1. Introduction
Pulmonary arterial hypertension (PAH) is a progressive cardiopulmonary disease that interferes with the ability of patients to exercise or perform routine duties [
1]. According to the 6th World Symposium on Pulmonary Hypertension in France in 2018, PAH occurs when the mean pulmonary artery pressure (mPAP) exceeds 20 mmHg at rest [
2]. Pathogenesis of PAH includes vascular remodeling giving rise to narrow pulmonary arteries, increasing pulmonary vascular resistance (PVR), leading to right ventricular failure and death [
2]. PAH is a multi-factorial disease due to its complex etiology [
1]. Symptoms of the disease include nonproductive cough, dyspnea, syncope, fatigue, and peripheral edema [
1]. Pediatric PAH is increasingly recognized to have different etiologies, clinical presentations, and outcomes than adult PAH [
3]. In 2010–2013, the incidence rate of pediatric PAH in the United States was estimated to be 4.8 to 8.1 per million children a year, and the annual prevalence ranged from 25.7 to 32.6 per million children [
3]. The management of Pediatric PAH is still challenging despite the recent advances since the pediatric population possesses variable growth, development, and metabolism characteristics, which complicates the extrapolation of conclusions from the adult data to pediatrics [
2].
A number of physiological pathways are involved in the pathogenesis of PAH, including the endothelin, nitric oxide (NO), and prostacyclin pathways [
3]. Accordingly, various drug classes based on targeted pulmonary vasodilator therapy demonstrated beneficial therapeutic efficacy via hemodynamic and functional improvement, including (i) endothelin receptor antagonists, (ii) phosphodiesterase 5 inhibitors (PDE5-Is), and (iii) and prostacyclin analogs [
2]. PDE5-Is exert their vasodilating effect in the pulmonary vasculature through inhibition of phosphodiesterase 5 (PDE5), the enzyme that breaks down the secondary messenger cyclic guanosine monophosphate (cGMP), which regulates smooth muscle contraction, resulting eventually in vasodilatation [
3]. Among the available PDE5-Is, both sildenafil and tadalafil (TD) were approved by the Food and Drug Administration (FDA) for the treatment of PAH in adults, as these medications were shown to enhance the exercise capacity and delay the clinical worsening of the case [
2].
On the other hand, neither of the two drugs was approved by the FDA for treating PAH in children [
2]. Despite the extensive use of sildenafil as an off-label cure for pediatric PAH since 2005 [
4], the FDA released a strong warning in August 2012 against its chronic use for children aged 1–17 years [
2]. This warning was announced after a long-term clinical study of oral sildenafil (STARTS-2), which proved that children administered a high dose of sildenafil are at higher risk of death, while those receiving low doses do not demonstrate obvious improvement in the exercise ability [
2]. Therefore, much attention was directed to TD for treating PAH in children. TD offers many advantages for treating PAH in children, where it is a long-acting PDE5-I administered once daily [
2]. Furthermore, reported data showed that it could be well tolerated by children, as it offers a favorable adverse effect profile and provides better clinical improvement than sildenafil [
5].
Recent reports demonstrated that many therapies developed for treating PAH were delivered through the pulmonary route [
6]. Concerning the delivery of PDE5-Is, the oral route is associated with a number of adverse effects, such as headache, flushing, rare systemic hypotension, erection, and transient color vision disturbance [
2]. In the current study, the pulmonary route has been proposed for delivery of TD via nebulization for treatment of pediatric PAH, as direct delivery of medications to the lungs via inhalation is believed to offer many advantages, such as the local delivery provides direct access to the airways; thus high drug concentrations can be attained in the target organ, ensuring high efficacy [
6]. In addition, rapid drug absorption is secured via this route of administration due to the high vascularization, huge absorptive lung surface area, and excellent permeability, resulting in a fast onset of action [
6]. The targeted drug delivery reduces the systemic side effects and offers the potential to reduce both the medication dose and the frequency of administration, thus improving patient compliance [
6]. Moreover, pulmonary drug delivery avoids first-pass metabolism due to the relatively low enzymatic activity of the lungs [
6].
Many efforts have been made to overcome the limitations accompanying the conventional treatment using anti-PAH drugs and improve their efficacy through employing novel drug delivery systems, including liposomes, polymeric nanoparticles, solid lipid nanoparticles (SLNs), nanostructured lipid carriers (NLCs), micelles and nano-erythrosomes [
6,
7]. However, nanoemulsions (NEs) were chosen in the current study as they proved efficacy in the pulmonary delivery of poorly soluble drugs [
8,
9,
10,
11,
12,
13,
14,
15,
16]. Since TD is a poorly soluble class II drug [
17], whose bioavailability is greatly affected by its rate and extent of dissolution, NEs were proposed as the delivery system of choice due to their high drug solubilizing capacity [
10] and different forms of emulsions manifested superior solubilization of TD [
18,
19,
20,
21,
22]. NEs are heterogeneous colloidal systems of oil and aqueous medium stabilized by surfactant (SAA) molecules [
23]. These systems are kinetically stable during long-term storage without apparent flocculation or coalescence owing to their nanometer-sized droplets [
23]. The reduced droplet size (<100 nm) is also advantageous for improving the drug bioavailability via facilitating mucosal drug penetration [
10]. In addition, concerning pulmonary drug delivery, it was reported that the inhalation and aerosolization performances of NEs are superior to those of commercial suspensions and other related formulations, such as nanoparticles, liposomes, and micelles [
10].
Despite the aforementioned merits of NEs, many systems are destroyed upon dilution with the aqueous phase, followed by drug precipitation and uncontrolled absorption [
24]. Aqueous dilution with saline is required before nebulization to adjust the tonicity of the formulations and prevent aerosol-induced cough [
25]. Our research group successfully developed intranasal TD-NEs for the treatment of erectile dysfunction based on the following ingredients (System A): Capmul-MCM-EP, Labrasol, Transcutol HP, and water [
17], but unfortunately, these NEs turn turbid upon aqueous dilution, followed by precipitation of the dissolved drug.
On this basis, the current work endeavors for the first time—to the best of our knowledge—to develop TD-NEs that can resist aqueous dilution to be used for the treatment of pediatric PAH via nebulization using a jet nebulizer. Nanoparticles, microspheres, and nanocomposites of TD have been previously developed as inhalable dry powders for treating PAH [
26,
27,
28]. However, it has been reported that dry powder formulations encounter problems during powder processing and re-dispersion (adhesion and strong agglomeration), which makes them unsuitable for efficient inhalation [
10]. On the other hand, the administration of liquid formulations via nebulization is much more convenient, where the generated aerosol enables deep deposition in the lower respiratory tract [
29].
In the context of the study objectives, several tests were carried out to modulate system A into a more stable system via screening various excipients. Since high excipient concentration in the formulation can induce dose-related irritancy or toxicity problems during pulmonary administration [
11], the screening method aimed to select the NE excipients at their minimal optimal concentration. Then, different formulations of variable composition were fabricated and optimized via assessment of their physicochemical properties in drug solubilization, pH, viscosity, globule size, zeta potential, morphology, and stability. In addition, the suitability of the engineered formulations for pulmonary application was evaluated by studying nebulization, sterilization, and in-vitro and in-vivo toxicity studies.
2. Materials and Methods
2.1. Materials
Tadalafil (TD) was purchased from Radiant Pharma (Mumbai, India). Labrasol® (Caprylocaproyl macrogol-8 glycerides EP, hydrophilic–lipophilic balance (HLB) = 14), Transcutol® HP (Highly purified Diethylene glycol monoethyl ether EP/NF) and LabrafacTM-lipophile WL1349 (Medium chain fatty acid triglyceride JPE) were kind gifts from Gattefossé (Lyon, France). Capmul® MCM EP (Glycerol monocaprylocaprate, a mixture of medium chain length monoglyceride (60%) and diglyceride (35%), consisting of 83% w/w caprylic acid (C8) and 17% w/w capric acid (C10)) was donated as a gift from Abitec Corporation (Janesville, WI, USA). Cremophor® EL (Polyoxyl 35 castor oil USP, HLB = 12–14), Cremophor® RH40 (Polyoxyl 40 hydrogenated castor oil USP/NF, HLB = 14–16), and Poloxamer-407 (Lutrol® F 127, HLB = 18–23) was obtained from BASF (Ludwigshafen, Germany). Tween-80 (HLB = 15) and propylene glycol were provided by El-Nasr Pharmaceutical Chemicals (Cairo, Egypt). Polyethylene glycol 400 (PEG 400) was purchased from Sisco Research Laboratories Pvt. Ltd. (Mumbai, India). Human lung adenocarcinoma epithelial cell line (A549) was obtained from the American Type Culture Collection (ATCC) (Saint Cloud, MN, USA) through the Tissue Culture Unit, Egyptian Organization for Biological Products and Vaccines, Vacsera (Cairo, Egypt). Dulbecco’s Modified Eagle’s Medium (DMEM) was purchased from Lonza (Biowhittaker, Belgium) supplemented with 10% fetal bovine serum (FBS) (Lonza, Biowhittaker, Belgium), 1% (v/v) L-glutamine (Lonza, Biowhittaker, Belgium) and 1 g/L glucose (Lonza, Biowhittaker, Belgium). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) was purchased from Serva (Heidelberg, Germany). Ketamine® (Ketamine hydrochloride 50 mg/mL) was purchased from Sigmatec Pharmaceutical Industries (Giza, Egypt). Xyla-ject® (Xylazine hydrochloride 20 mg/mL) was purchased from Adwia Pharmaceuticals (Cairo, Egypt). Lactate dehydrogenase (LDH) and protein cytotoxicity detection kits were obtained from Roche Diagnostics (Indianapolis, IN, USA). All other chemicals and reagents were of analytical grade.
2.2. Screening of Surfactants (SAAs) and Co-Surfactants (Co-SAAs): Evaluation of Dispersion Properties
In order to produce a stable NE which can resist dilution with water, the nanoemulsifying properties of different SAAs and Co-SAAs were evaluated by visual assessment. The oily phase was mixed with the SAAs or SAA mixtures (Smixs) in a 1:3 (w/w) ratio, heated in a water bath at 40–50 °C and mixed with the aid of a vortex mixer (VM-300, Gemmy Industrial Corp., Taipei, Taiwan) to form a homogenous mixture. The oil-SAA mixture, 250 mg, was dispersed into 5 mL of distilled water with gentle mixing. Nanoemulsification was evaluated by visual observation of the final mixture (clear/turbid) after being left overnight at room temperature (RT) for equilibrium. Co-SAAs were screened by mixing the selected Smix with the Co-SAA in a 2:1 (w/w) ratio, followed by the addition of the oily phase to this mixture in 1:3 (w/w) ratio, heating, and vortex to obtain a homogenous mixture. They were assessed in the same manner as explained before.
2.3. Construction of Pseudo-Ternary Phase Diagrams
Pseudo-ternary phase diagrams of two systems (B and C) were developed to identify the NE existence region via employing the modified titration method. The following combination of ingredients was used in the preparation of these systems:
System B: Capmul-MCM-EP:Labrafac-lipophile WL 1349 (1:1) (w/w) as oily phase, Labrasol:Cremophor EL (1:1) (w/w) as Smix and water as the aqueous phase.
System C: Capmul-MCM-EP:Labrafac-lipophile WL 1349 (1:1) (w/w) as oily phase, Labrasol:Poloxamer-407 (2:1) (w/w) as Smix and water as the aqueous phase. The (2:1) (w/w) Smix of Labrasol with Poloxamer-407 was prepared by heating the two ingredients until a homogenous mixture was obtained. A Smix of equal weights of Labrasol and Poloxamer-407 was not employed as a Smix due to the high viscosity of the mixture and the difficulty in withdrawing an accurate volume.
In transparent glass vials, nine dissimilar combinations of oily phase and water (1:9, 2:8, 3:7, 4:6, 5:5, 6:4, 7:3, 8:2, 9:1) were prepared on a weight basis, and they were slowly titrated with the selected Smix under continuous stirring at RT till reaching clarity of the system. Afterward, the systems were allowed to equilibrate overnight at RT and were checked visually for clarity. In addition, their percentage transmittance (%T) was measured at 630 nm using ELx800TM Absorbance Microplate Reader (BioTek Instruments Inc., Winooski, VT, USA) against distilled water as a blank. The amount of Smix added till an opaque/transparent transition occurred was used to determine the phase domains. Phase diagrams were mapped using Sigma Plot® software (version 13, Systat Software Inc., San Jose, CA, USA).
2.4. Preparation of TD-Loaded NE Formulations
2.4.1. Preparation of Placebo NEs
NE formulations of various compositions were selected from the phase diagrams for further studies. Placebo NEs were formed spontaneously upon mixing the calculated weights of the oily phase, aqueous phase, and SAAs. Formulations of system B were formed at RT, whereas those of system C were prepared by dissolving the calculated weight of Poloxamer-407 in the calculated weight of water in an ice bath, followed by the addition of Labrasol under continuous stirring, then the oil mixture was added at the end. The stability of NEs towards aqueous dilution was evaluated by visual inspection of the clarity of the formulations following 50-fold dilution with distilled water after 2-h storage at RT.
2.4.2. Rationale for Dose Calculation
The dose of TD to be administered via nebulization for the treatment of pediatric PAH was calculated following an example reported by Sawatdee et al. in which the blood concentration was multiplied by the extravascular lung fluid volume [
30]. The pharmacokinetics of TD following a single oral dose of 40 mg in healthy adult male Japanese and Caucasian volunteers displayed a C
max of 446 ng/mL and 562 ng/mL, respectively [
31]. The mean value of C
max for both races was 504 ng/mL. The reported lung fluid volume was 3–10 mL [
30]. By considering both values, the TD dose varied between 1.5 µg and 5.04 µg. The percentage of nebulized dose delivered to the lung was reported to be around 10%, up to 30% in some reports [
32]. In this estimation, the percentage of nebulized doses reaching the lungs was 10%. So, the adult dose of TD required for treating PAH through nebulization ranged between 15 µg and 50.4 µg. Half the adult dose was considered in the current investigation for the treatment of pediatric PAH. Therefore, the calculated dose of TD for the treatment of pediatric PAH via nebulization ranged between 7.5 µg to 25.2 µg.
2.4.3. Determination of Drug-Loading Capacity of NEs
An excess amount of TD was dissolved in the oil/SAA phase of NEs by sonication (Elmasonic S 40 (H), Elma-Hans Schmidbauer GmbH & Co. KG, Singen, Germany) at ambient temperature (30 ± 2 °C) until no more drug dissolves, then the calculated weight of water was added to each mixture. Afterward, the prepared NEs were left for 24 h for equilibrium at ambient temperature, filtered through 0.45 µm cellulose acetate syringe filter (Minisart®, Sartorius, Goettingen, Germany), and the filtrates were properly diluted with methanol. The amount of drug dissolved was analyzed spectrophotometrically (UV-160A, Shimadzu, Kyoto, Japan) at λmax 285 nm against a blank. TD-loaded formulations were prepared by dissolving TD in pre-formed placebo NEs to produce an NE of a final drug concentration of 2 mg/mL.
2.4.4. Dispersion Stability of TD-Loaded NEs
The equilibrium solubility of TD was determined after 50-fold dilution of the drug-loaded NEs with distilled water at RT. An aliquot was taken immediately after dilution, followed by compensation with an equal volume, then another sample was withdrawn after 2 h to test for drug precipitation. TD concentration was measured spectrophotometrically after filtration and dilution with methanol. The amount of drug remaining after 2h dilution was calculated as a percentage relative to the original concentration determined after immediate dilution.
2.5. Physicochemical Characterization of Selected NE Formulations
The isotropic nature of NEs was confirmed using cross-polarized light microscopy (Olympus CX31PF, Olympus Corporation, Tokyo, Japan). Transparency of NEs was verified by measuring the refractive index using Abbe refractometer (Abbe 5 refractometer, Bellingham + Stanley Ltd., Kent, UK) and %T at 630 nm using ELx800TM Absorbance Microplate Reader (BioTek Instruments Inc., Winooski, VT, USA). The pH of the formulations was measured at 25 ± 2 °C using a calibrated pH meter (Mettler-Toledo S220 SevenCompact™ pH/Ion meters, Mettler Toledo Co, Greifensee, Switzerland) after 50-fold dilution with saline. The viscosity of 50-fold saline-diluted NEs was measured at 25 ± 2 °C using a Brookfield viscometer (Brookfield Engineering Laboratories Inc., Stoughton, MA, USA) equipped with spindle 2 at 100 rpm. Globule size and polydispersity index (PdI) of the formulations were determined using Malvern zetasizer Nano ZS (Malvern Instruments, Malvern, UK) after 50-fold dilution with either water or saline. Zeta potential was measured using Malvern zetasizer Nano ZS (Malvern Instruments, Malvern, UK) after 50-fold dilution with saline. The morphology of TD-loaded NEs was studied using transmission electron microscopy (TEM) (model JEM-100CX, JEOL, Tokyo, Japan) after 50-fold dilution with saline. The thermodynamic stability of TD-loaded NEs was evaluated via accelerated stability studies in terms of physical stability (phase separation, creaming, cracking, and drug precipitation) and globule size after subjecting the NEs to centrifugation (4000 rpm for 15 min) and three freeze-thaw cycles (alternating cycles of storage at −4 °C and +25 °C for 24 h at each temperature).
2.6. Nebulization Performance of TD-Loaded NEs
An aliquot (4 mL) of 50-fold saline-diluted TD-loaded NEs was placed in the nebulizer cup of a Microlux
® jet nebulizer (AS109N, Medel, Parma, Italy) and nebulized for 20 min. The stability of NEs towards nebulization was assessed in terms of globule size and morphology using TEM. In addition, the aerosol output and aerosol output rate were determined on a gravimetric basis using the following equations [
10]:
2.7. Sterilization of NE Formulations
TD-loaded NEs were sterilized using two techniques: autoclave (steam sterilization) using a portable Gallenkamp autoclave at 121 °C for 15 min and filtration through 0.22 µm cellulose acetate syringe filter (Minisart®, Sartorius, Goettingen, Germany). The stability of NEs upon sterilization by each technique was evaluated in terms of the percentage of drug remaining and globule size after 50-fold dilution with filtered distilled water.
2.8. In-Vitro Cytotoxicity Assessment Using MTT Assay
Biocompatibility of the formulations was tested on the A549 cell line using an MTT assay. Cells were propagated in a humidified air atmosphere (37 °C, 95% relative humidity (RH), and 5% CO
2) in DMEM that was fortified with 10% FBS, 1% (
v/
v) L-glutamine and 1 g/L glucose. Then, the cells were seeded into 96-well microtiter plates at a cell density of 5 × 10
3 cells/well and incubated for 24 h. Afterward, the media were replaced by 100 µL of fresh DMEM, where in the case of the negative control wells, only DMEM was added, while in the other wells, the cells were exposed to various concentrations of previously sterilized TD-loaded NE formulations after adequate dilution with DMEM. After 3 h of incubation, the media were replaced by fresh DMEM, and cells were left to grow overnight in the incubator. On the next day, a stock solution of 5 mg/mL of MTT solution (in PBS) was prepared, diluted to 1 mg/mL with serum-free culture medium, and a 120 µL was added to each well after removal of the old medium. Following incubation for 3 h, the medium was removed by slow aspiration and replaced by 100 µL of dimethyl sulfoxide (DMSO). Then the plates were incubated for 5 min at 37°C followed by shaking for 10 min to dissolve the formed purple-colored formazan crystals. Absorbance was measured at 570 nm using xMarkTM Microplate Absorbance Spectrophotometer (Smart Spec, Bio-Rad Laboratories, Hercules, CA, USA). The viability of the NE-treated cells was expressed as a percentage relative to the control, according to the following equation:
where ([Abs] test) is the absorbance of each concentration of the test substance, ([Abs] control) is the absorbance obtained from the untreated cells. The last reading was supposed to correspond to 100% cell viability. The results are presented as the means ± standard deviation (S.D.) of triplicate samples.
2.9. In-Vivo Lung Toxicity Evaluation
2.9.1. Experimental Animals
Male Sprague-Dawley rats (average weight 200 g) were used for the in-vivo toxicity study. Animals were housed in the animal house of the faculty of medicine (Alexandria University, Alexandria, Egypt) under controlled temperature (21 ± 1 °C), humidity (65% RH), and lighting (10 h light/14 h dark cycle). Two weeks were given for the animals to adapt to their environment before commencing the experiment. Standard chow and water ad libitum were allowed throughout the study duration. The study protocol was approved by the ethical committee of Alexandria University. Experimental procedures were conducted according to the ethical guidelines of Alexandria University and the European Council Law for animal care (EEC Directive of 1986, 86/609/EEC) to ensure minimal animal suffering.
2.9.2. Animal Dose Calculation
Two steps were followed to determine the proper dose of TD for treating PAH in rats following oro-tracheal administration. The first step was the conversion of human pediatric oral dose to rat oral dose. Unfortunately, no dose of TD has been approved by the FDA for treating pediatric PAH. However, a study reported that a quarter or half of the adult dose was safe and effective for treating pediatric patients [
33]. Thus, half the adult dose (20 mg) was selected in the present study as the pediatric oral dose and it was extrapolated to the rat oral dose based on the standard table of Paget and Barnes [
34]. The second calculation step was the conversion of the oral dose to the pulmonary dose. Pulmonary administration was reported to decrease the required dose by 40–100 times compared to the oral route [
30]. Based on this information, a rat weighing 200 g should administer a pulmonary dose of TD ranging between 3.6 µg to 9 µg.
2.9.3. Study Design
The safety of the selected formulations was evaluated following oro-tracheal delivery into the rat lungs through biochemical analysis of bronchoalveolar lavage (BAL) and histopathological examination of lung slices. On the day of the experiment, animals were randomly assigned into five groups (n = 5). Animals of one of the groups were administered saline as a negative control. On the other hand, those in the other groups administered the two selected formulations at the two dosing levels after appropriate dilution with saline as follows: C4TD(3.6) group [C4TD (3.6 µg)], C4TD(9) group [C4TD (9 µg)], P2TD(3.6) group [P2TD (3.6 µg)] and P2TD(9) group [P2TD (9 µg)].
2.9.4. Oro-Tracheal Administration
Prior to the procedure, animals were intraperitoneally anesthetized with a cocktail of ketamine (80 mg/kg) and xylazine (10 mg/kg). Animals of the control group were administered 100 µL saline through the oro-tracheal route. By adopting the same technique, animals of the other groups were administered 100 µL of the diluted formulations, which included the calculated drug doses. Rats were held tightly at a 45° angle. For ultimate oropharyngeal exposure, the mouth was kept open by blunt forceps and another was used to help displace the tongue. A light source was also adjusted for optimal illumination of the rat’s trachea. When the trachea became visible, a syringe equipped with a 22-gauge intravenous catheter was introduced into the trachea after pushing it against the soft palate. Afterward, the catheter was inserted approximately at the bottom of the trachea when the tracheal cartilage ring was sensed, and the liquid sample was injected gently. Then, the catheter was slowly removed, and the rat was held vertically for one min to prevent reflux or regurgitation and to allow the administered fluid to distribute in the lungs. The animals were euthanized via ether inhalation 24 h post-oro-tracheal administration, BAL was performed, and the lungs were collected for histopathological examination.
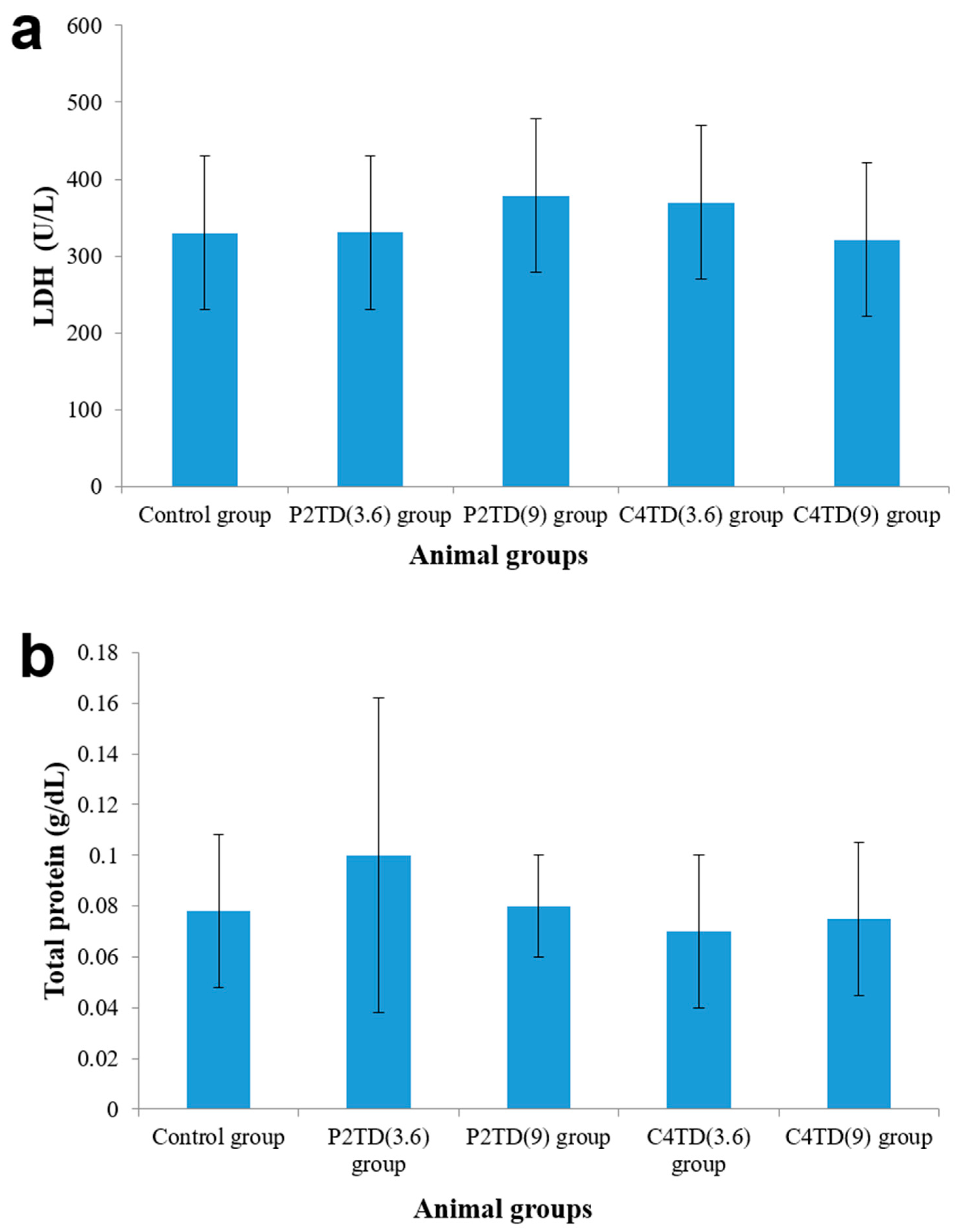
2.9.5. Biochemical Assessment of BAL Fluid
BAL was performed by injecting and withdrawing 4 mL aliquot of sterile saline into the lungs through the trachea three consecutive times. The collected BAL fluid was centrifuged for 5 min, 400 rpm, at 25 °C. The supernatant was analyzed for lactate dehydrogenase activity (LDH) and total protein content using commercial kits (Roche Diagnostics. Indianapolis, IN, USA). Statistical analysis was carried out using an unpaired student’s t-test. The significance level was set at p < 0.05.
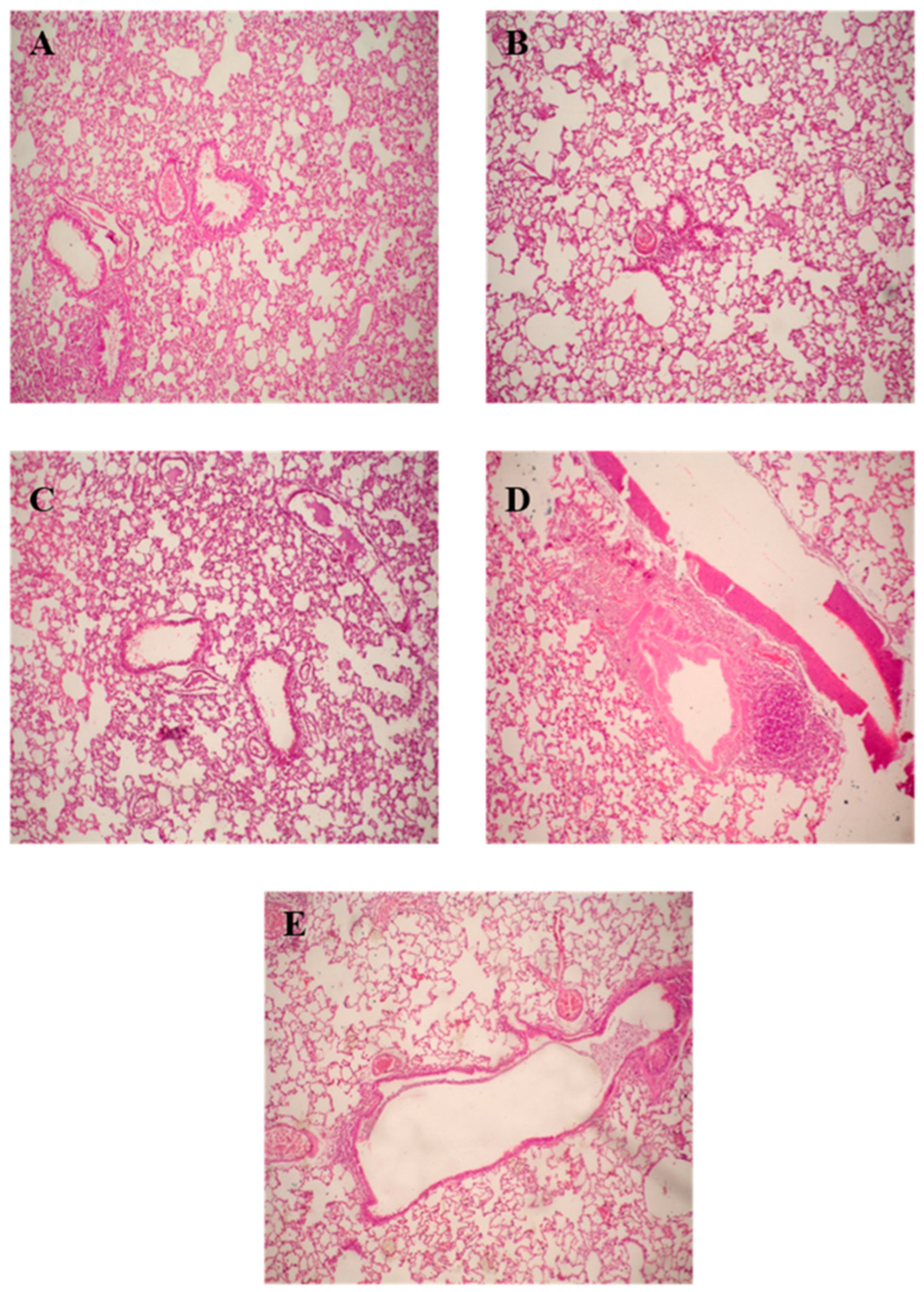
2.9.6. Histopathological Examination
After completion of lung lavage, the lungs were washed with saline and preserved in a 10% formalin solution, followed by washing and dehydration using alcohol. Samples were then cleared in xylene, embedded in paraffin at 56 °C, and dried in a hot air oven for 24 h. This was followed by the sectioning of paraffin tissue blocks at 4 µm by sledge microtome. The specimens were then deparaffinized and stained by hematoxylin and eosin (H & E) stains for histopathological inspection using light microscopy.
2.10. Long-Term Stability Study
A long-term stability study was performed for three months at different storage conditions, RT, refrigerator (2–8 °C), and 40 °C/75% RH. Samples were evaluated at 0, 1, 2, and 3 months for signs of physical instability (phase separation or drug precipitation) through visual examination. In addition, globule size, %T and drug content were determined.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}