
Unique Phenanthrenes from Juncus ensifolius and Their Antiproliferative and Synergistic Effects with the Conventional Anticancer Agent Doxorubicin against Human Cancer Cell Lines

 , ,
, ,  , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. General Procedures
2.2. Plant Material
2.3. Extraction and Isolation
2.4. Physical Characteristics of New Compounds
2.5. Antiproliferative Assays
2.5.1. Cell Lines
2.5.2. Antiproliferative Assay
2.5.3. Drug Combination Assay
3. Results and Discussion
3.1. Structure Elucidation of the Isolated Compounds
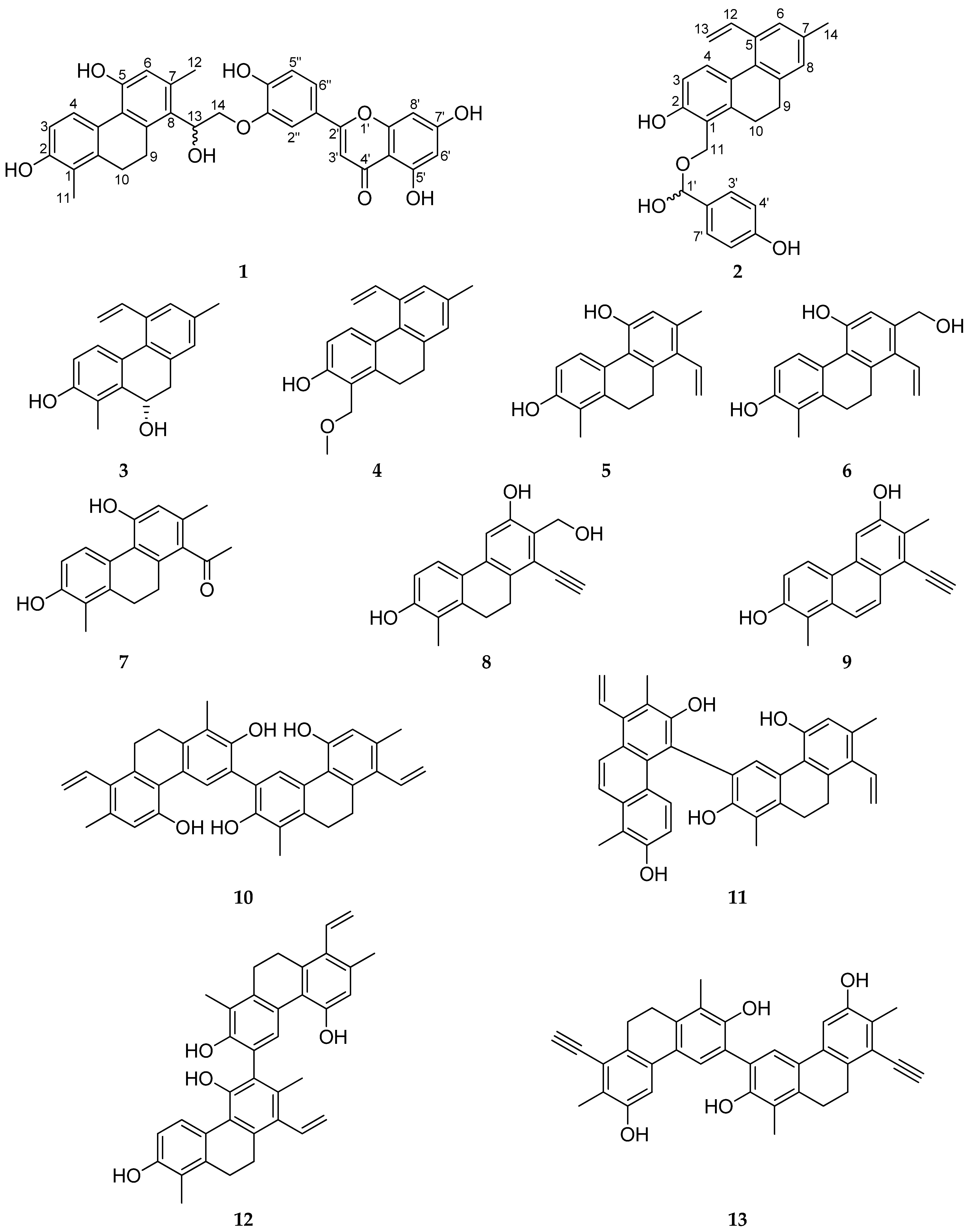
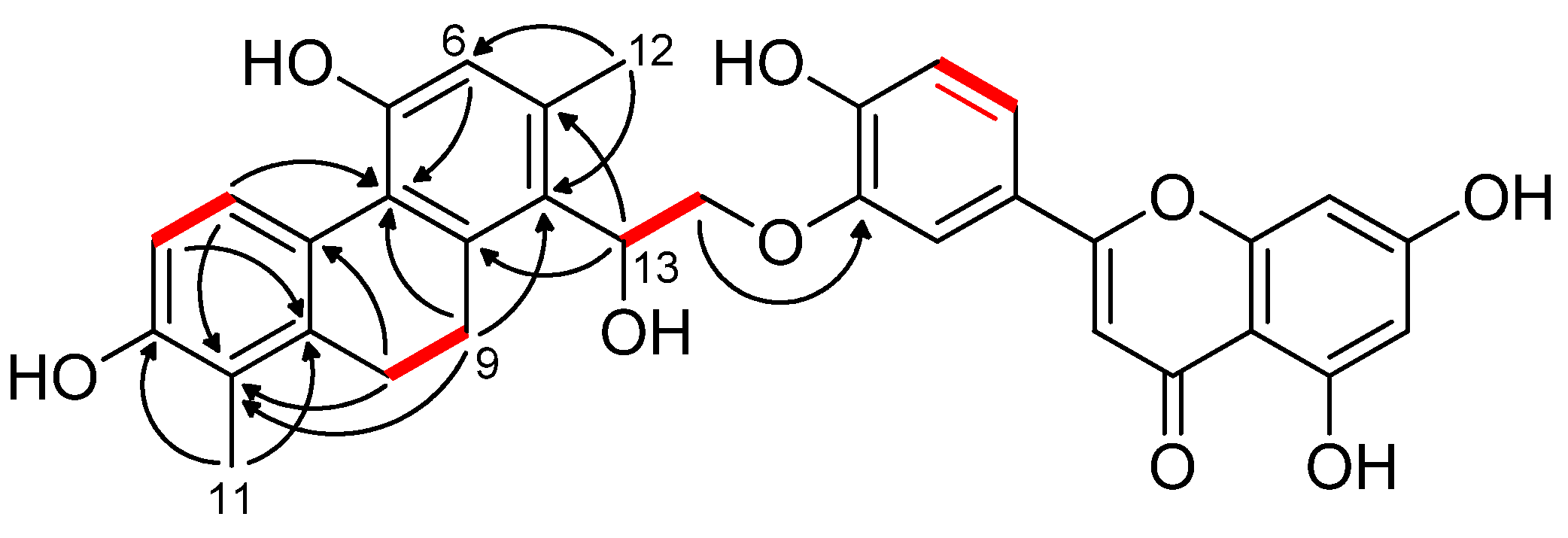
3.1.1. Ensifolin A (1)
3.1.2. Ensifolin B (2)
3.1.3. Ensifolin C (3)
3.1.4. Ensifolin D (4)
3.1.5. Ensifolin E (5)
3.1.6. Ensifolin F (6)
3.1.7. Ensifolin G (7)
3.1.8. Ensifolin H (8)
3.1.9. Ensifolin I (9)
3.1.10. Ensifolin J (10)
3.1.11. Ensifolin K (11)
3.1.12. Ensifolin L (12)
3.1.13. Ensifolin M (13)
3.2. Antiproliferative Activity of the Compounds
3.3. Drug Combination Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Juaid, N.; Amin, A.; Abdalla, A.; Reese, K.; Alamri, Z.; Moulay, M.; Abdu, S.; Miled, N. Anti-hepatocellular carcinoma biomolecules: Molecular targets insights. Int. J. Mol. Sci. 2021, 22, 10774. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, A.; Murali, C.; Amin, A. Safranal inhibits angiogenesis via targeting HIF-1a/VEGF machinery: In vitro and ex vivo insights. Front. Oncol. 2022, 11, 789172. [Google Scholar] [CrossRef]
- Hamza, A.A.; Heeba, G.H.; Hamza, S.; Abdalla, A.; Amin, A. Standardized extract of ginger ameliorates liver cancer by reducing proliferation and inducing apoptosis through inhibition oxidative stress/inflammation pathway. Biomed. Pharmacother. 2021, 134, 111102. [Google Scholar] [CrossRef] [PubMed]
- Kojima-Yuasa, A.; Huang, X.; Matsui-Yuasa, I. Synergistic anticancer activities of natural substances in human hepatocellular carcinoma. Diseases 2015, 3, 260–281. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Phan, H.; Yang, I.X. Improved chemotherapy for hepatocellular carcinoma. Anticancer Res. 2012, 32, 1379–1386. [Google Scholar] [PubMed]
- Lagoa, R.; Silva, J.; Rodrigues, J.R.; Bishayee, A. Advances in phytochemical delivery systems for improved anticancer activity. Biotechnol. Adv. 2020, 38, 107382. [Google Scholar] [CrossRef]
- Baig, B.; Halim, S.A.; Farrukh, A.; Greish, Y.; Amin, A. Current status of nanomaterial-based treatment for hepatocellular carcinoma. Biomed. Pharmacother. 2019, 116, 108852. [Google Scholar] [CrossRef]
- Vasas, A. Phenanthrenes from Orchidaceae and Their Biological Activities. In Orchids Phytochemistry, Biology and Horticulture; Merillon, J.M., Kodja, H., Eds.; Reference Series in Phytochemistry; Springer: Cham, Switzerland, 2021; pp. 1–41. [Google Scholar] [CrossRef]
- Bús, C.; Tóth, B.; Stefkó, D.; Hohmann, J.; Vasas, A. Family Juncaceae: Promising source of biologically active natural phenanthrenes. Phytochem. Rev. 2018, 17, 833–851. [Google Scholar] [CrossRef] [Green Version]
- Tóth, B.; Hohmann, J.; Vasas, A. Phenanthrenes: A promising group of plant secondary metabolites. J. Nat. Prod. 2018, 81, 661–678. [Google Scholar] [CrossRef]
- Liu, W.; Meng, M.; Zhang, B.; Du, L.; Pan, Y.; Yang, P.; Gu, Z.; Zhou, Q.; Cao, Z. Dehydroeffusol effectively inhibits human gastric cancer cell-mediated vasculogenic mimicry with low toxicity. Toxicol. Appl. Pharmacol. 2015, 287, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Han, H.; Fu, S.; Yang, P.; Gu, Z.; Zhou, Q.; Cao, Z. Dehydroeffusol inhibits gastric cancer cell growth and tumorigenicity by selectively inducing tumor-suppressive endoplasmic reticulum stress and a moderate apoptosis. Biochem. Pharmacol. 2016, 104, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.Y.; Schelz, Z.; Tóth, B.; Vasas, A.; Ocsovszki, I.; Chang, F.R.; Hohmann, J.; Zupkó, I.; Wang, H.C. Investigation of natural phenanthrenes and the antiproliferative potential of juncusol in cervical cancer cell lines. Phytomedicine 2019, 58, 152770. [Google Scholar] [CrossRef] [PubMed]
- Bús, C.; Kulmány, Á.; Kúsz, N.; Gonda, T.; Zupkó, I.; Mándi, A.; Kurtán, T.; Tóth, B.; Hohmann, J.; Hunyadi, A.; et al. Oxidized juncuenin B analogues with increased antiproliferative activity on human adherent cell lines: Semisynthesis and biological evaluation. J. Nat. Prod. 2020, 83, 3250–3261. [Google Scholar] [CrossRef] [PubMed]
- Tóth, B.; Chang, F.R.; Hwang, T.L.; Szappanos, Á.; Mándi, A.; Hunyadi, A.; Kurtán, T.; Jakab, G.; Hohmann, J.; Vasas, A. Screening of Luzula species native to the Carpathian Basin for anti-inflammatory activity and bioactivity-guided isolation of compounds from Luzula luzuloides (Lam.) Dandy & Wilmott. Fitoterapia 2017, 116, 131–138. [Google Scholar]
- Juncus ensifolius. Invasive Species Compendium. Available online: https://www.cabi.org/isc/datasheet/115030#147582B0-EA1E-4137-A35A-F2A22FA1DBF2 (accessed on 9 January 2022).
- Reagan, A.B. Plants used by the Hoh and Quileute Indians. Trans. Kans. Acad. Sci. 1936, 37, 55–70. [Google Scholar] [CrossRef]
- Gunther, E. Ethnobotany of Western Washington (Revised Edition); University of Washington Press: Seattle, WA, USA, 1973. [Google Scholar]
- Bacher, F.; Wittmann, C.; Nové, M.; Spengler, G.; Marć, M.A.; Enyedy, E.A.; Darvasiová, D.; Rapta, P.; Reinere, T.; Arion, V.B. Novel latonduine derived proligands and their copper(II) complexes show cytotoxicity in the nanomolar range in human colon adenocarcinoma cells and in vitro cancer selectivity. Dalton Trans. 2019, 48, 10464. [Google Scholar] [CrossRef] [Green Version]
- CompuSyn; Version 1.00; ComboSyn, Inc.: Paramus, NJ, USA, 2005.
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Chou, T.C. Drug Combination Studies and their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Stefkó, D.; Kúsz, N.; Csorba, A.; Jakab, G.; Bérdi, P.; Zupkó, I.; Hohmann, J.; Vasas, A. Phenanthrenes from Juncus atratus with antiproliferative activity. Tetrahedron 2019, 75, 116–120. [Google Scholar] [CrossRef]
- Bús, C.; Kúsz, N.; Jakab, G.; Senobar Tahaei, S.A.; Zupkó, I.; Endrész, V.; Bogdanov, A.; Burián, K.; Csupor-Löffler, B.; Hohmann, J.; et al. Phenanthrenes from Juncus compressus Jacq. with promising antiproliferative and anti-HSV-2 activities. Molecules 2018, 23, 2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.C.; Pai, Y.F.; Tsai, T.H. Isolation of luteolin and luteolin-7-O-glucoside from Dendranthema morifolium Ramat Tzvel and their pharmacokinetics in rats. J. Agric. Food Chem. 2015, 63, 7700–7706. [Google Scholar] [CrossRef] [PubMed]
- Gainche, M.; Ripoche, I.; Senejoux, F.; Cholet, J.; Ogeron, C.; Decombat, C.; Danton, O.; Delort, L.; Vareille-Delarbre, M.; Berry, A.; et al. Anti-inflammatory and cytotoxic potential of new phenanthrenoids from Luzula sylvatica. Molecules 2020, 25, 2372. [Google Scholar] [CrossRef] [PubMed]
- Resnick, S.M.; Gibson, D.T. Regio- and stereospecific oxidation of 9,10-dihydroanthracene and 9,10-dihydrophenanthrene by naphthalene dioxygenase: Structure and absolute stereochemistry of metabolites. Appl. Environ. Microbiol. 1996, 62, 3355–3359. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.Y.; Ke, C.Q.; Tang, C.P.; Yuan, D.; Ye, Y. 9,10-Dihydrophenanthrenes and phenanthrenes from Juncus setchuensis. J. Nat. Prod. 2009, 72, 1209–1212. [Google Scholar] [CrossRef] [PubMed]
- Della Greca, M.; Fiorentino, A.; Monaco, P.; Pinto, G.; Pollio, A.; Previtera, L. Action of antialgal compounds from Juncus effusus L. on Selenastrum capricornutum. J. Chem. Ecol. 1996, 22, 587–603. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.J.U.; Duarte, N.; Reis, M.; Madureira, A.M.; Molnar, J. Euphorbia and Momordica metabolites for overcoming multidrug resistance. Phytochem. Rev. 2014, 13, 915–935. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Position | 1 a | 2 b | 3 a | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | |
| 1 | 121.1, C | 117.5, C | 123.5, C | |||
| 1a | 139.5, C | 135.9, C | 139.4, C | |||
| 2 | 155.0, C | 152.4, C | 155.9, C | |||
| 3 | 6.66, d (8.6) | 112.8, CH | 6.86, d (8.6) | 114.1, CH | 6.76, d (8.4) | 114.3, CH |
| 4 | 7.97, d (8.6) | 128.1, CH | 7.53, d (8.6) | 129.3, CH | 7.38, d (8.4) | 129.2, CH |
| 4a | 125.8, C | 127.6, C | 126.4, C | |||
| 5a | 123.2, C | 130.6, C | 132.0, C | |||
| 5 | 155.3, C | 135.3, C | 136.0, C | |||
| 6 | 6.67, s | 118.5, CH | 7.25, br s | 127.7, CH | 7.21, br s | 128.5, CH |
| 7 | 136.8, C | 136.3, C | 136.8, C | |||
| 8 | 122.1, C | 7.02, br s | 127.9, CH | 7.03, br s | 130.5, CH | |
| 8a | 140.9, C | 138.5, C | 134.8, C | |||
| 9 | 2.77, m 2.87, m | 27.8, CH2 | 2.73, m (2H) | 29.6, CH2 | 2.84, dd (16.0, 3.0) 3.05, dd (16.0, 2.8) | 39.1, CH2 |
| 10 | 2.64, m (2H) | 26.5, CH2 | 2.54, m (2H) | 24.1, CH2 | 5.10, br t (2.8) | 64.3, CH |
| 11 | 2.18, s (3H) | 11.6, CH3 | 5.09, d (14.5) 5.19, d (14.5) | 66.2, CH2 | 2.33 *, s (3H) | 11.1, CH3 |
| OCH3–11 | ||||||
| 12 | 2.38, s (3H) | 20.7, CH3 | 6.98, dd (17.4, 10.9) | 138.9, CH | 7.02, dd (17.4, 10.9) | 140.9, CH |
| 13 | 5.65, dd (9.9, 2.9) | 75.6, CH | 5.28, dd (10.9, 1.3) 5.72, dd (17.4, 1.3) | 114.7, CH2 | 5.21, dd (10.9, 1.4) 5.66, dd (17.4, 1.4) | 113.6, CH2 |
| 14 | 4.32, dd (11.9, 2.9) 4.42, dd (11.9, 9.9) | 67.1, CH2 | 2.37, s (3H) | 21.2, CH3 | 2.33 *, s (3H) | 21.1, CH3 |
| Position | 4 | 5 | 6 | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | |
| 1 | 121.9, C | 121.2, C | 121.2, C | |||
| 1a | 142.8, C | 139.5, C | 139.7, C | |||
| 2 | 156.5, C | 154.7, C | 154.9, C | |||
| 3 | 6.71, d (8.5) | 113.0, CH | 6.65, d (8.6) | 112.7, CH | 6.65, d (8.6) | 112.7, CH |
| 4 | 7.40, d (8.5) | 131.6, CH | 7.98, d (8.6) | 127.9, CH | 8.03, d (8.6) | 128.1, CH |
| 4a | 127.2, C | 126.5, C | 126.2, C | |||
| 5a | 132.4, C | 121.7, C | 123.0, C | |||
| 5 | 135.9, C | 153.8, C | 154.2, C | |||
| 6 | 7.22, br s | 128.0, CH | 6.61, s | 117.0, CH | 6.91, s | 114.9, CH |
| 7 | 136.8, C | 135.6, C | 138.54 #, C | |||
| 8 | 7.00, br s | 128.5, CH | 129.4, C | 128.5, C | ||
| 8a | 140.0, C | 138.4, C | 138.57 #, C | |||
| 9 | 2.66, m (2H) | 31.0, CH2 | 2.72, m (2H) | 28.5, CH2 | 2.74, m (2H) | 28.2, CH2 |
| 10 | 2.77, m (2H) | 26.5, CH2 | 2.60, m (2H) | 26.7, CH2 | 2.63, m (2H) | 26.6, CH2 |
| 11 | 4.66, s (2H) | 66.5, CH2 | 2.18, s (3H) | 11.7, CH3 | 2.19, s (3H) | 11.7, CH3 |
| OCH3–11 | 3.40, s (3H) | 58.1, CH3 | ||||
| 12 | 6.92, dd (17.5, 10.9) | 140.3, CH | 2.22, s (3H) | 20.8, CH3 | 4.58, s (2H) | 63.3, CH2 |
| 13 | 5.21, dd (10.9, 1.6) 5.68, dd (17.5, 1.6) | 113.8, CH2 | 6.72, dd (17.9, 11.3) | 136.5, CH | 6.79, dd (17.8, 11.3) | 135.5, CH |
| 14 | 2.33, s (3H) | 21.1, CH3 | 5.09, dd (17.9, 2.3) 5.48, dd (11.3, 2.3) | 119.5, CH2 | 5.17, dd (17.8, 2.2) 5.51, dd (11.3, 2.2) | 120.1, CH2 |
| Position | 7 a | 8 a | 9 b | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | |
| 1 | 121.5, C | 122.7, C | 121.5, C | |||
| 1a | 139.1, C | 138.7, C | 139.1, C | |||
| 2 | 155.1, C | 156.5, C | 155.1, C | |||
| 3 | 6.66, d (8.6) | 112.9, CH | 6.72, d (8.4) | 114.1, CH | 6.66, d (8.6) | 112.9, CH |
| 4 | 8.03, d (8.6) | 128.1, CH | 7.38, d (8.4) | 123.2, CH | 8.03, d (8.6) | 128.1, CH |
| 4a | 125.5, C | 127.0, C | 125.5, C | |||
| 5a | 121.8, C | 137.5, C | 121.8, C | |||
| 5 | 155.5, C | 7.16, s | 112.2, CH | 155.5, C | ||
| 6 | 6.61, s | 117.4, CH | 156.1, C | 6.61, s | 117.4, CH | |
| 7 | 132.4, C | 127.2, C | 132.4, C | |||
| 8 | 134.3, C | 122.2, C | 134.3, C | |||
| 8a | 135.8, C | 131.3, C | 135.8, C | |||
| 9 | 2.55, m (2H) | 28.3, CH2 | 2.94, m (2H) | 27.2, CH2 | 2.55, m (2H) | 28.3, CH2 |
| 10 | 2.68, m (2H) | 26.3, CH2 | 2.76, m (2H) | 26.1, CH2 | 2.68, m (2H) | 26.3, CH2 |
| 11 | 2.18, s (3H) | 11.7, CH3 | 2.19, s (3H) | 11.5, CH3 | 2.18, s (3H) | 11.7, CH3 |
| 12 | 2.17, s (3H) | 19.1, CH3 | 4.93, s (2H) | 60.1, CH2 | 2.17, s (3H) | 19.1, CH3 |
| 13 | 211.4, C | 81.1, C | 211.4, C | |||
| 14 | 2.46, s (3H) | 33.1, CH3 | 3.85, s | 86.4, CH | 2.46, s (3H) | 33.1, CH3 |
| Position | 10 | 13 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | |
| 1, 1′ | 122.9, C | 124.5, C | ||
| 1a, 1a′ | 139.0, C | 137.7, C | ||
| 2, 2′ | 151.4, C | 153.3 +, C | ||
| 3, 3′ | 124.9, C | 126.8, C | ||
| 4, 4′ | 8.12, s | 130.3, CH | 7.48, s | 125.3, CH |
| 4a, 4a′ | 127.4, C | 128.3 +, C | ||
| 5a, 5a′ | 121.5, C | 134.9, C | ||
| 5, 5′ | 153.9, C | 7.17, s | 111.3, CH | |
| 6, 6′ | 6.60, s | 117.0, CH | 155.0, C | |
| 7, 7′ | 136.0, C | 126.4, C | ||
| 8, 8′ | 129.5, C | 123.0, C | ||
| 8a, 8a′ | 138.6, C | 131.2, C | ||
| 9, 9′ | 2.82, m (2H) | 28.5, CH2 | 2.99, m (2H) | 27.5, CH2 |
| 10, 10′ | 2.70, m (2H) | 26.9, CH2 | 2.84, m (2H) | 26.4, CH2 |
| 11, 11′ | 2.32, s (3H) | 12.4, CH3 | 2.32, s (3H) | 12.3, CH3 |
| 12, 12′ | 2.23, s (3H) | 20.8, CH3 | 2.33, s (3H) | 14.1, CH3 |
| 13, 13′ | 6.76, dd (17.9, 11.3) | 136.5, CH | 82.2, C | |
| 14, 14′ | 5.13, dd (17.9, 2.2) 5.52, dd (11.3, 2.2) | 119.6, CH2 | 3.85, s | 86.1, C |
| Position | 11 | 12 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | |
| 1 | 117.4, C | 121.2, C | ||
| 1a | 134.9, C | 139.82 #, C | ||
| 2 | 153.47 #, C | 154.9, C | ||
| 3 | 6.53, d (9.4) | 115.4, CH | 6.64, d (8.6) | 112.7, CH |
| 4 | 7.56, d (9.4) | 127.1, CH | 8.02, d (8.6) | 128.2, CH |
| 4a | 125.82, C | 126.5, C | ||
| 5a | 131.0, C | 122.1, C | ||
| 5 | 118.6, C | 151.3, C | ||
| 6 | 153.37 #, C | 125.3, C | ||
| 7 | 123.3, C | 135.4, C | ||
| 8 | 138.8, C | 130.49, C | ||
| 8a | 125.5, C | 137.9, C | ||
| 9 | 8.04, d (9.5) | 125.80, CH | 2.87, m (2H) | 28.4, CH2 |
| 10 | 7.72, d (9.5) | 120.9, CH | 2.69 *, m (2H) | 26.8, CH2 |
| 11 | 2.47, s (3H) | 11.2, CH3 | 2.21, s (3H) | 11.7, CH3 |
| 12 | 2.44, s (3H) | 14.5, CH3 | 2.03, s (3H) | 18.5, CH3 |
| 13 | 7.19, dd (17.9, 11.4) | 136.9, CH | 6.82, dd (17.8, 11.2) | 137.3, CH |
| 14 | 5.41, dd (17.9, 2.2) 5.85, dd (11.4, 2.2) | 121.8, CH2 | 5.16, dd (17.8, 2.4) 5.54, dd (11.2, 2.4) | 119.9, CH2 |
| 1′ | 123.4, C | 122.7, C | ||
| 1a′ | 140.5, C | 139.89 #, C | ||
| 2′ | 152.2, C | 152.6, C | ||
| 3′ | 123.6, C | 120.8, C | ||
| 4′ | 7.96, s | 130.0, CH | 7.92, s | 130.38, CH |
| 4a′ | 128.6, C | 127.3, C | ||
| 5a′ | 121.2, C | 121.4, C | ||
| 5′ | 153.9, C | 153.9, C | ||
| 6′ | 6.55, s | 117.0, CH | 6.61, s | 117.1, CH |
| 7′ | 136.2, C | 136.0, C | ||
| 8′ | 129.4, C | 129.5, C | ||
| 8a′ | 138.4, C | 138.5, C | ||
| 9′ | 2.77, m 3.05, ddd (15.1, 6.9, 4.3) | 28.6, CH2 | 2.78 *, m (2H | 28.8, CH2 |
| 10′ | 2.67, m 2.94, ddd (11.1, 6.9, 4.3) | 27.1, CH2 | 2.67 *, m 2.77 *, m | 27.0, CH2 |
| 11′ | 2.32, s (3H) | 12.4, CH3 | 2.30, s (3H) | 12.3, CH3 |
| 12′ | 2.22, s (3H) | 20.8, CH3 | 2.23, s (3H) | 20.8, CH3 |
| 13′ | 6.78, dd (17.9, 11.3) | 136.4, CH | 6.76, dd (17.8, 11.3) | 136.4, CH |
| 14′ | 5.16, dd (17.9, 2.2) 5.54, dd (11.3, 2.2) | 119.7, CH2 | 5.13, dd (17.8, 2.2) 5.52, dd (11.3, 2.2) | 119.6, CH2 |
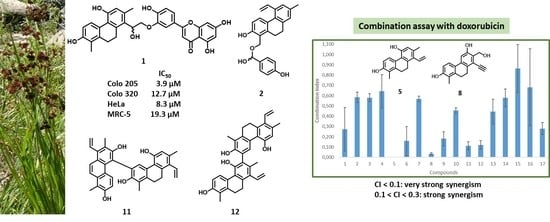
| Compound | IC50 (µM) ± SD | SI MRC-5/COLO 205 | SI MRC-5/COLO 320 | SI MRC-5/HeLa | |||
|---|---|---|---|---|---|---|---|
| COLO 205 | COLO 320 | HeLa | MRC-5 | ||||
| 1 | 3.86 ± 0.08 | 12.71 ± 0.05 | 8.25 ±0.51 | 19.29 ± 0.54 | 5.00 | 1.52 | 2.34 |
| 2 | 45.64 ± 0.50 | 37.24 ± 0.11 | 33.49 ± 0.29 | 51.87 ± 0.14 | 1.14 | 1.39 | 1.55 |
| 3 | >100 | >100 | >100 | >100 | |||
| 4 | 65.61 ± 0.78 | 61.56 ± 9.95 | 19.40 ± 0.33 | >100 | >1.52 | >1.62 | >5.15 |
| 5 | 31.23 ± 0.66 | 25.17 ± 0.92 | 27.46 ± 1.19 | 44.31 ± 0.61 | 1.42 | 1.76 | 1.61 |
| 6 | >100 | 93.71 ± 0.14 | 74.32 ± 2.98 | >100 | >1.07 | >1.35 | |
| 7 | >100 | >100 | 75.57 ± 0.94 | >100 | >1.32 | ||
| 8 | >100 | 63.46 ± 2.70 | 12.31 ± 0.13 | >100 | >1.58 | >8.12 | |
| 9 | 18.21 ± 0.28 | 18.52 ± 0.06 | 24.09 ± 0.11 | 49.14 ± 0.83 | 2.70 | 2.65 | 2.04 |
| 10 | 44.48 ± 1.22 | 42.76 ± 1.28 | 33.54 ± 1.89 | 57.75 ± 1.32 | 1.30 | 1.35 | 1.72 |
| 11 | 31.38 ± 0.72 | 37.84 ± 1.05 | 29.53 ± 0.31 | 33.16 ± 0.05 | 1.06 | 0.88 | 1.12 |
| 12 | 26.91 ± 1.19 | 37.36 ± 2.13 | 30.22 ± 0.21 | 50.36 ± 1.30 | 1.87 | 1.35 | 1.67 |
| 13 | 42.72 ± 0.92 | 37.27 ± 0.55 | 31.51 ± 0.53 | 72.54 ± 1.56 | 1.70 | 1.95 | 2.30 |
| 14 | 32.92 ± 0.59 | 52.36 ± 0.77 | 58.09 ± 1.20 | 60.89 ± 0.25 | 1.85 | 1.16 | 1.05 |
| 15 | 37.08 ± 0.57 | 30.54 ± 0.93 | 6.67 ± 0.03 | 35.85 ± 1.23 | 0.97 | 1.17 | 5.37 |
| 16 | 34.42 ± 0.57 | 32.48 ± 0.75 | 6.65 ± 0.10 | 26.03 ± 0.85 | 0.76 | 0.80 | 3.91 |
| 17 | 56.73 ± 0.75 | 57.66 ± 0.92 | 10.56 ± 0.09 | >100 | >1.76 | >1.73 | >9.47 |
| 18 | >100 | >100 | >100 | >100 | |||
| 19 | >100 | >100 | >100 | >100 | |||
| DMSO | >1% | >1% | >1% | >1% | |||
| cisplatin | 41.67 ± 1.62 | 2.14 ± 0.32 | 3.62 ± 0.16 | 2.36 ± 0.33 | |||
| doxorubicin | 1.36 ± 0.36 | 0.22 ± 0.004 | 0.04 ± 0.004 | 0.53 ± 0.06 | |||
| Compound | CI | SD | Ratio | Interaction |
|---|---|---|---|---|
| 1 | 0.272 | 0.2124 | 9.28:1 | strong synergism |
| 2 | 0.584 | 0.0510 | 23.2:1 | synergism |
| 3 | 0.580 | 0.0387 | 13.92:1 | synergism |
| 4 | 0.643 | 0.1623 | 55.68:1 | synergism |
| 5 | 0.001 | 0.0003 | 9.28:1 | very strong synergism |
| 6 | 0.159 | 0.1414 | 23.2:1 | strong synergism |
| 7 | 0.568 | 0.0268 | 46.4:1 | synergism |
| 8 | 0.033 | 0.0106 | 185.6:1 | very strong synergism |
| 9 | 0.180 | 0.0675 | 9.28:1 | strong synergism |
| 10 | 0.454 | 0.0269 | 38.4:1 | synergism |
| 11 | 0.112 | 0.0387 | 10.44:1 | strong synergism |
| 12 | 0.120 | 0.0418 | 11.6:1 | strong synergism |
| 13 | 0.445 | 0.1202 | 46.4:1 | synergism |
| 14 | 0.579 | 0.0855 | 92.8:1 | synergism |
| 15 | 0.864 | 0.2338 | 27.84:1 | slight synergism |
| 16 | 0.682 | 0.3743 | 13.92:1 | synergism |
| 17 | 0.279 | 0.0574 | 13.92:1 | strong synergism |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stefkó, D.; Kúsz, N.; Szemerédi, N.; Barta, A.; Spengler, G.; Berkecz, R.; Hohmann, J.; Vasas, A. Unique Phenanthrenes from Juncus ensifolius and Their Antiproliferative and Synergistic Effects with the Conventional Anticancer Agent Doxorubicin against Human Cancer Cell Lines. Pharmaceutics 2022, 14, 608. https://doi.org/10.3390/pharmaceutics14030608
Stefkó D, Kúsz N, Szemerédi N, Barta A, Spengler G, Berkecz R, Hohmann J, Vasas A. Unique Phenanthrenes from Juncus ensifolius and Their Antiproliferative and Synergistic Effects with the Conventional Anticancer Agent Doxorubicin against Human Cancer Cell Lines. Pharmaceutics. 2022; 14(3):608. https://doi.org/10.3390/pharmaceutics14030608
Chicago/Turabian StyleStefkó, Dóra, Norbert Kúsz, Nikoletta Szemerédi, Anita Barta, Gabriella Spengler, Róbert Berkecz, Judit Hohmann, and Andrea Vasas. 2022. "Unique Phenanthrenes from Juncus ensifolius and Their Antiproliferative and Synergistic Effects with the Conventional Anticancer Agent Doxorubicin against Human Cancer Cell Lines" Pharmaceutics 14, no. 3: 608. https://doi.org/10.3390/pharmaceutics14030608
APA StyleStefkó, D., Kúsz, N., Szemerédi, N., Barta, A., Spengler, G., Berkecz, R., Hohmann, J., & Vasas, A. (2022). Unique Phenanthrenes from Juncus ensifolius and Their Antiproliferative and Synergistic Effects with the Conventional Anticancer Agent Doxorubicin against Human Cancer Cell Lines. Pharmaceutics, 14(3), 608. https://doi.org/10.3390/pharmaceutics14030608