
The Effect of Liposomal Diallyl Disulfide and Oxaliplatin on Proliferation of Colorectal Cancer Cells: In Vitro and In Silico Analysis

 ,
,  ,
,  , ,
, ,  ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Methods
2.1. Reagents
2.2. Molecular Docking Studies
2.3. Preparation of DADS- or OXA-Entrapped Liposomes
2.4. Characterization of Liposomes
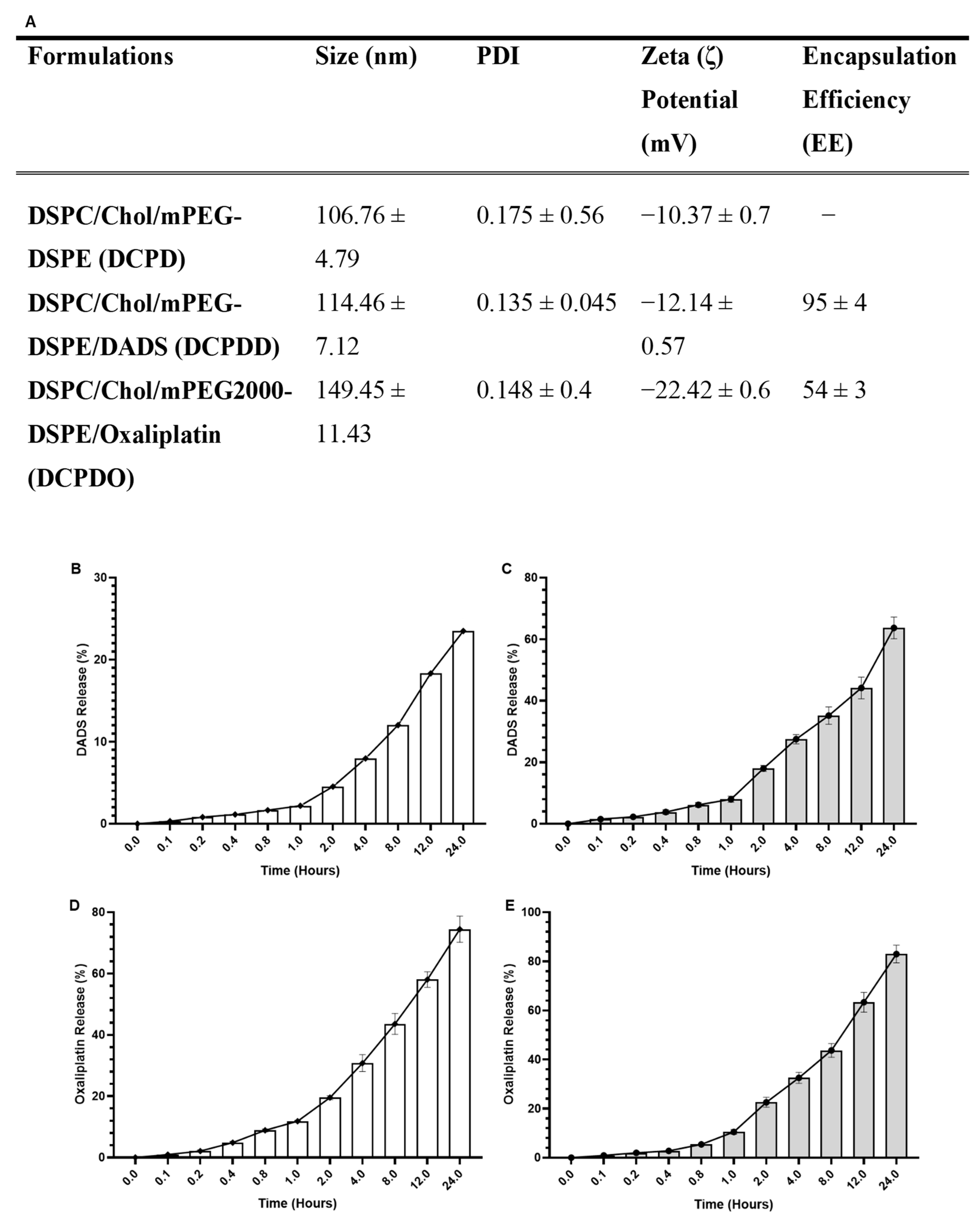
2.4.1. The Size, Zeta (ζ) Potential (mV), Poly Dispersity Index (PDI), and Entrapment Efficiency (EE) of DADS- and OXA-Containing and Empty Liposomes
2.4.2. In Vitro Stability of DCPDD and DCPDO and Release Kinetics of DADS and OXA
2.5. Cell Cytotoxicity Assay
2.6. Determination of Intracellular ROS Generation by Flow Cytometry
2.7. Annexin V-FITC/PI Apoptosis Assay
2.8. Statistical Analysis
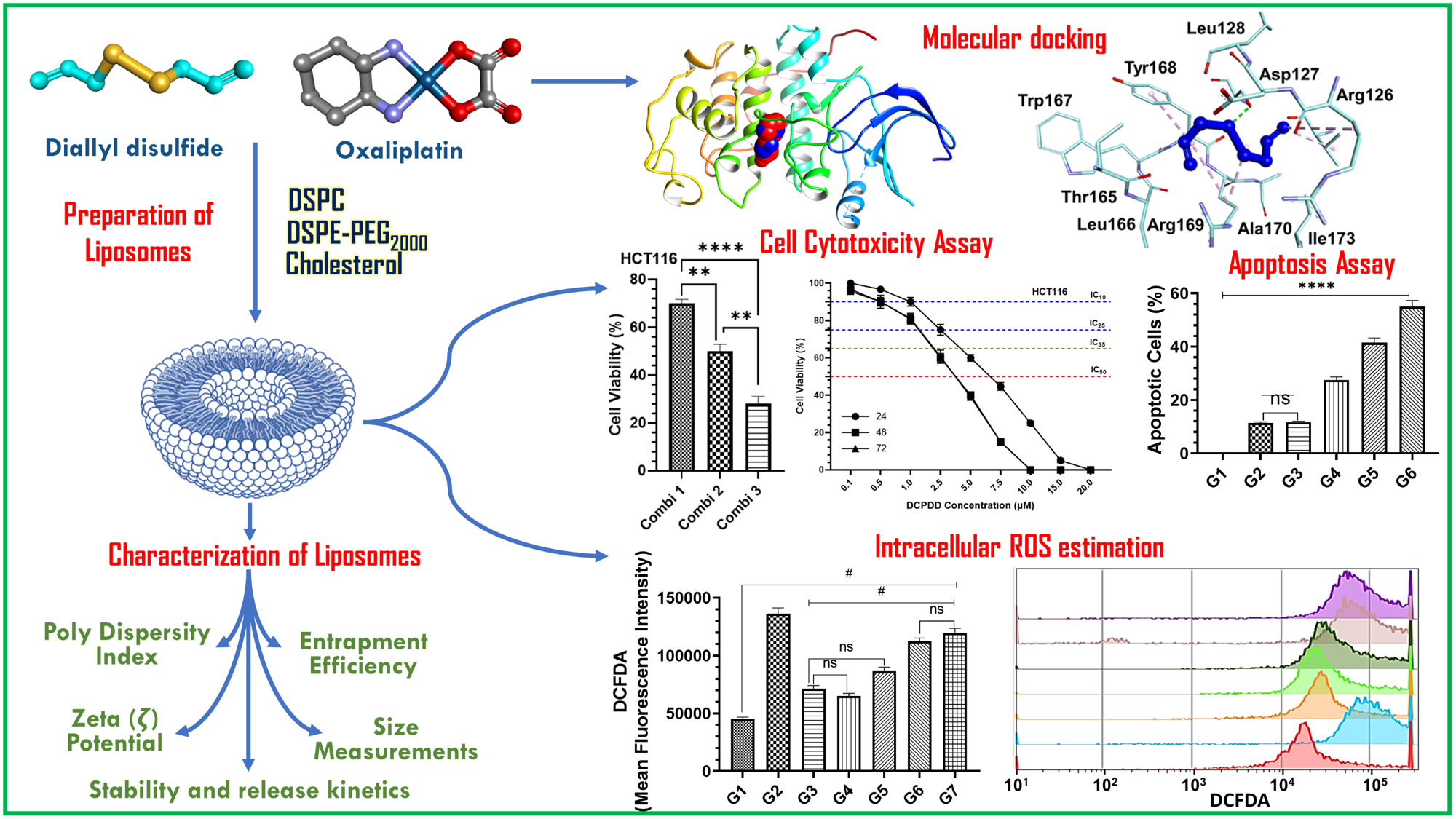
3. Results
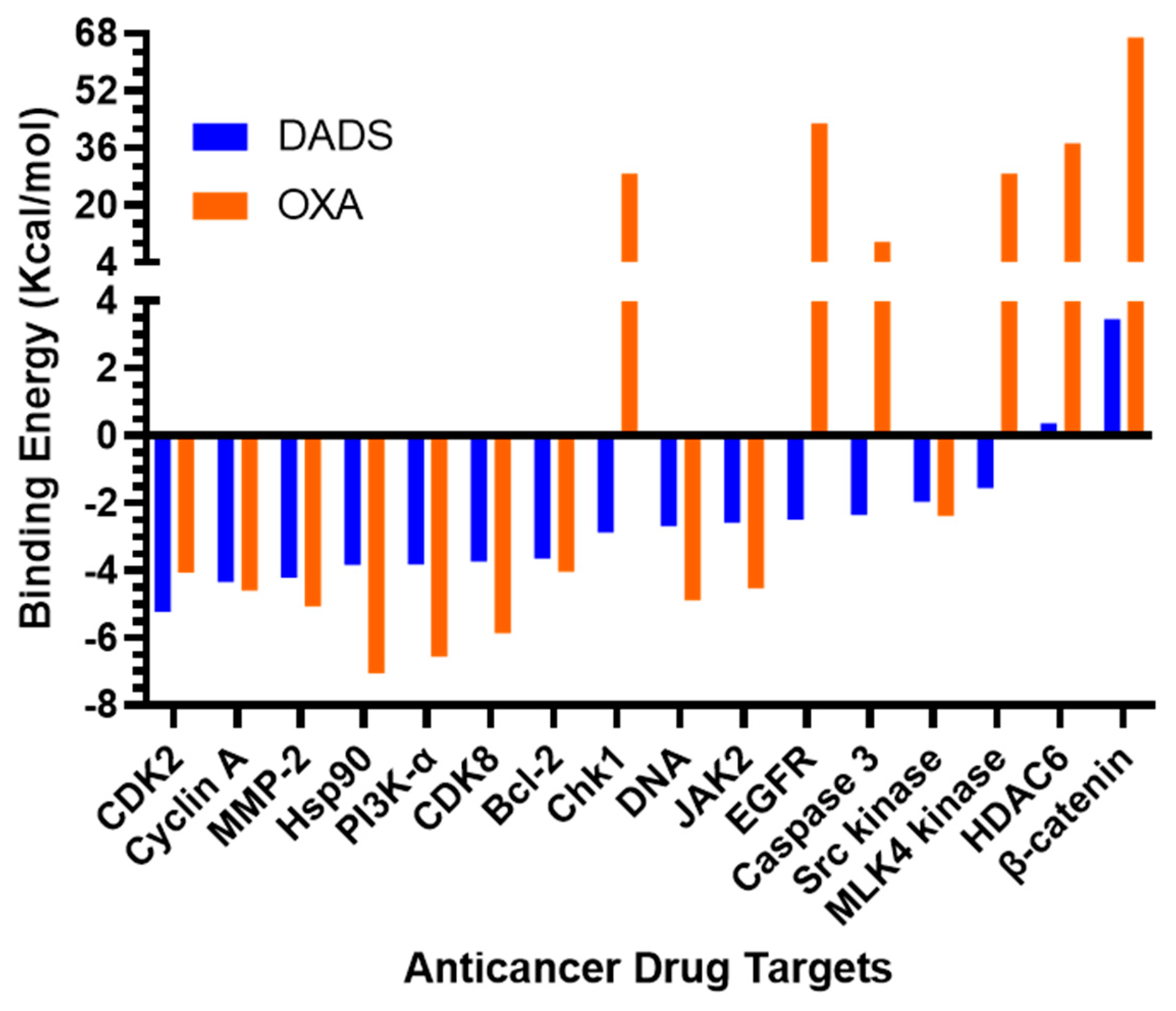
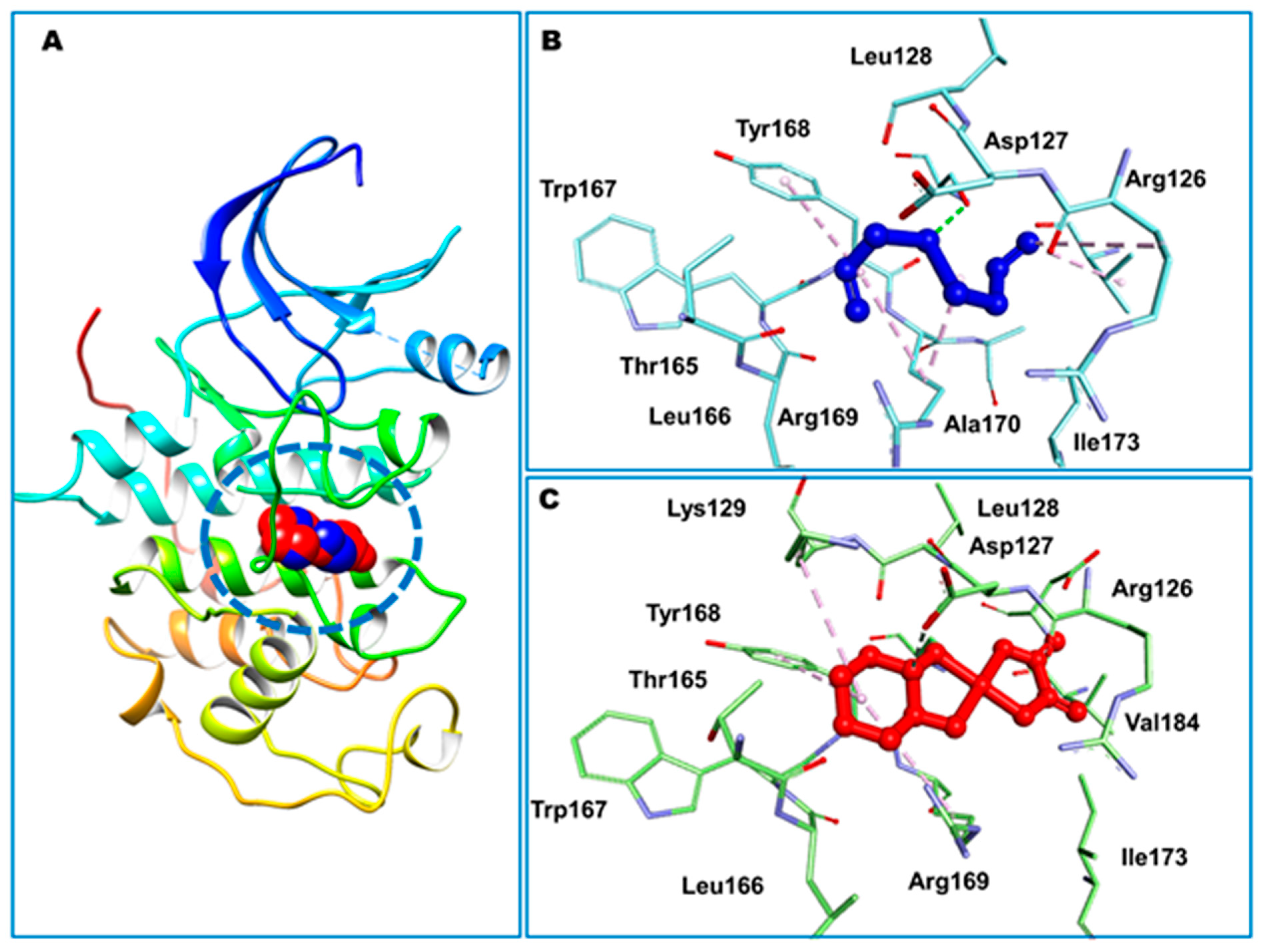
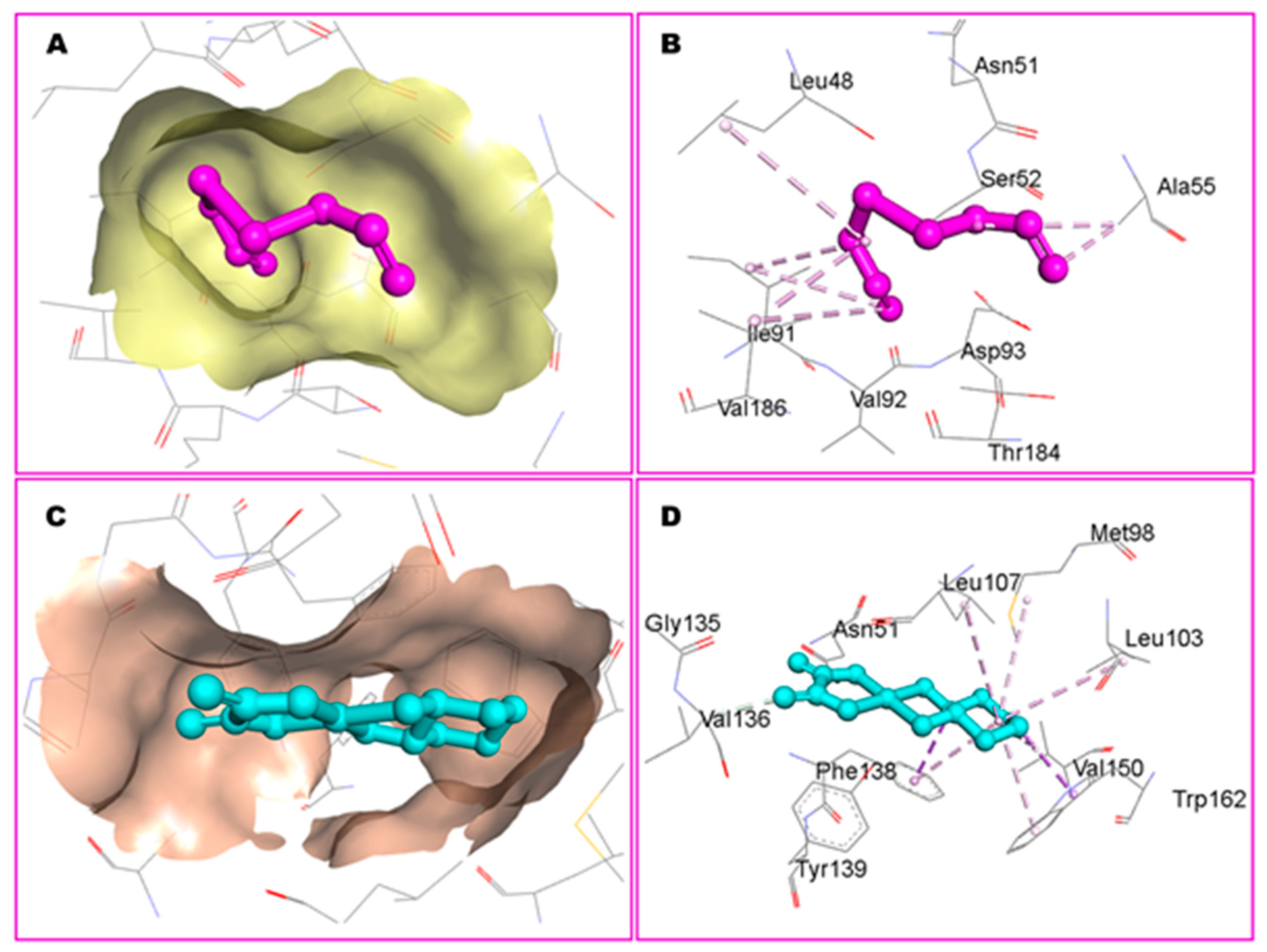
3.1. Molecular Docking Studies
3.2. Characterization of Liposomes
3.2.1. Size, PDI, ζ Potential, and EE
3.2.2. In Vitro Stability of DCPDD and DCPDO and Release Kinetics of DADS and OXA
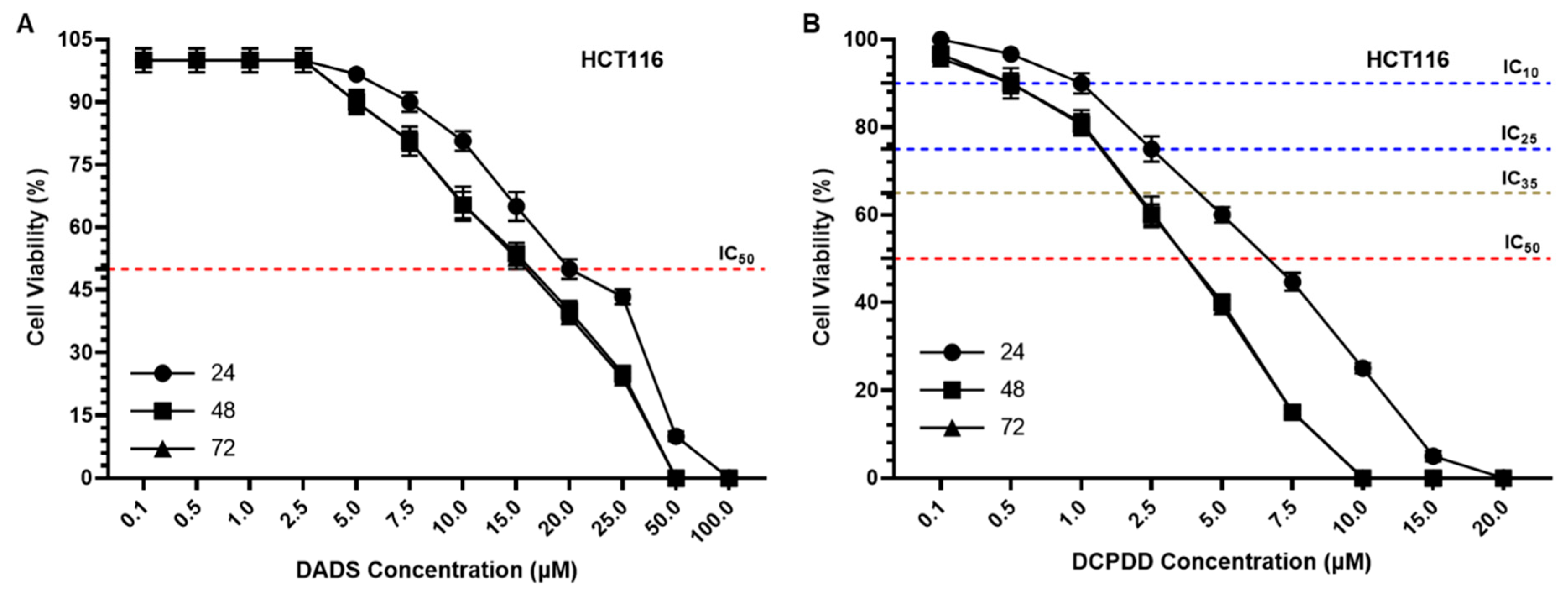
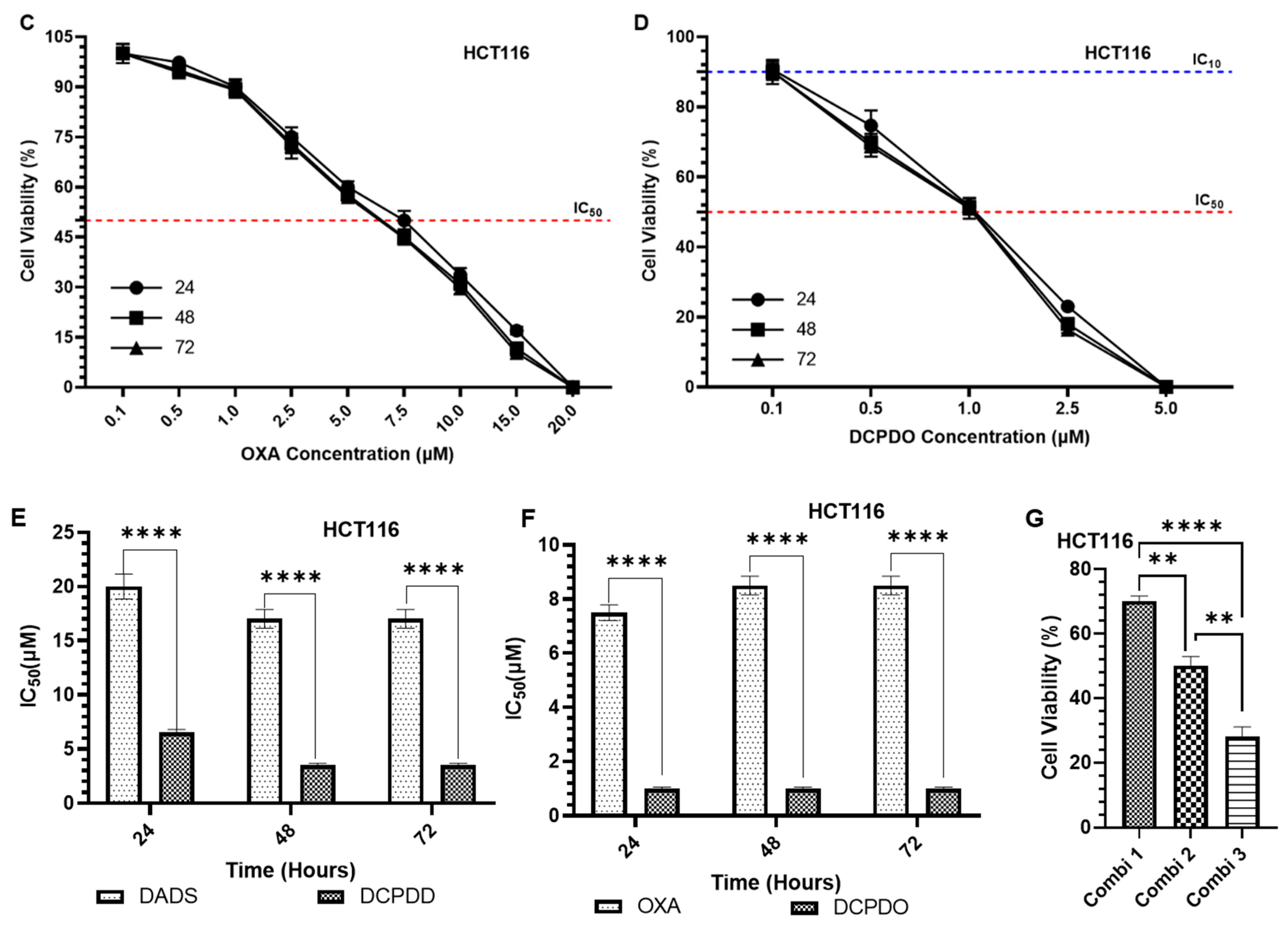
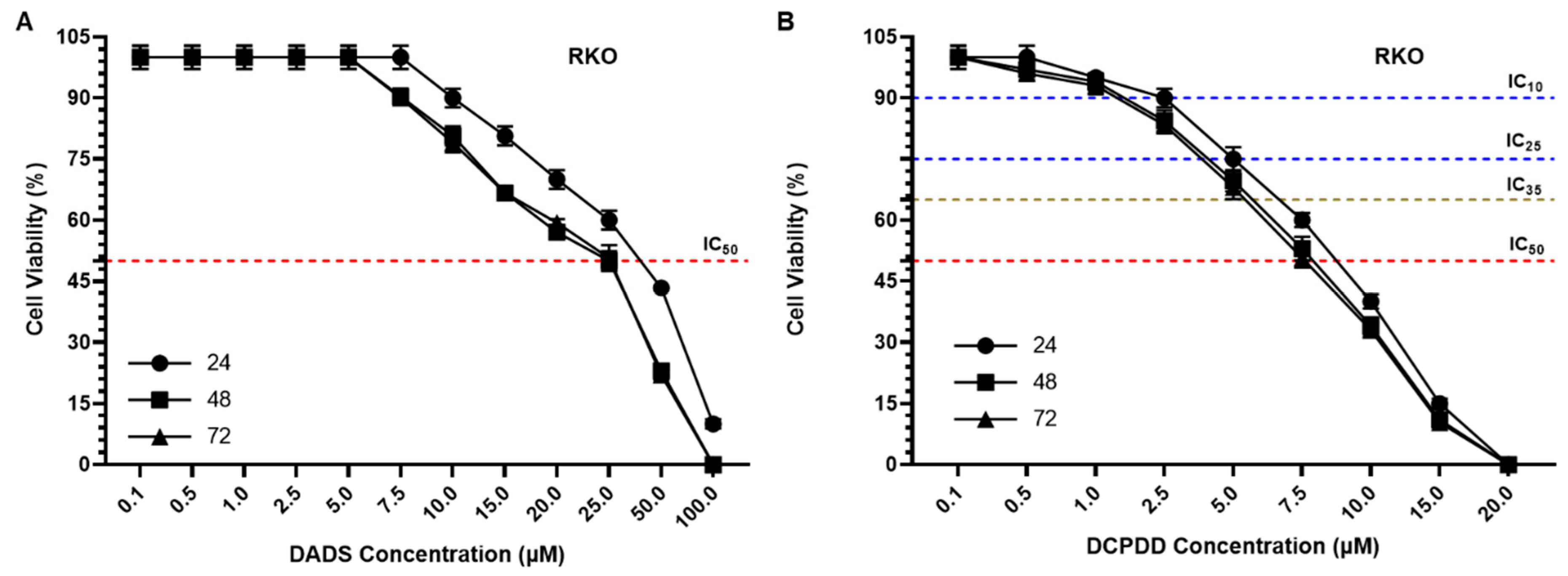
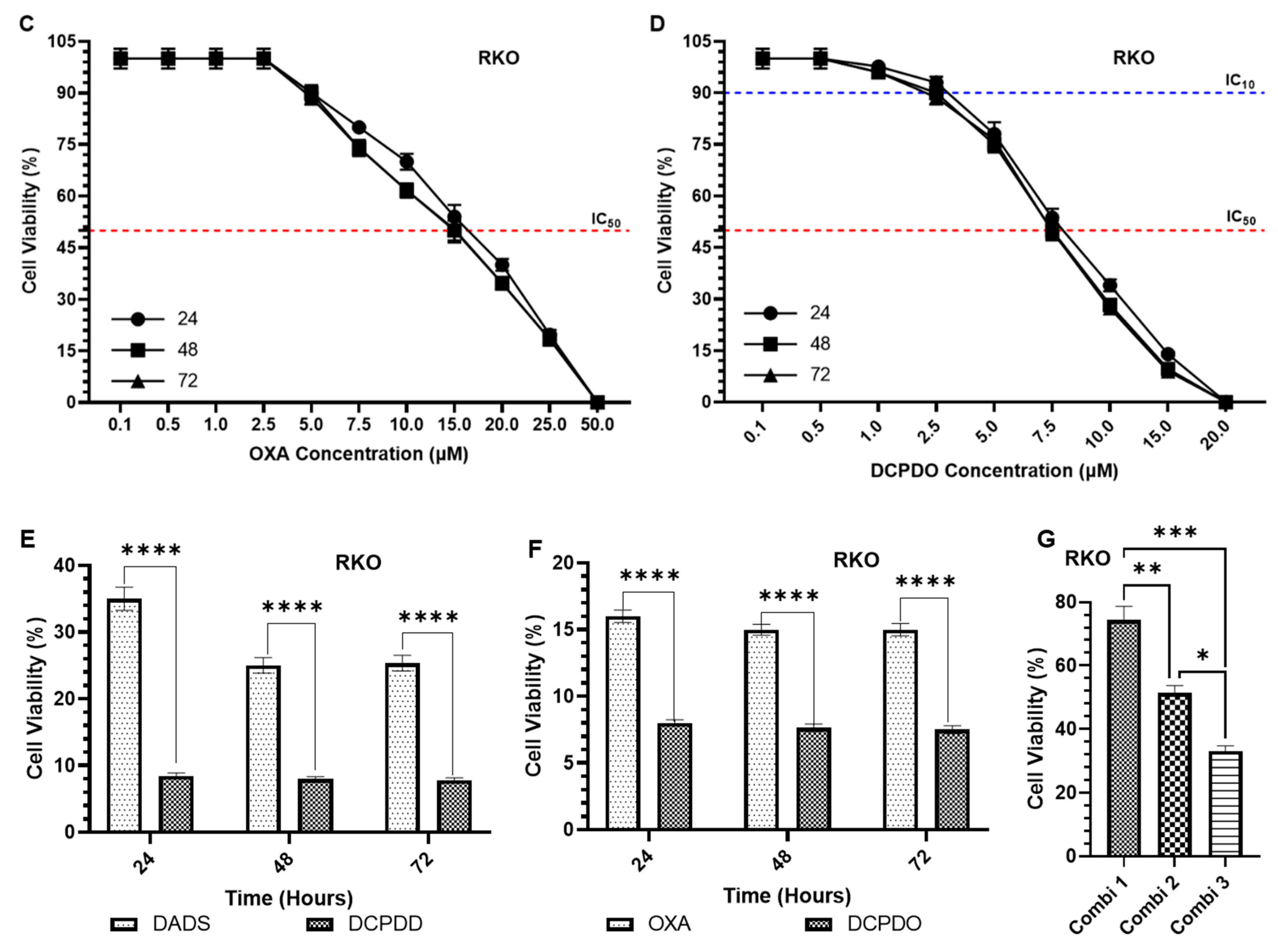
3.3. Effect of DADS and OXA on Cellular Proliferation and IC10, IC25, IC35, and IC50 at Varying Doses of DADS, OXA, DCPDD, and DCPDO in HCT116 and RKO Colon Cancer Cell Lines
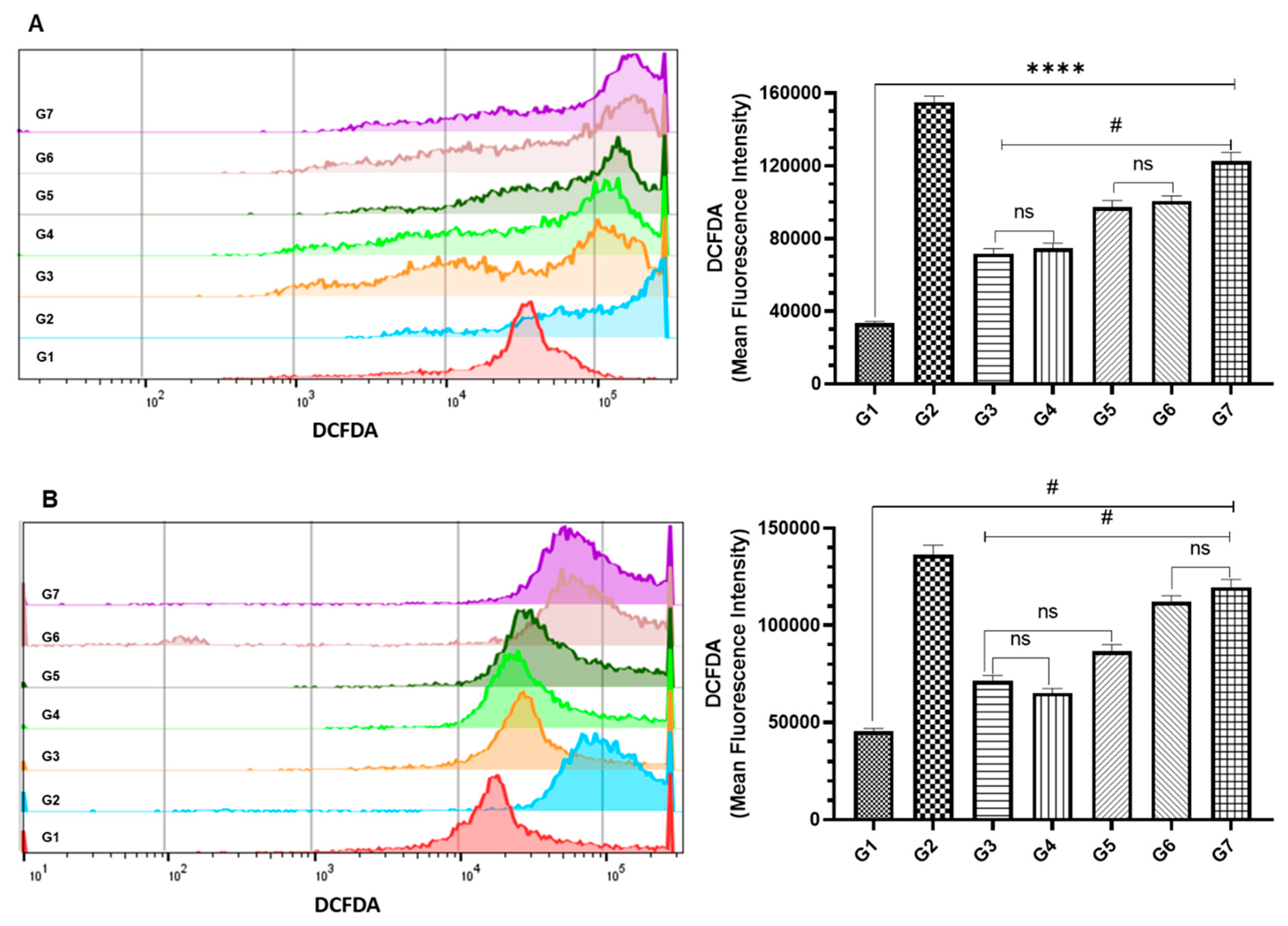
3.4. Effect of DCPDD and DCPDO on the Intracellular ROS by Flow Cytometry in HCT116 and RKO Colon Cancer Cell Lines
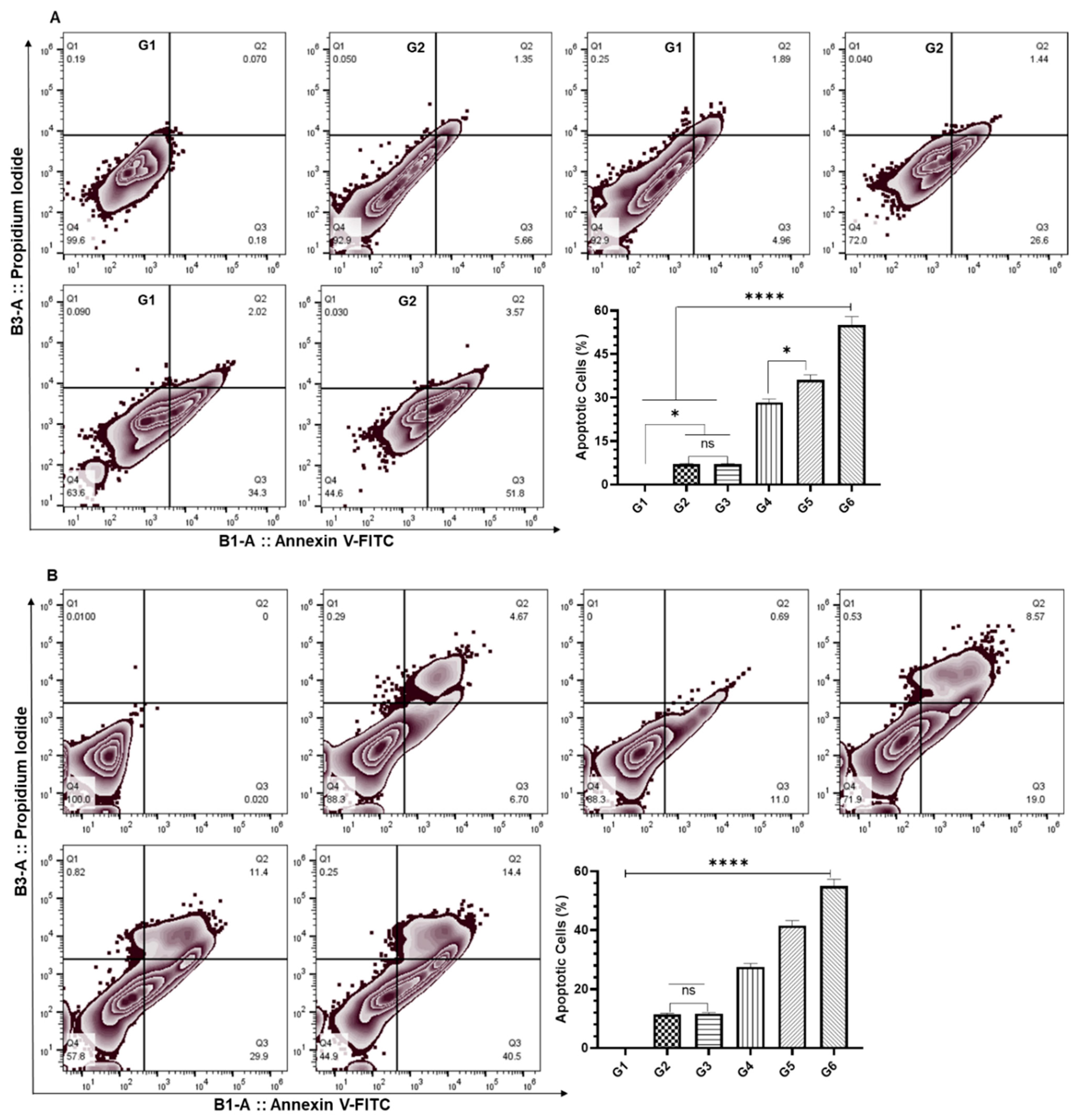
3.5. Effect of DCPDD and DCPDO on the Induction of Apoptosis by Flow Cytometry in HCT116 and RKO Colon Cancer Cell Lines
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Lieberman, D.; Anderson, J.C.; Burke, C.A.; Dominitz, J.; Kaltenbach, T.; Robertson, D.J.; Shaukat, A.; Syngal, S.; Rex, D.K. Recommendations for Follow-Up After Colonoscopy and Polypectomy: A Consensus Update by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2020, 158, 1131–1153.e5. [Google Scholar] [CrossRef]
- Song, E.M.; Park, B.; Ha, C.-A.; Hwang, S.W.; Park, S.H.; Yang, D.-H.; Ye, B.D.; Myung, S.-J.; Yang, S.-K.; Kim, N.; et al. Endoscopic diagnosis and treatment planning for colorectal polyps using a deep-learning model. Sci. Rep. 2020, 10, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, A.L.; Faria, G.; Araújo, A.; Monteiro, M.P. Impact of adiposity on staging and prognosis of colorectal cancer. Crit. Rev. Oncol. 2020, 145, 102857. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, E.K.; Vauthey, J.-N.; Ellis, L.M.; Ellis, V.; Pollock, R.; Broglio, K.R.; Hess, K.; Curley, S.A. Recurrence and Outcomes Following Hepatic Resection, Radiofrequency Ablation, and Combined Resection/Ablation for Colorectal Liver Metastases. Ann. Surg. 2004, 239, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Meyerhardt, J.A.; Mayer, R.J. Systemic Therapy for Colorectal Cancer. N. Engl. J. Med. 2005, 352, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Atanackovic, D.; Bokemeyer, C. Current standards and new trends in the primary treatment of colorectal cancer. Eur. J. Cancer 2011, 47, S312–S314. [Google Scholar] [CrossRef]
- Biller, L.H.; Schrag, D. Diagnosis and Treatment of Metastatic Colorectal Cancer. JAMA J. Am. Med Assoc. 2021, 325, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Zaborowski, A.M.; Winter, D.C.; Lynch, L. The therapeutic and prognostic implications of immunobiology in colorectal cancer: A review. Br. J. Cancer 2021, 125, 1341–1349. [Google Scholar] [CrossRef]
- Sánchez-Gundín, J.; Fernández-Carballido, A.M.; Martínez-Valdivieso, L.; Barreda-Hernández, D.; Torres-Suárez, A.I. New Trends in the Therapeutic Approach to Metastatic Colorectal Cancer. Int. J. Med Sci. 2018, 15, 659–665. [Google Scholar] [CrossRef] [Green Version]
- Van Der Stok, E.P.; Spaander, M.C.W.; Grünhagen, D.J.; Verhoef, E.P.V.D.S.D.J.G.C.; Kuipers, M.C.W.S.E.J. Surveillance after curative treatment for colorectal cancer. Nat. Rev. Clin. Oncol. 2017, 14, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, O.; Inanc, M.; Turkmen, E.; Karaca, H.; Berk, V.; Duran, A.O.; Ozaslan, E.; Ucar, M.; Hacibekiroglu, I.; Eker, B.; et al. Clinicopathological Characteristics and Prognosis of Patients According to Recurrence Time After Curative Resection for Colorectal Cancer. Asian Pac. J. Cancer Prev. 2014, 15, 9277–9281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, A.; Saraf, S.; Verma, A.; Panda, P.; Jain, S.K. Novel targeting approaches and signaling pathways of colorectal cancer: An insight. World J. Gastroenterol. 2018, 24, 4428–4435. [Google Scholar] [CrossRef] [PubMed]
- Piawah, S.; Venook, A.P. Targeted therapy for colorectal cancer metastases: A review of current methods of molecularly targeted therapy and the use of tumor biomarkers in the treatment of metastatic colorectal cancer. Cancer 2019, 125, 4139–4147. [Google Scholar] [CrossRef] [PubMed]
- Meropol, N.J.; Feng, Y.; Grem, J.L.; Mulcahy, M.F.; Catalano, P.J.; Kauh, J.S.; Hall, M.J.; Saltzman, J.N.; George, T.J.; Zangmeister, J.; et al. Phase 2 study of treatment selection based on tumor thymidylate synthase expression in previously untreated patients with metastatic colorectal cancer: A trial of the ECOG-ACRIN Cancer Research Group (E4203). Cancer 2017, 124, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Price, T.J.; Tang, M.; Gibbs, P.; Haller, D.G.; Peeters, M.; Arnold, D.; Segelov, E.; Roy, A.; Tebbutt, N.; Pavlakis, N.; et al. Targeted therapy for metastatic colorectal cancer. Expert Rev. Anticancer. Ther. 2018, 18, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, K.; Zhu, X.; Zheng, X.; Wang, C.; Niu, X.; Jiang, T.; Ji, X.; Zhao, W.; Pang, L.; et al. Synergistic Inhibition of Drug-Resistant Colon Cancer Growth with PI3K/mTOR Dual Inhibitor BEZ235 and Nano-Emulsioned Paclitaxel via Reducing Multidrug Resistance and Promoting Apoptosis. Int. J. Nanomed. 2021, 16, 2173–2186. [Google Scholar] [CrossRef]
- Infante, J.R.; Reid, T.R.; Cohn, A.L.; Edenfield, W.J.; Cescon, T.P.; Hamm, J.T.; Malik, I.A.; Rado, T.A.; McGee, P.J.; Richards, D.A.; et al. Axitinib and/or bevacizumab with modified FOLFOX-6 as first-line therapy for metastatic colorectal cancer: A randomized phase 2 study. Cancer 2013, 119, 2555–2563. [Google Scholar] [CrossRef] [Green Version]
- Seymour, M.T.; Maughan, T.S.; A Ledermann, J.; Topham, C.; James, R.; Gwyther, S.J.; Smith, D.B.; Shepherd, S.; Maraveyas, A.; Ferry, D.R.; et al. Different strategies of sequential and combination chemotherapy for patients with poor prognosis advanced colorectal cancer (MRC FOCUS): A randomised controlled trial. Lancet 2007, 370, 143–152. [Google Scholar] [CrossRef]
- Koopman, M.; Antonini, N.F.; Douma, J.; Wals, J.; Honkoop, A.H.; Erdkamp, F.L.; de Jong, R.S.; Rodenburg, C.J.; Vreugdenhil, G.; Loosveld, O.J.; et al. Sequential versus combination chemotherapy with capecitabine, irinotecan, and oxaliplatin in advanced colorectal cancer (CAIRO): A phase III randomised controlled trial. Lancet 2007, 370, 135–142. [Google Scholar] [CrossRef]
- Chopra, B.; Dhingra, A.K. Natural products: A lead for drug discovery and development. Phytotherapy Res. 2021, 35, 4660–4702. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.; Pradhan, B.; Nayak, R.; Behera, C.; Das, S.; Patra, S.K.; Efferth, T.; Jena, M.; Bhutia, S.K. Dietary polyphenols in chemoprevention and synergistic effect in cancer: Clinical evidences and molecular mechanisms of action. Phytomedicine 2021, 90, 153554. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, M.; Zhao, R.; Wang, D.; Ma, Y.; Li, A. Plant Natural Products: Promising Resources for Cancer Chemoprevention. Molecules 2021, 26, 933. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- De Kok, T.M.; Van Breda, S.G.; Manson, M.M. Mechanisms of combined action of different chemopreventive dietary compounds: A review. Eur. J. Nutr. 2008, 47 (Suppl. 2), 51–59. [Google Scholar] [CrossRef] [PubMed]
- Block, E. Garlic and Other Alliums; The Royal Society of Chemistry: London, UK, 2010. [Google Scholar]
- Zhang, Y.; Liu, X.; Ruan, J.; Zhuang, X.; Zhang, X.; Li, Z. Phytochemicals of garlic: Promising candidates for cancer therapy. Biomed. Pharmacother. 2020, 123, 109730. [Google Scholar] [CrossRef]
- Iciek, M.; Kwiecień, I.; Włodek, L. Biological properties of garlic and garlic-derived organosulfur compounds. Environ. Mol. Mutagen. 2009, 50, 247–265. [Google Scholar] [CrossRef]
- He, H.; Ma, Y.; Huang, H.; Huang, C.; Chen, Z.; Chen, D.; Gu, Y.; Wang, X.; Chen, J. A comprehensive understanding about the pharmacological effect of diallyl disulfide other than its anti-carcinogenic activities. Eur. J. Pharmacol. 2021, 893, 173803. [Google Scholar] [CrossRef]
- Sengupta, A.; Ghosh, S.; Bhattacharjee, S. Allium vegetables in cancer prevention: An overview. Asian Pac. J. Cancer Prev. 2004, 5, 237–245. [Google Scholar]
- Knowles, L.M.; Milner, J.A. Possible mechanism by which allyl sulfides suppress neoplastic cell proliferation. J. Nutr. 2001, 131, 1061S–1066S. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Du, C.; Guo, N.; Teng, Y.; Meng, X.; Sun, H.; Li, S.; Yu, P.; Galons, H. Composition design and medical application of liposomes. Eur. J. Med. Chem. 2019, 164, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Borek, C. Antioxidant Health Effects of Aged Garlic Extract. J. Nutr. 2001, 131, 1010S–1015S. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-T.; Wong, P.-L.; Lii, C.-K.; Hse, H.; Sheen, L.-Y. Antidiabetic effect of garlic oil but not diallyl disulfide in rats with streptozotocin-induced diabetes. Food Chem. Toxicol. 2006, 44, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- De Greef, D.; Barton, E.M.; Sandberg, E.N.; Croley, C.R.; Pumarol, J.; Wong, T.L.; Das, N.; Bishayee, A. Anticancer potential of garlic and its bioactive constituents: A systematic and comprehensive review. Semin. Cancer Biol. 2021, 73, 219–264. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Sheen, L.; Chen, H.-W.; Tsai, S.-J.; Lii, C.-K. Effects of organosulfur compounds from garlic oil on the antioxidation system in rat liver and red blood cells. Food Chem. Toxicol. 2001, 39, 563–569. [Google Scholar] [CrossRef]
- Hashizume, Y.; Shirato, K.; Sato, S.; Matsumoto, A.; Tachiyashiki, K.; Imaizumi, K. Dose-dependent effects of diallyl disulfide on plasma glucose and free fatty acid levels in rats. J. Toxicol. Sci. 2013, 38, 879–884. [Google Scholar] [CrossRef] [Green Version]
- Druesne, N.; Pagniez, A.; Mayeur, C.; Thomas, M.; Cherbuy, C.; Duée, P.-H.; Latino-Martel, P.; Chaumontet, C. Diallyl disulfide (DADS) increases histone acetylation and p21waf1/cip1 expression in human colon tumor cell lines. Carcinogenesis 2004, 25, 1227–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.-S.; Ham, Y.-A.; Choi, J.-H.; Kim, J. Effects of allyl sulfur compounds and garlic extract on the expression of Bcl-2, Bax, and p53 in non small cell lung cancer cell lines. Exp. Mol. Med. 2000, 32, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.-P.; Wang, G.-H.; Ling, H.; Su, Q.; Yang, Y.-H.; Song, Y.; Tang, R.-J.; Liu, Y.; Huang, C. Diallyl disulfide-induced G2/M arrest of human gastric cancer MGC803 cells involves activation of p38 MAP kinase pathways. World J. Gastroenterol. 2004, 10, 2731–2734. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.-C.; Hsu, S.-C.; Kuo, C.-L.; Yang, J.-S.; Ma, C.-Y.; Lu, H.-F.; Tang, N.-Y.; Hsia, T.-C.; Ho, H.-C.; Chung, J.-G. Diallyl sulfide, diallyl disulfide, and diallyl trisulfide inhibit migration and invasion in human colon cancer colo 205 cells through the inhibition of matrix metalloproteinase-2, -7, and -9 expressions. Environ. Toxicol. 2011, 28, 479–488. [Google Scholar] [CrossRef]
- Yin, X.; Feng, C.; Han, L.; Ma, Y.; Jiao, Y.; Wang, J.; Jia, L.; Jing, F.; Gao, X.; Zhang, Y. Diallyl disulfide inhibits the metastasis of type II esophageal-gastric junction adenocarcinoma cells via NF-κB and PI3K/AKT signaling pathways in vitro. Oncol. Rep. 2017, 39, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Siddhartha, V.T.; Pindiprolu, S.K.S.S.; Chintamaneni, P.K.; Tummala, S.; Kumar, S.N. RAGE receptor targeted bioconjuguate lipid nanoparticles of diallyl disulfide for improved apoptotic activity in triple negative breast cancer: In vitro studies. Artif. Cells Nanomed. Biotechnol. 2018, 46, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Kang, S.; Kim, D.Y.; You, S.; Park, D.; Oh, S.C.; Lee, D.-H. Diallyl disulfide (DADS) boosts TRAIL-Mediated apoptosis in colorectal cancer cells by inhibiting Bcl-2. Food Chem. Toxicol. 2019, 125, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Suangtamai, T.; Tanyong, D.I. Diallyl disulfide induces apoptosis and autophagy via mTOR pathway in myeloid leukemic cell line. Tumor Biol. 2016, 37, 10993–10999. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Guan, X.; Chao, R.; Huang, C.; Li, D.; Yang, P.; Liu, S.; Hasegawa, T.; Guo, J.; Li, M. Diallyl Disulfide Induces Apoptosis and Autophagy in Human Osteosarcoma MG-63 Cells through the PI3K/Akt/mTOR Pathway. Molecules 2019, 24, 2665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choromanska, A.; Kulbacka, J.; Saczko, J.; Surowiak, P. Effect of diallyl disulfide and garlic oil on different human astrocytoma cell lines. Biomed. Rep. 2020, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.; Zubair, S.; Farazuddin, M.; Ahmad, E.; Khan, A.; Zia, Q.; Malik, A.; Mohammad, O. Development, characterization and efficacy of niosomal diallyl disulfide in treatment of disseminated murine candidiasis. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Talluri, S.V.; Kuppusamy, G.; Karri, V.V.S.R.; Yamjala, K.; Wadhwani, A.; Madhunapantula, S.V.; Pindiprolu, S.S.S. Application of quality-by-design approach to optimize diallyl disulfide-loaded solid lipid nanoparticles. Artif. Cells Nanomed. Biotechnol. 2017, 45, 474–488. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Shukla, Y.; Kalra, N.; Alam, M.; Ahmad, M.G.; Hakim, S.R.; Owais, M. Potential of diallyl sulfide bearing pH-sensitive liposomes in chemoprevention against DMBA-induced skin papilloma. Mol. Med. 2007, 13, 443–451. [Google Scholar] [CrossRef]
- Allemailem, K.; Alnuqaydan, A.; Almatroudi, A.; Alrumaihi, F.; Aljaghwani, A.; Khalilullah, H.; Younus, H.; Khan, A.; Khan, M. Safety and Therapeutic Efficacy of Thymoquinone-Loaded Liposomes against Drug-Sensitive and Drug-Resistant Acinetobacter baumannii. Pharmaceutics 2021, 13, 677. [Google Scholar] [CrossRef]
- Khan, M.A.; Aldebasi, Y.H.; Alsuhaibani, S.A.; Alsahli, M.A.; Alzohairy, M.A.; Khan, A.; Younus, H. Therapeutic potential of thymoquinone liposomes against the systemic infection of Candida albicans in diabetic mice. PLoS ONE 2018, 13, e0208951. [Google Scholar] [CrossRef] [PubMed]
- González-Pastor, R.; Lancelot, A.; Morcuende-Ventura, V.; Anselmo, M.S.; Sierra, T.; Serrano, J.; Martin-Duque, P. Combination Chemotherapy with Cisplatin and Chloroquine: Effect of Encapsulation in Micelles Formed by Self-Assembling Hybrid Dendritic–Linear–Dendritic Block Copolymers. Int. J. Mol. Sci. 2021, 22, 5223. [Google Scholar] [CrossRef] [PubMed]
- Franco, M.S.; Oliveira, M.C. Liposomes Co- encapsulating Anticancer Drugs in Synergistic Ratios as an Approach to Promote Increased Efficacy and Greater Safety. Anti-Cancer Agents Med. Chem. 2019, 19, 17–28. [Google Scholar] [CrossRef]
- Liu, J.; Chi, D.; Pan, S.; Zhao, L.; Wang, X.; Wang, D.; Wang, Y. Effective co-encapsulation of doxorubicin and irinotecan for synergistic therapy using liposomes prepared with triethylammonium sucrose octasulfate as drug trapping agent. Int. J. Pharm. 2019, 557, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, E.C.; Dos Santos, N.; Dragowska, W.H.; Laskin, J.J.; Bally, M. The Formulation of Lipid-Based Nanotechnologies for the Delivery of Fixed Dose Anticancer Drug Combinations. Curr. Drug Deliv. 2005, 2, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Sesarman, A.; Muntean, D.; Abrudan, B.; Tefas, L.; Sylvester, B.; Licarete, E.; Rauca, V.; Luput, L.; Patras, L.; Banciu, M.; et al. Improved pharmacokinetics and reduced side effects of doxorubicin therapy by liposomal co-encapsulation with curcumin. J. Liposome Res. 2021, 31, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ahmed, B.; Mehta, K.; Kurzrock, R. Liposomal curcumin with and without oxaliplatin: Effects on cell growth, apoptosis, and angiogenesis in colorectal cancer. Mol. Cancer Ther. 2007, 6, 1276–1282. [Google Scholar] [CrossRef] [Green Version]
- Shahraki, N.; Mehrabian, A.; Amiri-Darban, S.; Moosavian, S.A.; Jaafari, M.R. Preparation and characterization of PEGylated liposomal Doxorubicin targeted with leptin-derived peptide and evaluation of their anti-tumor effects, in vitro and in vivo in mice bearing C26 colon carcinoma. Colloids Surfaces B Biointerfaces 2021, 200, 111589. [Google Scholar] [CrossRef]
- Mozar, F.S. Impact of PEGylated Nanoparticles on Tumor Targeted Drug Delivery. Curr. Pharm. Des. 2018, 24, 3283–3296. [Google Scholar] [CrossRef]
- Pozzi, D.; Colapicchioni, V.; Caracciolo, G.; Piovesana, S.; Capriotti, A.L.; Palchetti, S.; De Grossi, S.; Riccioli, A.; Amenitsch, H.; Laganà, A. Effect of polyethyleneglycol (PEG) chain length on the bio–nano-interactions between PEGylated lipid nanoparticles and biological fluids: From nanostructure to uptake in cancer cells. Nanoscale 2014, 6, 2782–2792. [Google Scholar] [CrossRef] [PubMed]
- Chow, T.-H.; Lin, Y.-Y.; Hwang, J.-J.; Wang, H.-E.; Tseng, Y.-L.; Wang, S.-J.; Liu, Re.; Lin, Wu.; Yang, Ch.; Ting, G. Improvement of biodistribution and therapeutic index via increase of polyethylene glycol on drug-carrying liposomes in an HT-29/luc xenografted mouse model. Anticancer Res. 2009, 29, 2111–2120. [Google Scholar] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, K.S.; Hussein, S.A.; Ali, A.; Korma, S.A.; Lipeng, Q.; Jinghua, C. Liposome: Composition, characterisation, preparation, and recent innovation in clinical applications. J. Drug Target. 2018, 27, 742–761. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.; Omri, A. The Effect of Different Lipid Components on the In Vitro Stability and Release Kinetics of Liposome Formulations. Drug Deliv. 2004, 11, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Crommelin, D.J. Influence of Lipid Composition and Ionic Strength on the Physical Stability of Liposomes. J. Pharm. Sci. 1984, 73, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Briuglia, M.-L.; Rotella, C.M.; McFarlane, A.; Lamprou, D.A. Influence of cholesterol on liposome stability and on in vitro drug release. Drug Deliv. Transl. Res. 2015, 5, 231–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magarkar, A.; Dhawan, V.; Kallinteri, P.; Viitala, T.; Elmowafy, M.; Róg, T.; Bunker, A. Cholesterol level affects surface charge of lipid membranes in saline solution. Sci. Rep. 2014, 4, 5005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papahadjopoulos, D.; Cowden, M.; Kimelberg, H. Role of cholesterol in membranes effects on phospholipid-protein interactions, membrane permeability and enzymatic activity. Biochimica et Biophysica Acta 1973, 330, 8–26. [Google Scholar] [CrossRef]
- Khan, A.; Aljarbou, A.N.; Aldebasi, Y.H.; Allemeilam, K.S.; A Alsahly, M.; Khan, S.; Alruwetei, A.M.; A Khan, M. Fatty Acid Synthase (FASN) siRNA-Encapsulated-Her-2 Targeted Fab’-Immunoliposomes for Gene Silencing in Breast Cancer Cells. Int. J. Nanomed. 2020, 15, 5575–5589. [Google Scholar] [CrossRef]
- Vargason, A.M.; Anselmo, A.C.; Mitragotri, S. The evolution of commercial drug delivery technologies. Nat. Biomed. Eng. 2021, 5, 951–967. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Neupane, Y.R.; Shafi, S.; Mangla, B.; Kohli, K. PEGylated liposomes as an emerging therapeutic platform for oral nanomedicine in cancer therapy: In vitro and in vivo assessment. J. Mol. Liq. 2020, 303, 112649. [Google Scholar] [CrossRef]
- Yang, C.; Liu, H.Z.; Fu, Z.X.; Lu, W.D. Oxaliplatin long-circulating liposomes improved therapeutic index of colorectal carcinoma. BMC Biotechnol. 2011, 11, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheraga, N.; Ouahab, A.; Shen, Y.; Huang, N.-P. Characterization and Pharmacokinetic Evaluation of Oxaliplatin Long-Circulating Liposomes. BioMed Res. Int. 2021, 2021, 5949804. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Yu, F.; Yang, Y.; Cheng, X.; Liu, Y.; Zhang, H.; Zhao, S.; Yang, Z.; Li, M.; Li, Z.; et al. Preparation and Evaluation of Oxaliplatin Thermosensitive Liposomes with Rapid Release and High Stability. PLoS ONE 2016, 11, e0158517. [Google Scholar] [CrossRef]
- Abu Lila, A.S.; Doi, Y.; Nakamura, K.; Ishida, T.; Kiwada, H. Sequential administration with oxaliplatin-containing PEG-coated cationic liposomes promotes a significant delivery of subsequent dose into murine solid tumor. J. Control. Release 2010, 142, 167–173. [Google Scholar] [CrossRef]
- William-Faltaos, S.; Rouillard, D.; Lechat, P.; Bastian, G. Cell cycle arrest and apoptosis induced by oxaliplatin (L-OHP) on four human cancer cell lines. Anticancer. Res. 2006, 26, 2093–2099. [Google Scholar] [PubMed]
- Song, J.-D.; Lee, S.K.; Kim, K.M.; Park, S.E.; Park, S.-J.; Kim, K.H.; Ahn, S.C.; Park, Y.C. Molecular mechanism of diallyl disulfide in cell cycle arrest and apoptosis in HCT-116 colon cancer cells. J. Biochem. Mol. Toxicol. 2009, 23, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Odom, R.Y.; Dansby, M.Y.; Rollins-Hairston, A.M.; Jackson, K.M.; Kirlin, W.G. Phytochemical Induction of Cell Cycle Arrest by Glutathione Oxidation and Reversal by N-Acetylcysteine in Human Colon Carcinoma Cells. Nutr. Cancer 2009, 61, 332–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.-S.; Chen, G.-W.; Hsia, T.-C.; Ho, H.-C.; Ho, C.-C.; Lin, M.-W.; Lin, S.-S.; Yeh, R.-D.; Ip, S.-W.; Lu, H.-F.; et al. Diallyl disulfide induces apoptosis in human colon cancer cell line (COLO 205) through the induction of reactive oxygen species, endoplasmic reticulum stress, caspases casade and mitochondrial-dependent pathways. Food Chem. Toxicol. 2009, 47, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-J.; Kassie, F.; Mersch-Sundermann, V. The role of reactive oxygen species (ROS) production on diallyl disulfide (DADS) induced apoptosis and cell cycle arrest in human A549 lung carcinoma cells. Mutat. Res. Mol. Mech. Mutagen. 2005, 579, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Tummala, S.; Kumar, M.N.S.; Pindiprolu, S.K. Improved anti-tumor activity of oxaliplatin by encapsulating in anti-DR5 targeted gold nanoparticles. Drug Deliv. 2016, 23, 3505–3519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Liu, H.-Z.; Fu, Z.-X. Effects of PEG-liposomal oxaliplatin on apoptosis, and expression of Cyclin A and Cyclin D1 in colorectal cancer cells. Oncol. Rep. 2012, 28, 1006–1012. [Google Scholar] [CrossRef]
- Zalba, S.; Navarro-Blasco, I.; Troconiz, I.; de Ilarduya, C.T.; Garrido, M.J. Application of different methods to formulate PEG-liposomes of oxaliplatin: Evaluation in vitro and in vivo. Eur. J. Pharm. Biopharm. 2012, 81, 273–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azam, F. Elucidation of Teicoplanin Interactions with Drug Targets Related to COVID-19. Antibiotics 2021, 10, 856. [Google Scholar] [CrossRef] [PubMed]
- Pantsar, T.; Poso, A. Binding Affinity via Docking: Fact and Fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef] [Green Version]
- Tadesse, S.; Anshabo, A.T.; Portman, N.; Lim, E.; Tilley, W.; Caldon, C.E.; Wang, S. Targeting CDK2 in cancer: Challenges and opportunities for therapy. Drug Discov. Today 2020, 25, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.; Lang, S.A.; Stoeltzing, O. Heat-shock protein 90 (Hsp90) as a molecular target for therapy of gastrointestinal cancer. Anticancer Res. 2009, 29, 2031–2042. [Google Scholar] [PubMed]
- Zhang, H.; Burrows, F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J. Mol. Med. 2004, 82, 488–499. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Colon Cancer Cells | Formulations | |||
|---|---|---|---|---|
| DADS | DCPDD | OXA | DCPDO | |
| HCT116 | 0.1–100.0 μM | 0.1–20 μM | 0.1–20 μM | 0.1–5 μM |
| RKO | 0.1–100.0 μM | 0.1–20 μM | 0.1–20 μM | 0.1–20 μM |
| Colon Cancer Cells | Formulations | |||
|---|---|---|---|---|
| DCPDD IC10 | DCPDD IC25 | DCPDD IC35 | DCPDO IC10 | |
| HCT116 | 0.5 μM | 0.95 μM | 2.95 μM | 0.1 μM |
| RKO | 1.6 μM | 4.0 μM | 4.75 μM | 2.5 μM |
| S.N. | Targets | PDB Code | Binding Energy (kcal/mol) | |
|---|---|---|---|---|
| DADS | OXA | |||
| 1 | Apoptosis regulator Bcl-2 | 4LXD | −3.65 | −4.04 |
| 2 | β-catenin | 3SL9 | 3.46 | 66.91 |
| 3 | Caspase 3 | 1RE1 | −2.36 | 9.81 |
| 4 | Cyclin dependent kinase 2 (CDK2) | 2A4L | −5.23 | −2.11 |
| 5 | Cyclin dependent kinase 8 (CDK8) | 3RGF | −3.73 | −5.87 |
| 6 | Checkpoint Kinase 1 (Chk1) | 2R0U | −2.87 | 28.89 |
| 7 | Cyclin A | 6GUE | −4.34 | −4.6 |
| 8 | Double-stranded DNA | 1AIO | −2.68 | −4.89 |
| 9 | Epidermal growth factor receptor (EGFR) | 1M17 | −2.5 | 42.87 |
| 10 | Histone deacetylase 6 (HDAC6) | 5WGI | 0.37 | 37.34 |
| 11 | Heat shock protein 90 (Hsp90) | 4BQG | −3.84 | −7.06 |
| 12 | Janus kinase 2 (JAK2) | 3KCK | −2.58 | −4.54 |
| 13 | Mixed-lineage kinase (MLK4) | 4UYA | −1.56 | 28.89 |
| 14 | Matrix metalloproteinase-2 (MMP-2) | 1HOV | −4.22 | −5.07 |
| 15 | Phosphatidylinositol-3 kinase alpha (PI3K-α) | 3ZIM | −3.83 | −6.56 |
| 16 | Src kinase | 2BDF | −1.96 | −2.38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alrumaihi, F.; Khan, M.A.; Babiker, A.Y.; Alsaweed, M.; Azam, F.; Allemailem, K.S.; Almatroudi, A.A.; Ahamad, S.R.; AlSuhaymi, N.; Alsugoor, M.H.; et al. The Effect of Liposomal Diallyl Disulfide and Oxaliplatin on Proliferation of Colorectal Cancer Cells: In Vitro and In Silico Analysis. Pharmaceutics 2022, 14, 236. https://doi.org/10.3390/pharmaceutics14020236
Alrumaihi F, Khan MA, Babiker AY, Alsaweed M, Azam F, Allemailem KS, Almatroudi AA, Ahamad SR, AlSuhaymi N, Alsugoor MH, et al. The Effect of Liposomal Diallyl Disulfide and Oxaliplatin on Proliferation of Colorectal Cancer Cells: In Vitro and In Silico Analysis. Pharmaceutics. 2022; 14(2):236. https://doi.org/10.3390/pharmaceutics14020236
Chicago/Turabian StyleAlrumaihi, Faris, Masood Alam Khan, Ali Yousif Babiker, Mohammed Alsaweed, Faizul Azam, Khaled S. Allemailem, Ahmad A. Almatroudi, Syed Rizwan Ahamad, Naif AlSuhaymi, Mahdi H. Alsugoor, and et al. 2022. "The Effect of Liposomal Diallyl Disulfide and Oxaliplatin on Proliferation of Colorectal Cancer Cells: In Vitro and In Silico Analysis" Pharmaceutics 14, no. 2: 236. https://doi.org/10.3390/pharmaceutics14020236
APA StyleAlrumaihi, F., Khan, M. A., Babiker, A. Y., Alsaweed, M., Azam, F., Allemailem, K. S., Almatroudi, A. A., Ahamad, S. R., AlSuhaymi, N., Alsugoor, M. H., Algefary, A. N., & Khan, A. (2022). The Effect of Liposomal Diallyl Disulfide and Oxaliplatin on Proliferation of Colorectal Cancer Cells: In Vitro and In Silico Analysis. Pharmaceutics, 14(2), 236. https://doi.org/10.3390/pharmaceutics14020236