | NCT00822809 | C | ||||
| Gastric | 3 | NCT04222114 | R | ||||
| Ertumaxomab | Quadroma | CD3/HER2 | Solid tumor | 1/2 | NCT01569412 | T | Neovii Biotech/ Fresenius Biotech North America (Boston, MA, USA) |
| Breast | 2 | NCT00452140 | T | ||||
| Breast | 2 | NCT00351858 | T | ||||
| Breast | 2 | NCT00522457 | T | ||||
| MEDI-565 | BITE | CD3/CEA | GI | 1 | NCT01284231 | C | MedImmune LLC (Gaithersburg, MD, USA) |
| GI | 1 | NCT02291614 | T | ||||
| RO6958688 | CrossMab | CD3/CEA | Solid tumors | 1 | NCT02324257 | C | Hoffmann-La Roche (Basel, Switzerland) |
| Solid tumors | 1 | NCT02650713 | C | ||||
| CR | 1 | NCT03866239 | ANR | ||||
| CR | 1/2 | NCT04826003 | R | ||||
| NSCL | 1/2 | NCT03337698 | R | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liguori, L.; Polcaro, G.; Nigro, A.; Conti, V.; Sellitto, C.; Perri, F.; Ottaiano, A.; Cascella, M.; Zeppa, P.; Caputo, A.; et al. Bispecific Antibodies: A Novel Approach for the Treatment of Solid Tumors. Pharmaceutics 2022, 14, 2442. https://doi.org/10.3390/pharmaceutics14112442
Liguori L, Polcaro G, Nigro A, Conti V, Sellitto C, Perri F, Ottaiano A, Cascella M, Zeppa P, Caputo A, et al. Bispecific Antibodies: A Novel Approach for the Treatment of Solid Tumors. Pharmaceutics. 2022; 14(11):2442. https://doi.org/10.3390/pharmaceutics14112442
Chicago/Turabian StyleLiguori, Luigi, Giovanna Polcaro, Annunziata Nigro, Valeria Conti, Carmine Sellitto, Francesco Perri, Alessandro Ottaiano, Marco Cascella, Pio Zeppa, Alessandro Caputo, and et al. 2022. "Bispecific Antibodies: A Novel Approach for the Treatment of Solid Tumors" Pharmaceutics 14, no. 11: 2442. https://doi.org/10.3390/pharmaceutics14112442
APA StyleLiguori, L., Polcaro, G., Nigro, A., Conti, V., Sellitto, C., Perri, F., Ottaiano, A., Cascella, M., Zeppa, P., Caputo, A., Pepe, S., & Sabbatino, F. (2022). Bispecific Antibodies: A Novel Approach for the Treatment of Solid Tumors. Pharmaceutics, 14(11), 2442. https://doi.org/10.3390/pharmaceutics14112442