

The Diphtheria Toxin Translocation Domain Impairs Receptor Selectivity in Cancer Cell-Targeted Protein Nanoparticles

 , ,
, ,  and
and 
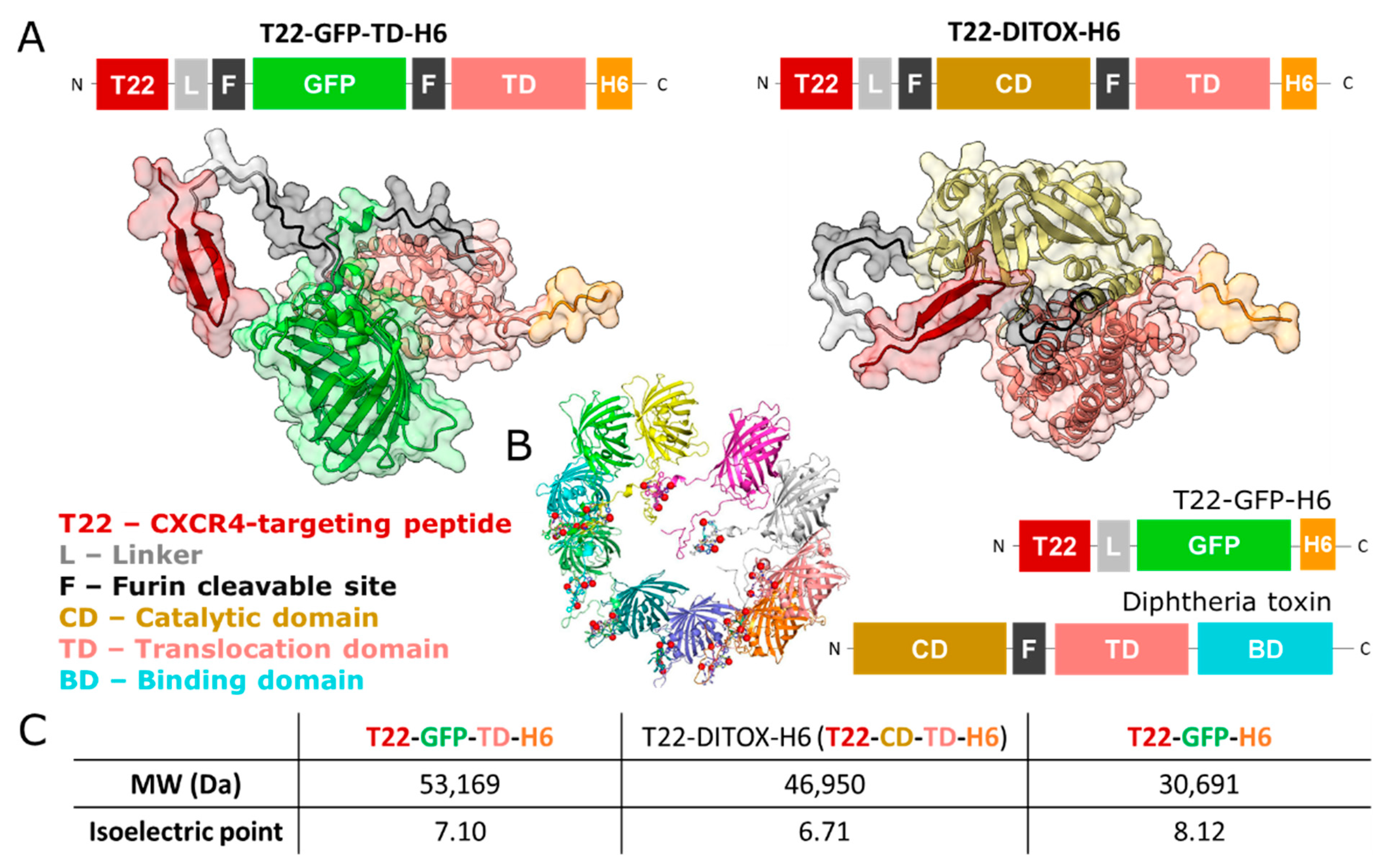{kind=link}
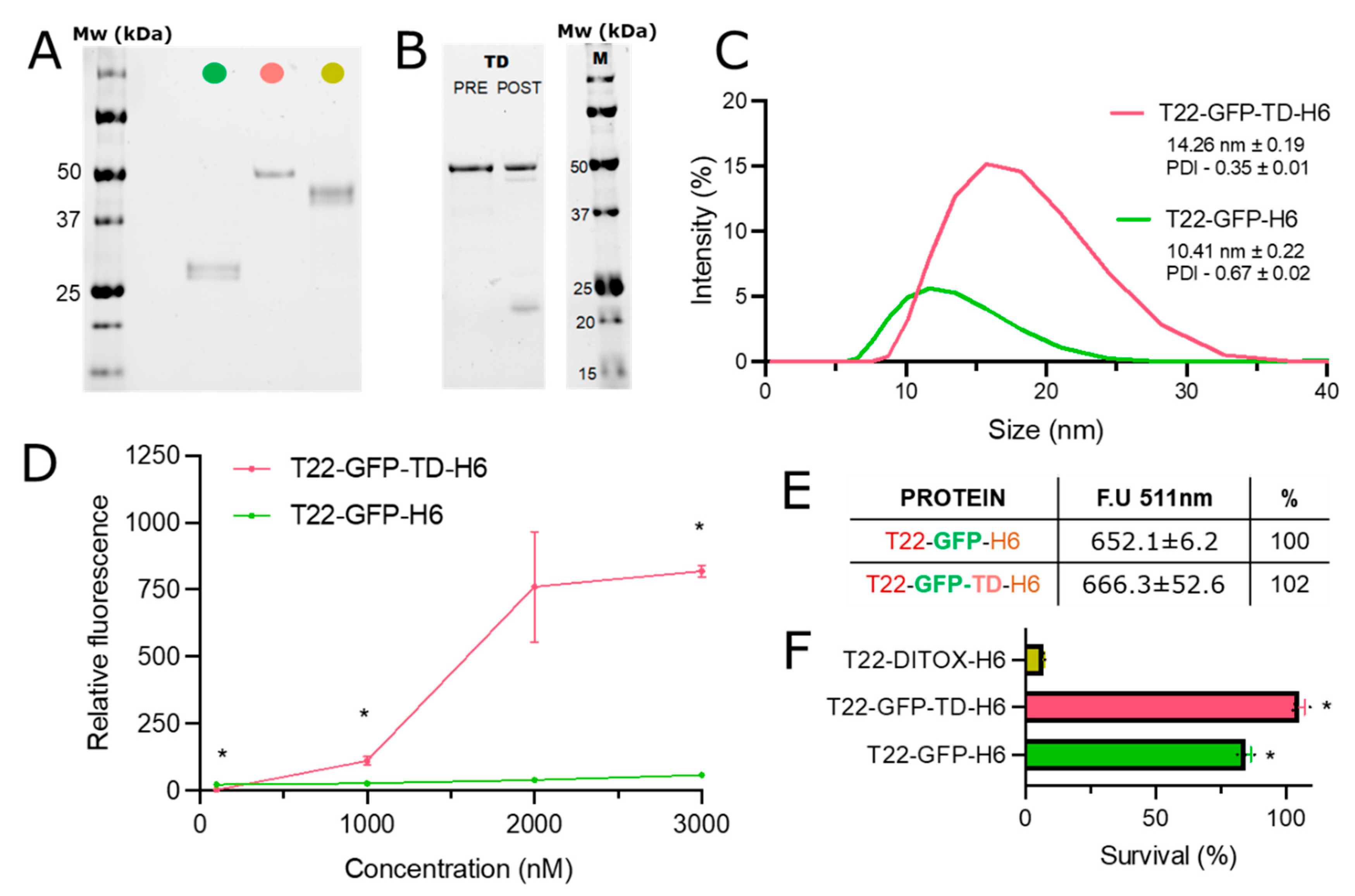{kind=link}
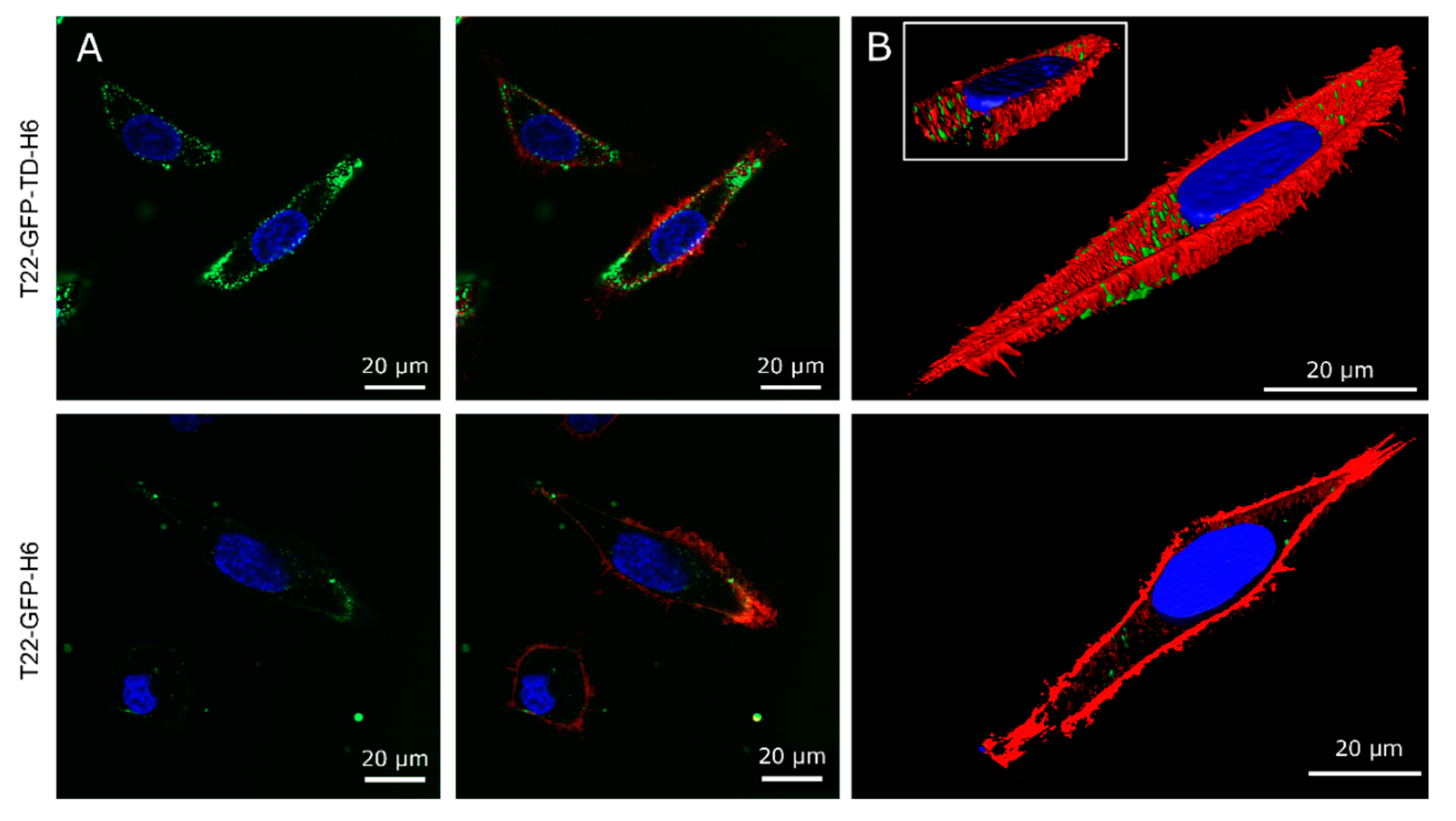{kind=link}
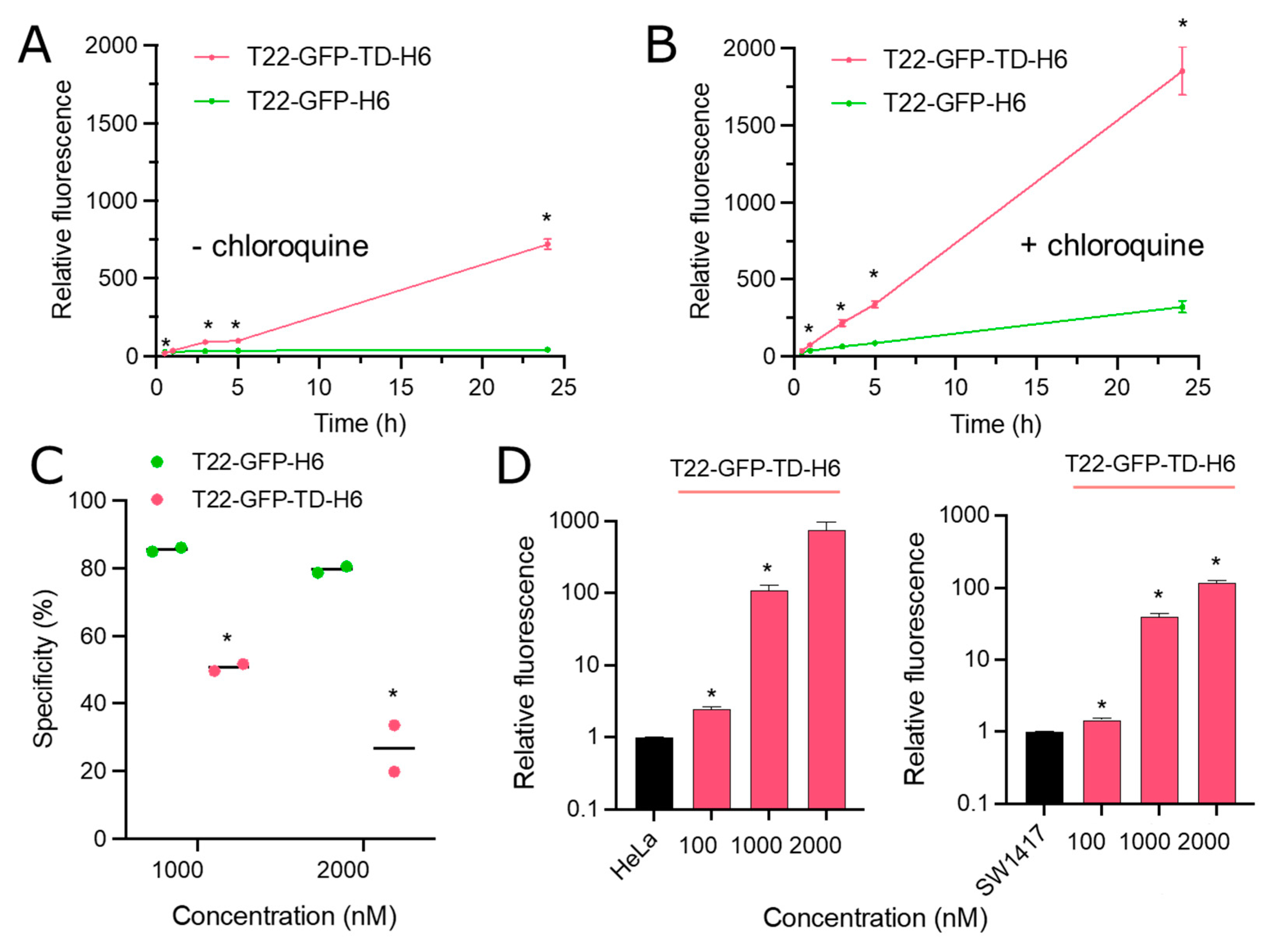{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genetic Design and Protein Production and Purification
2.2. Three-Dimensional Protein Modelling and Visualization
2.3. Protein Purity, Integrity and Concentration
2.4. Size Distribution and Fluorescence Emission
2.5. Cell Culture and Confocal Laser Scanning Microscopy
2.6. Cell Cytometry
2.7. Cell Viability
2.8. Proteolytic Analysis
2.9. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Z.; Zhi, K.; Ding, Z.; Sun, Y.; Li, S.; Li, M.; Pu, K.; Zou, J. Emergence in protein derived nanomedicine as anticancer therapeutics: More than a tour de force. Semin. Cancer Biol. 2021, 69, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M.; Paquette, C. Therapeutic recombinant proteins: Trends in US approvals 1982 to 2002. Curr. Opin. Mol. Ther. 2003, 5, 139–147. [Google Scholar]
- Lagasse, H.A.; Alexaki, A.; Simhadri, V.L.; Katagiri, N.H.; Jankowski, W.; Sauna, Z.E.; Kimchi-Sarfaty, C. Recent advances in (therapeutic protein) drug development. F1000Research 2017, 6, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, E.; Mangues, R.; Villaverde, A. Functional recruitment for drug delivery through protein-based nanotechnologies. Nanomedicine 2016, 11, 1333–1336. [Google Scholar] [CrossRef] [Green Version]
- Serna, N.; Sanchez-Garcia, L.; Unzueta, U.; Diaz, R.; Vazquez, E.; Mangues, R.; Villaverde, A. Protein-Based Therapeutic Killing for Cancer Therapies. Trends Biotechnol. 2018, 36, 318–335. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, E.; Ferrer-Miralles, N.; Mangues, R.; Corchero, J.L.; Schwartz, S., Jr.; Villaverde, A. Modular protein engineering in emerging cancer therapies. Curr. Pharm. Des. 2009, 15, 893–916. [Google Scholar] [CrossRef]
- Sanchez-Garcia, L.; Martin, L.; Mangues, R.; Ferrer-Miralles, N.; Vazquez, E.; Villaverde, A. Recombinant pharmaceuticals from microbial cells: A 2015 update. Microb. Cell Factories 2016, 15, 33. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, F.R. Recombinant expression systems in the pharmaceutical industry. Appl. Microbiol. Biotechnol. 2004, 65, 363–372. [Google Scholar] [CrossRef]
- Akbari, B.; Farajnia, S.; Ahdi Khosroshahi, S.; Safari, F.; Yousefi, M.; Dariushnejad, H.; Rahbarnia, L. Immunotoxins in cancer therapy: Review and update. Int. Rev. Immunol. 2017, 36, 207–219. [Google Scholar] [CrossRef]
- Tourdot, S.; Hickling, T.P. Nonclinical immunogenicity risk assessment of therapeutic proteins. Bioanalysis 2019, 11, 1631–1643. [Google Scholar] [CrossRef]
- Mazor, R.; Pastan, I. Immunogenicity of Immunotoxins Containing Pseudomonas Exotoxin A: Causes, Consequences, and Mitigation. Front. Immunol. 2020, 11, 1261. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Chen, X.; Vicini, P.; Rup, B.; Hickling, T.P. Therapeutic outcomes, assessments, risk factors and mitigation efforts of immunogenicity of therapeutic protein products. Cell. Immunol. 2015, 295, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; Onda, M.; Pastan, I. Immunogenicity of therapeutic recombinant immunotoxins. Immunol. Rev. 2016, 270, 152–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii-Watabe, A.; Shibata, H.; Nishimura, K.; Hosogi, J.; Aoyama, M.; Nishimiya, K.; Saito, Y. Immunogenicity of therapeutic protein products: Current considerations for anti-drug antibody assay in Japan. Bioanalysis 2018, 10, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release Off. J. Control. Release Soc. 2011, 151, 220–228. [Google Scholar] [CrossRef]
- Shete, H.K.; Prabhu, R.H.; Patravale, V.B. Endosomal escape: A bottleneck in intracellular delivery. J. Nanosci. Nanotechnol. 2014, 14, 460–474. [Google Scholar] [CrossRef]
- Munsell, E.V.; Ross, N.L.; Sullivan, M.O. Journey to the Center of the Cell: Current Nanocarrier Design Strategies Targeting Biopharmaceuticals to the Cytoplasm and Nucleus. Curr. Pharm. Des. 2016, 22, 1227–1244. [Google Scholar] [CrossRef]
- Ahmad, A.; Khan, J.M.; Haque, S. Strategies in the design of endosomolytic agents for facilitating endosomal escape in nanoparticles. Biochimie 2019, 160, 61–75. [Google Scholar] [CrossRef]
- Tran, S.; DeGiovanni, P.J.; Piel, B.; Rai, P. Cancer nanomedicine: A review of recent success in drug delivery. Clin. Transl. Med. 2017, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Casanova, I.; Unzueta, U.; Arroyo-Solera, I.; Cespedes, M.V.; Villaverde, A.; Mangues, R.; Vazquez, E. Protein-driven nanomedicines in oncotherapy. Curr. Opin. Pharmacol. 2019, 47, 1–7. [Google Scholar] [CrossRef]
- Perez-Herrero, E.; Fernandez-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Selby, L.I.; Johnston, A.P.R.; Such, G.K. The Endosomal Escape of Nanoparticles: Toward More Efficient Cellular Delivery. Bioconjugate Chem. 2019, 30, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Pei, D.; Buyanova, M. Overcoming Endosomal Entrapment in Drug Delivery. Bioconjugate Chem. 2019, 30, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Lonn, P.; Kacsinta, A.D.; Cui, X.S.; Hamil, A.S.; Kaulich, M.; Gogoi, K.; Dowdy, S.F. Enhancing Endosomal Escape for Intracellular Delivery of Macromolecular Biologic Therapeutics. Sci. Rep. 2016, 6, 32301. [Google Scholar] [CrossRef]
- López-Laguna, H.; Cubarsi, R.; Unzueta, U.; Mangues, R.; Vázquez, E.; Villaverde, A. Endosomal escape of protein nanoparticles engineered through humanized histidine-rich peptides. Sci. China Mater. 2020, 63, 644–653. [Google Scholar] [CrossRef] [Green Version]
- Uchida, T. CHAPTER 1—Diphtheria toxin: Biological activity. In Molecular Aspects of Cellular Regulation; Cohen, P., Van Heyningen, S., Eds.; Elsevier: Amsterdam, The Netherlands, 1982; Volume 2, pp. 1–31. [Google Scholar]
- Simon, N.C.; Aktories, K.; Barbieri, J.T. Novel bacterial ADP-ribosylating toxins: Structure and function. Nat. Rev. Microbiol. 2014, 12, 599–611. [Google Scholar] [CrossRef] [Green Version]
- Collier, R.J. Understanding the mode of action of diphtheria toxin: A perspective on progress during the 20th century. Toxicon Off. J. Int. Soc. Toxinol. 2001, 39, 1793–1803. [Google Scholar] [CrossRef]
- Gilyazova, D.G.; Rosenkranz, A.A.; Gulak, P.V.; Lunin, V.G.; Sergienko, O.V.; Khramtsov, Y.V.; Timofeyev, K.N.; Grin, M.A.; Mironov, A.F.; Rubin, A.B.; et al. Targeting cancer cells by novel engineered modular transporters. Cancer Res. 2006, 66, 10534–10540. [Google Scholar] [CrossRef] [Green Version]
- Auger, A.; Park, M.; Nitschke, F.; Minassian, L.M.; Beilhartz, G.L.; Minassian, B.A.; Melnyk, R.A. Efficient Delivery of Structurally Diverse Protein Cargo into Mammalian Cells by a Bacterial Toxin. Mol. Pharm. 2015, 12, 2962–2971. [Google Scholar] [CrossRef]
- Ladokhin, A.S.; Vargas-Uribe, M.; Rodnin, M.V.; Ghatak, C.; Sharma, O. Cellular Entry of the Diphtheria Toxin Does Not Require the Formation of the Open-Channel State by Its Translocation Domain. Toxins 2017, 9, 299. [Google Scholar] [CrossRef] [Green Version]
- Ratts, R.; Murphy, J.R. Diphtheria toxin, diphtheria-related fusion protein toxins, and the molecular mechanism of their action against eukaryotic cells. In Microbial Protein Toxins; Schmitt, M.J., Schaffrath, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 1–20. [Google Scholar]
- Park, S.G.; Choi, B.; Bae, Y.; Lee, Y.G.; Park, S.A.; Chae, Y.C.; Kang, S. Selective and Effective Cancer Treatments using Target-Switchable Intracellular Bacterial Toxin Delivery Systems. Adv. Ther. 2020, 3, 2000043. [Google Scholar] [CrossRef]
- Zhao, S.; Karp, J.M.; Joshi, N. Toxin-Mediated siRNA Delivery. Trends Pharmacol. Sci. 2020, 41, 511–513. [Google Scholar] [CrossRef] [PubMed]
- Serna, N.; Pallarès, V.; Unzueta, U.; Garcia-Leon, A.; Voltà-Durán, E.; Sánchez-Chardi, A.; Parladé, E.; Rueda, A.; Casanova, I.; Falgàs, A.; et al. Engineering non-antibody human proteins as efficient scaffolds for selective, receptor-targeted drug delivery. J. Control. Release 2022, 343, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Cano-Garrido, O.; Alamo, P.; Sanchez-Garcia, L.; Falgas, A.; Sanchez-Chardi, A.; Serna, N.; Parlade, E.; Unzueta, U.; Roldan, M.; Volta-Duran, E.; et al. Biparatopic Protein Nanoparticles for the Precision Therapy of CXCR4(+) Cancers. Cancers 2021, 13, 2929. [Google Scholar] [CrossRef] [PubMed]
- Salan, A.I.; Marasescu, P.C.; Camen, A.; Ciuca, E.M.; Matei, M.; Florescu, A.M.; Padureanu, V.; Margaritescu, C. The prognostic value of CXCR4, alpha-SMA and WASL in upper lip basal cell carcinomas. Rom. J. Morphol. Embryol. Rev. Roum. Morphol. Embryol. 2018, 59, 839–849. [Google Scholar]
- Cabrero-de Las Heras, S.; Martinez-Balibrea, E. CXC family of chemokines as prognostic or predictive biomarkers and possible drug targets in colorectal cancer. World J. Gastroenterol. 2018, 24, 4738–4749. [Google Scholar] [CrossRef] [PubMed]
- Tamamura, H.; Arakaki, R.; Funakoshi, H.; Imai, M.; Otaka, A.; Ibuka, T.; Nakashima, H.; Murakami, T.; Waki, M.; Matsumoto, A.; et al. Effective lowly cytotoxic analogs of an HIV-cell fusion inhibitor, T22 ([Tyr5,12, Lys7]-polyphemusin II). Bioorganic Med. Chem. 1998, 6, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Unzueta, U.; Cespedes, M.V.; Ferrer-Miralles, N.; Casanova, I.; Cedano, J.; Corchero, J.L.; Domingo-Espin, J.; Villaverde, A.; Mangues, R.; Vazquez, E. Intracellular CXCR4(+) cell targeting with T22-empowered protein-only nanoparticles. Int. J. Nanomed. 2012, 7, 4533–4544. [Google Scholar] [CrossRef] [Green Version]
- Tamamura, H.; Imai, M.; Ishihara, T.; Masuda, M.; Funakoshi, H.; Oyake, H.; Murakami, T.; Arakaki, R.; Nakashima, H.; Otaka, A.; et al. Pharmacophore identification of a chemokine receptor (CXCR4) antagonist, T22 ([Tyr(5,12),Lys7]-polyphemusin II), which specifically blocks T cell-line-tropic HIV-1 infection. Bioorganic Med. Chem. 1998, 6, 1033–1041. [Google Scholar] [CrossRef]
- Sala, R.; Rioja-Blanco, E.; Serna, N.; Sánchez-García, L.; Álamo, P.; Alba-Castellón, L.; Casanova, I.; López-Pousa, A.; Unzueta, U.; Céspedes, M.V.; et al. GSDMD-dependent pyroptotic induction by a multivalent CXCR4-targeted nanotoxin blocks colorectal cancer metastases. Drug Delivery 2022, 29, 1384–1397. [Google Scholar] [CrossRef]
- Pallares, V.; Unzueta, U.; Falgas, A.; Sanchez-Garcia, L.; Serna, N.; Gallardo, A.; Morris, G.A.; Alba-Castellon, L.; Alamo, P.; Sierra, J.; et al. An Auristatin nanoconjugate targeting CXCR4+ leukemic cells blocks acute myeloid leukemia dissemination. J. Hematol. Oncol. 2020, 13, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serna, N.; Falgas, A.; Garcia-Leon, A.; Unzueta, U.; Nunez, Y.; Sanchez-Chardi, A.; Martinez-Torro, C.; Mangues, R.; Vazquez, E.; Casanova, I.; et al. Time-Prolonged Release of Tumor-Targeted Protein-MMAE Nanoconjugates from Implantable Hybrid Materials. Pharmaceutics 2022, 14, 192. [Google Scholar] [CrossRef] [PubMed]
- Pallares, V.; Unzueta, U.; Falgas, A.; Avino, A.; Nunez, Y.; Garcia-Leon, A.; Sanchez-Garcia, L.; Serna, N.; Gallardo, A.; Alba-Castellon, L.; et al. A multivalent Ara-C-prodrug nanoconjugate achieves selective ablation of leukemic cells in an acute myeloid leukemia mouse model. Biomaterials 2022, 280, 121258. [Google Scholar] [CrossRef] [PubMed]
- Cespedes, M.V.; Unzueta, U.; Avino, A.; Gallardo, A.; Alamo, P.; Sala, R.; Sanchez-Chardi, A.; Casanova, I.; Mangues, M.A.; Lopez-Pousa, A.; et al. Selective depletion of metastatic stem cells as therapy for human colorectal cancer. EMBO Mol. Med. 2018, 10, e8772. [Google Scholar] [CrossRef]
- Lopez-Laguna, H.; Volta-Duran, E.; Parlade, E.; Villaverde, A.; Vazquez, E.; Unzueta, U. Insights on the emerging biotechnology of histidine-rich peptides. Biotechnol. Adv. 2022, 54, 107817. [Google Scholar] [CrossRef]
- Sanchez-Garcia, L.; Serna, N.; Alamo, P.; Sala, R.; Cespedes, M.V.; Roldan, M.; Sanchez-Chardi, A.; Unzueta, U.; Casanova, I.; Mangues, R.; et al. Self-assembling toxin-based nanoparticles as self-delivered antitumoral drugs. J. Control. Release Off. J. Control. Release Soc. 2018, 274, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Laguna, H.; Unzueta, U.; Conchillo-Sole, O.; Sanchez-Chardi, A.; Pesarrodona, M.; Cano-Garrido, O.; Volta, E.; Sanchez-Garcia, L.; Serna, N.; Saccardo, P.; et al. Assembly of histidine-rich protein materials controlled through divalent cations. Acta Biomater. 2019, 83, 257–264. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Volta-Duran, E.; Serna, N.; Sanchez-Garcia, L.; Avino, A.; Sanchez, J.M.; Lopez-Laguna, H.; Cano-Garrido, O.; Casanova, I.; Mangues, R.; Eritja, R.; et al. Design and engineering of tumor-targeted, dual-acting cytotoxic nanoparticles. Acta Biomater. 2021, 119, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.H.; Lee, D.Y.; Cha, W.; Kim, B.H.; Sung, M.W.; Kim, K.H.; Ahn, S.H. Antitumor effect of CXCR4 antagonist AMD3100 on the tumorigenic cell line of BHP10-3 papillary thyroid cancer cells. Head Neck 2016, 38, 1479–1486. [Google Scholar] [CrossRef]
- Kim, H.Y.; Hwang, J.Y.; Kim, S.W.; Lee, H.J.; Yun, H.J.; Kim, S.; Jo, D.Y. The CXCR4 Antagonist AMD3100 Has Dual Effects on Survival and Proliferation of Myeloma Cells In Vitro. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2010, 42, 225–234. [Google Scholar] [CrossRef]
- Donzella, G.A.; Schols, D.; Lin, S.W.; Este, J.A.; Nagashima, K.A.; Maddon, P.J.; Allaway, G.P.; Sakmar, T.P.; Henson, G.; De Clercq, E.; et al. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat. Med. 1998, 4, 72–77. [Google Scholar] [CrossRef]
- Al-Bari, M.A.A. Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases. Pharmacol. Res. Perspect. 2017, 5, e00293. [Google Scholar] [CrossRef]
- Fredericksen, B.L.; Wei, B.L.; Yao, J.; Luo, T.; Garcia, J.V. Inhibition of endosomal/lysosomal degradation increases the infectivity of human immunodeficiency virus. J. Virol. 2002, 76, 11440–11446. [Google Scholar] [CrossRef] [Green Version]
- Rosenkranz, A.A.; Vaidyanathan, G.; Pozzi, O.R.; Lunin, V.G.; Zalutsky, M.R.; Sobolev, A.S. Engineered modular recombinant transporters: Application of new platform for targeted radiotherapeutic agents to alpha-particle emitting 211 At. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Falgàs, A.; Pallarès, V.; Unzueta, U.; Núñez, Y.; Sierra, J.; Gallardo, A.; Alba-Castellón, L.; Mangues, M.A.; Álamo, P.; Villaverde, A.; et al. Specific Cytotoxic Effect of an Auristatin Nanoconjugate Towards CXCR4(+) Diffuse Large B-Cell Lymphoma Cells. Int. J. Nanomed. 2021, 16, 1869–1888. [Google Scholar] [CrossRef]
- Sánchez-García, L.; Sala, R.; Serna, N.; Álamo, P.; Parladé, E.; Alba-Castellón, L.; Voltà-Durán, E.; Sánchez-Chardi, A.; Unzueta, U.; Vázquez, E.; et al. A refined cocktailing of pro-apoptotic nanoparticles boosts anti-tumor activity. Acta Biomater. 2020, 113, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Serna, N.; Céspedes, M.V.; Sánchez-García, L.; Unzueta, U.; Sala, R.; Sánchez-Chardi, A.; Cortés, F.; Ferrer-Miralles, N.; Mangues, R.; Vázquez, E.; et al. Peptide-Based Nanostructured Materials with Intrinsic Proapoptotic Activities in CXCR4+ Solid Tumors. Adv. Funct. Mater. 2017, 27, 1700919. [Google Scholar] [CrossRef]
- Yu, M.; Wu, J.; Shi, J.; Farokhzad, O.C. Nanotechnology for protein delivery: Overview and perspectives. J. Control. Release Off. J. Control. Release Soc. 2016, 240, 24–37. [Google Scholar] [CrossRef]
- Manzari, M.T.; Shamay, Y.; Kiguchi, H.; Rosen, N.; Scaltriti, M.; Heller, D.A. Targeted drug delivery strategies for precision medicines. Nat. Rev. Mater. 2021, 6, 351–370. [Google Scholar] [CrossRef]
- Cespedes, M.V.; Unzueta, U.; Alamo, P.; Gallardo, A.; Sala, R.; Casanova, I.; Pavon, M.A.; Mangues, M.A.; Trias, M.; Lopez-Pousa, A.; et al. Cancer-specific uptake of a liganded protein nanocarrier targeting aggressive CXCR4+ colorectal cancer models. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Khramtsov, Y.V.; Rokitskaya, T.I.; Rosenkranz, A.A.; Trusov, G.A.; Gnuchev, N.V.; Antonenko, Y.N.; Sobolev, A.S. Modular drug transporters with diphtheria toxin translocation domain form edged holes in lipid membranes. J. Control. Release 2008, 128, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, M.; Hällbrink, M.; Prochiantz, A.; Langel, Ü. Cell-penetrating peptides. Trends Pharmacol. Sci. 2000, 21, 99–103. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voltà-Durán, E.; Sánchez, J.M.; Parladé, E.; Serna, N.; Vazquez, E.; Unzueta, U.; Villaverde, A. The Diphtheria Toxin Translocation Domain Impairs Receptor Selectivity in Cancer Cell-Targeted Protein Nanoparticles. Pharmaceutics 2022, 14, 2644. https://doi.org/10.3390/pharmaceutics14122644
Voltà-Durán E, Sánchez JM, Parladé E, Serna N, Vazquez E, Unzueta U, Villaverde A. The Diphtheria Toxin Translocation Domain Impairs Receptor Selectivity in Cancer Cell-Targeted Protein Nanoparticles. Pharmaceutics. 2022; 14(12):2644. https://doi.org/10.3390/pharmaceutics14122644
Chicago/Turabian StyleVoltà-Durán, Eric, Julieta M. Sánchez, Eloi Parladé, Naroa Serna, Esther Vazquez, Ugutz Unzueta, and Antonio Villaverde. 2022. "The Diphtheria Toxin Translocation Domain Impairs Receptor Selectivity in Cancer Cell-Targeted Protein Nanoparticles" Pharmaceutics 14, no. 12: 2644. https://doi.org/10.3390/pharmaceutics14122644
APA StyleVoltà-Durán, E., Sánchez, J. M., Parladé, E., Serna, N., Vazquez, E., Unzueta, U., & Villaverde, A. (2022). The Diphtheria Toxin Translocation Domain Impairs Receptor Selectivity in Cancer Cell-Targeted Protein Nanoparticles. Pharmaceutics, 14(12), 2644. https://doi.org/10.3390/pharmaceutics14122644