
Effects of Injection Volume and Route of Administration on Dolutegravir In Situ Forming Implant Pharmacokinetics

 ,
, 
Abstract
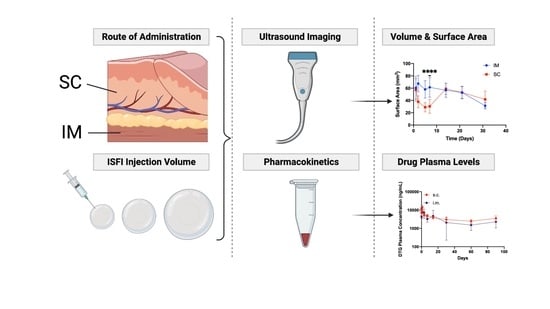
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. High-Performance Liquid Chromatography (HPLC)
2.2.2. Preparation of ISFI Formulations
2.2.3. In Vitro Drug Release from DTG ISFIs
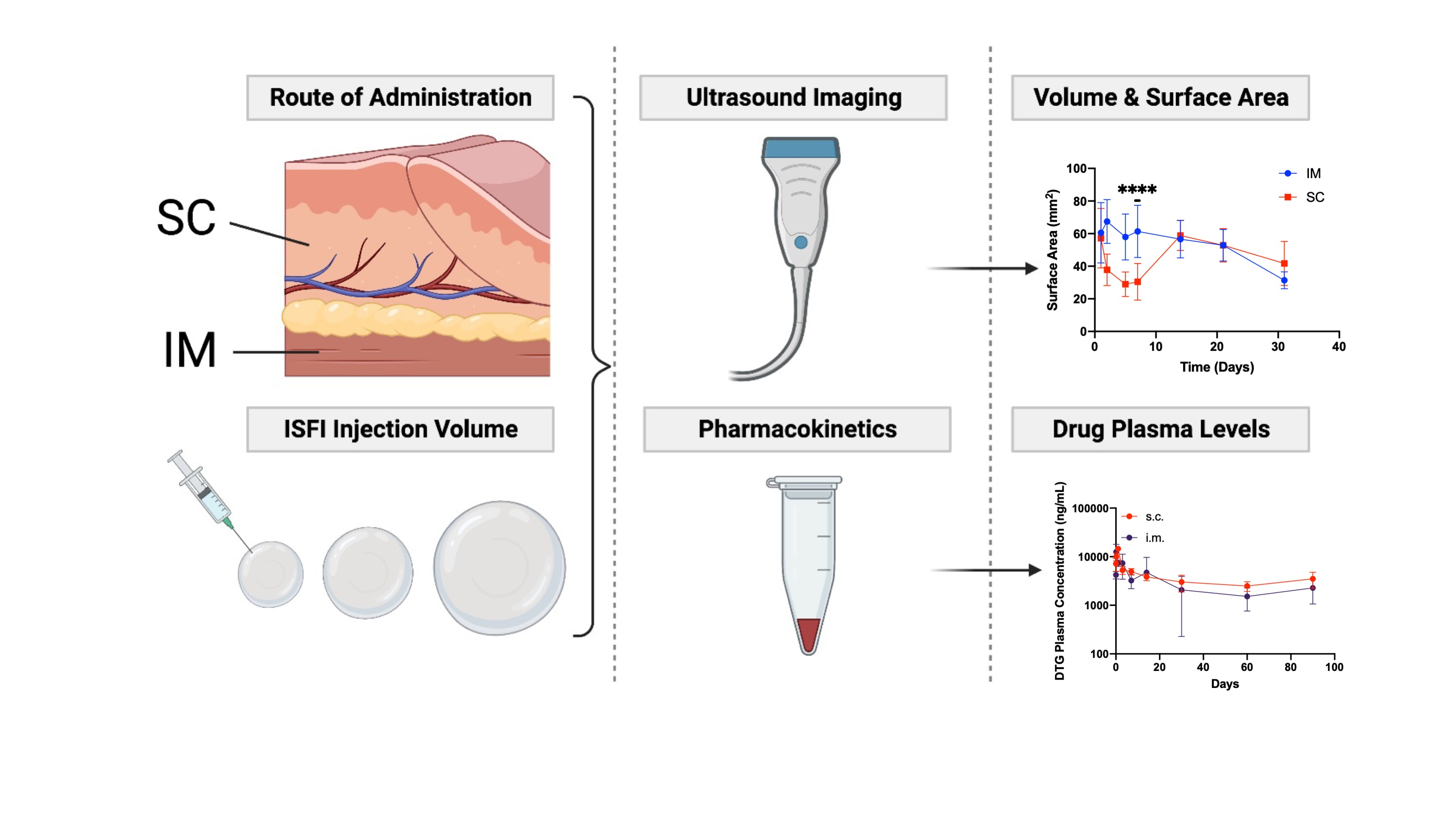
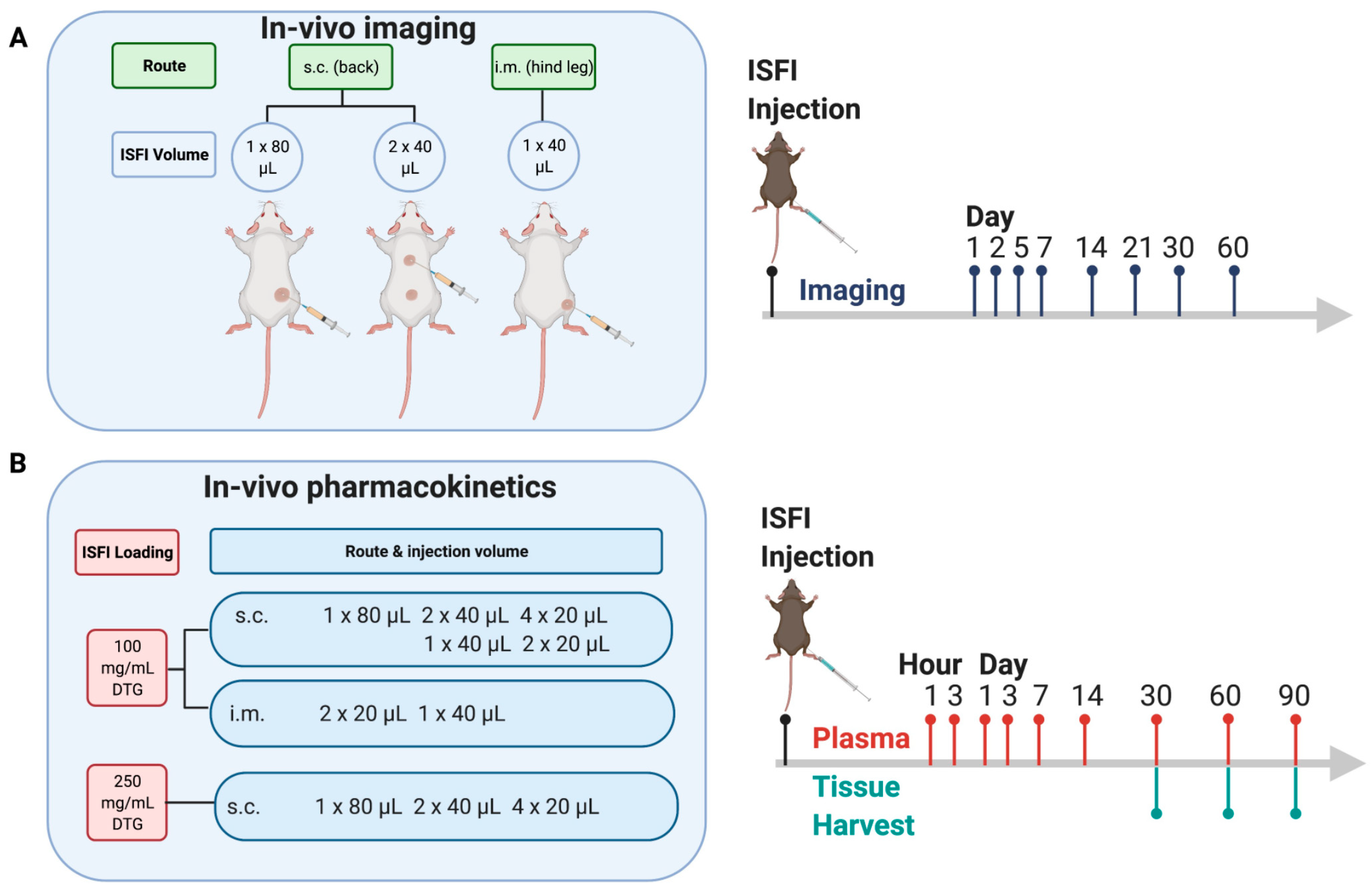
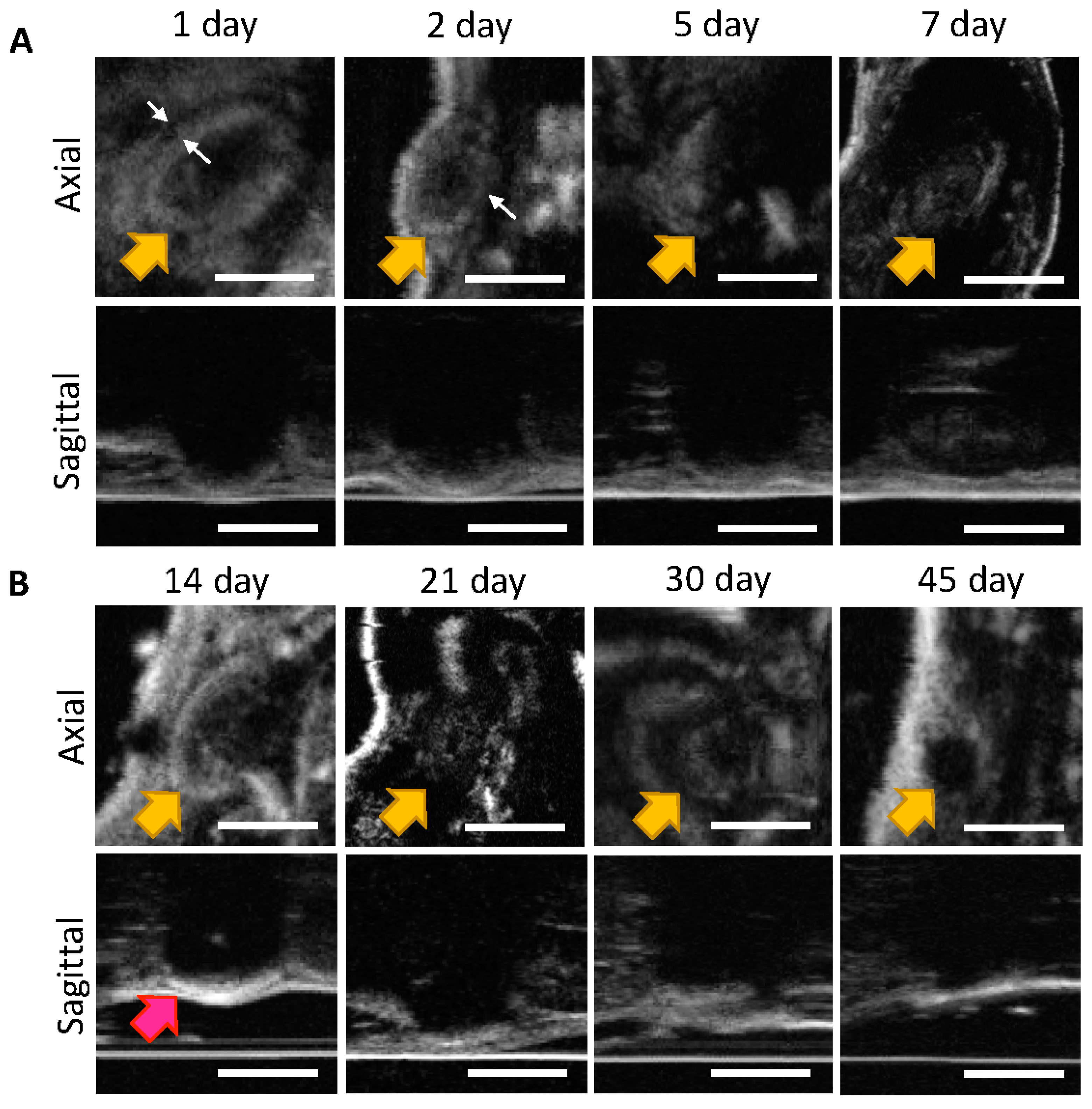
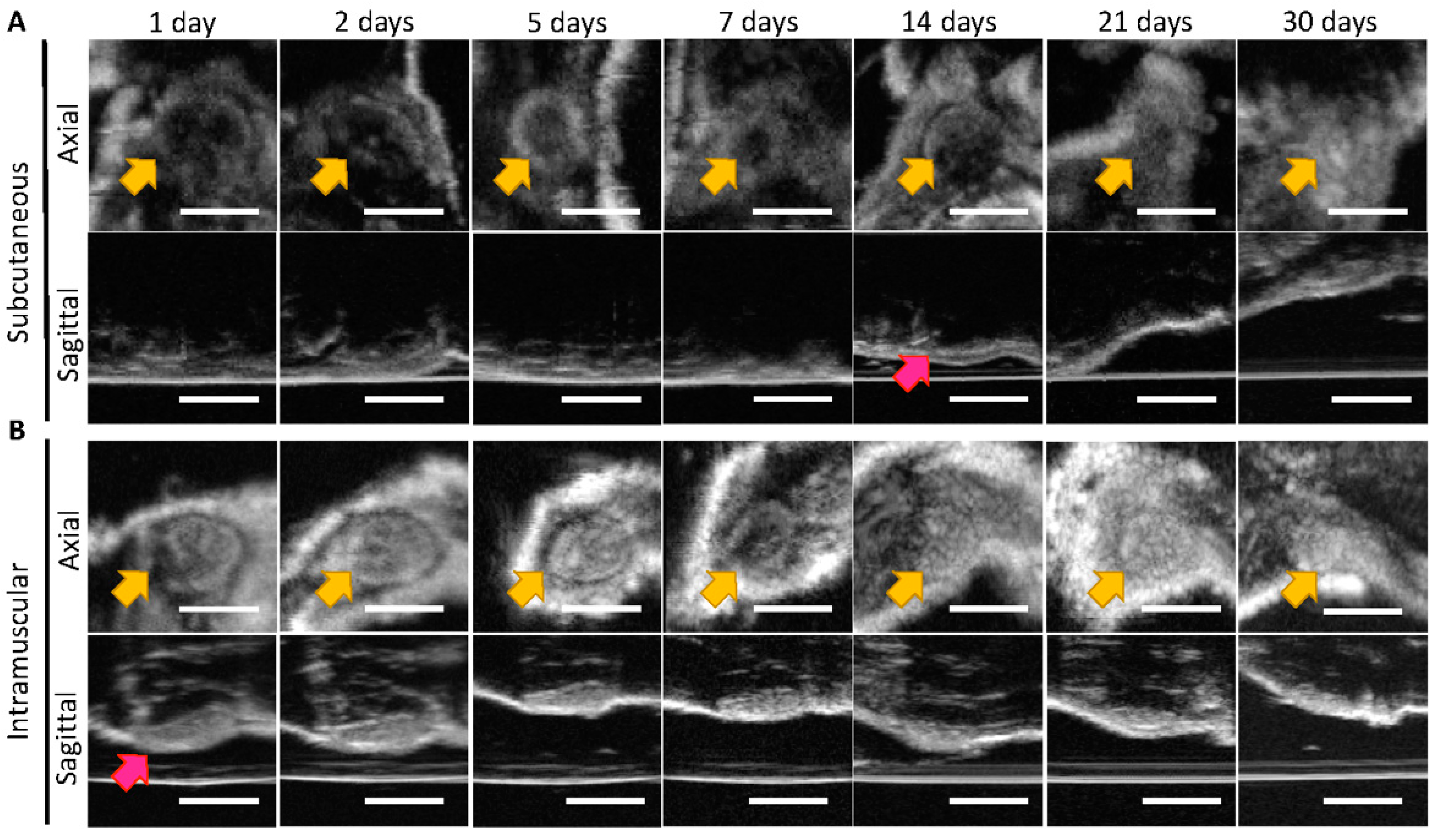
2.2.4. Ultrasound Imaging of ISFIs in Mice
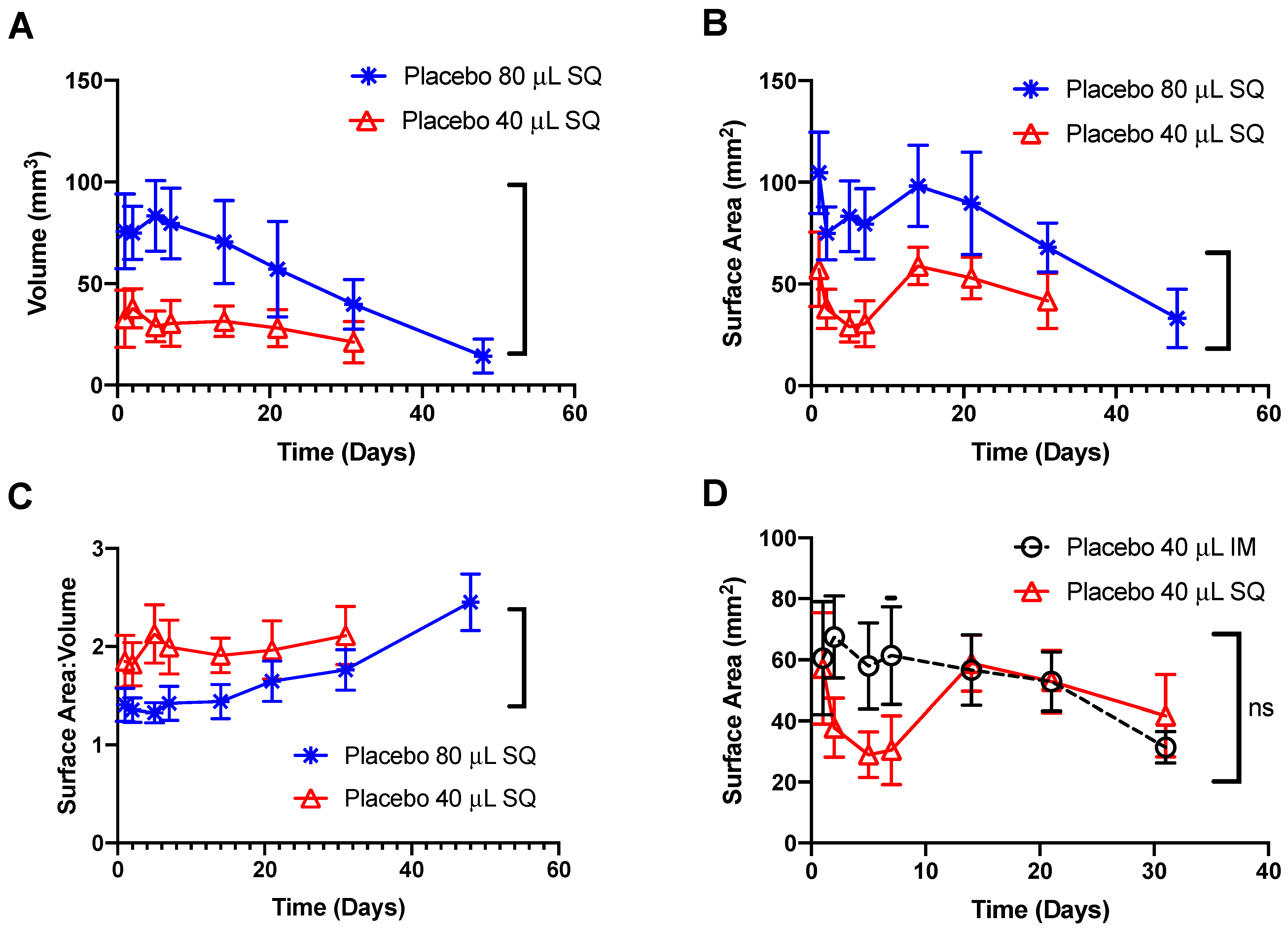
2.2.5. Statistical Analysis of Ultrasound Images
2.2.6. In Vivo Pharmacokinetic Studies
2.2.7. Mass Spectrometry of Plasma Samples
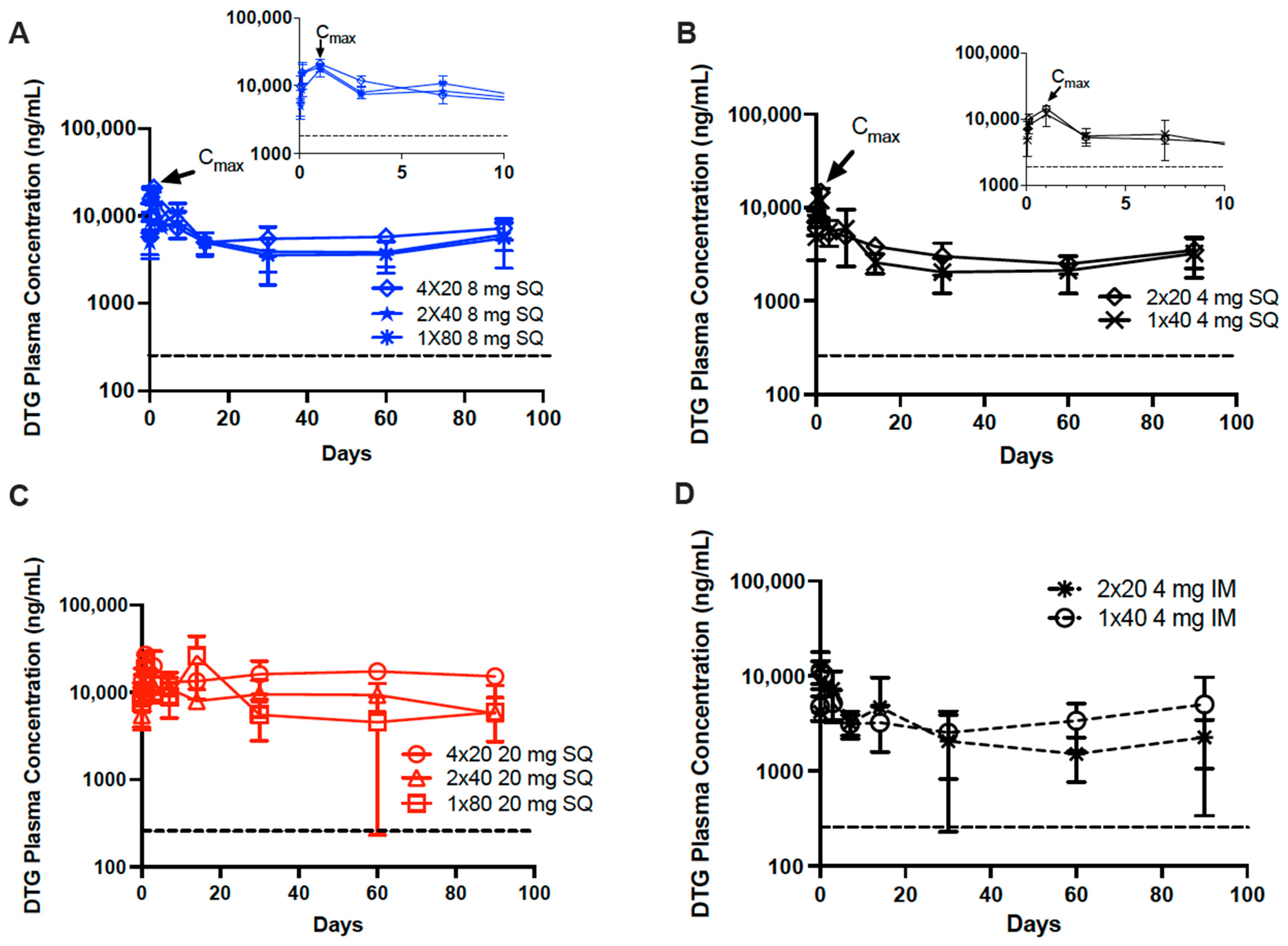
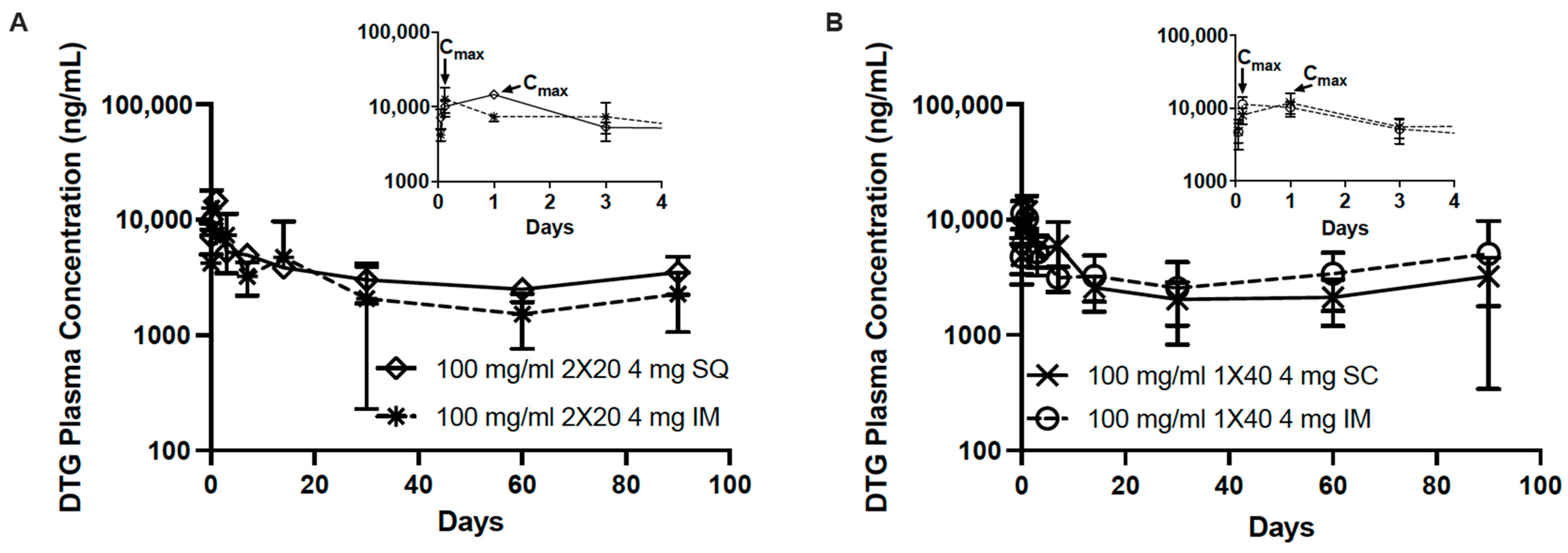
2.2.8. Noncompartmental Analysis of Pharmacokinetic Data
2.2.9. Deconvolution Analysis of Pharmacokinetic Data
2.2.10. Statistical Analysis of Pharmacokinetic Data
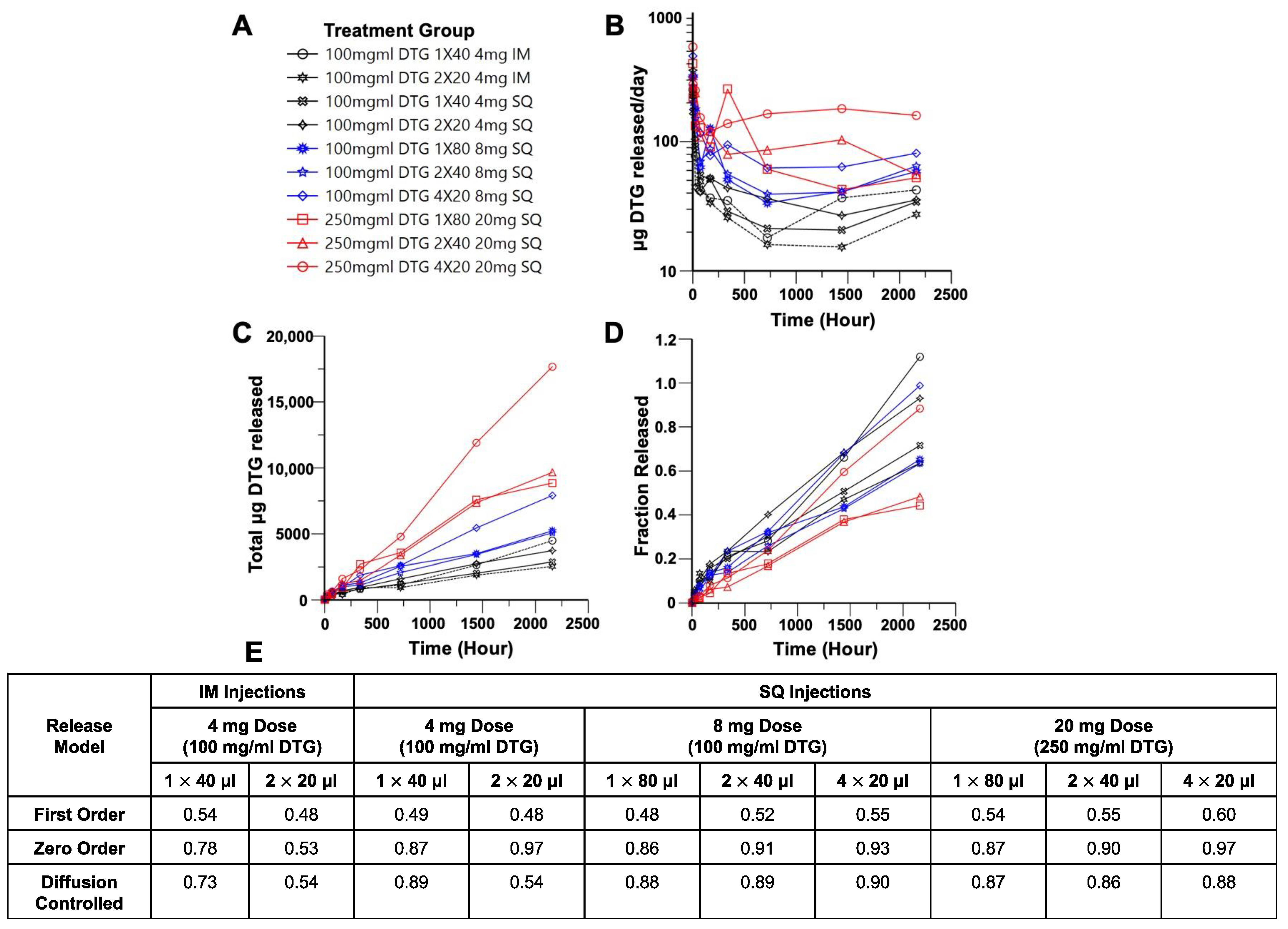
3. Results
3.1. In Vivo Imaging of Placebo ISFIs
3.2. In Vivo Pharmacokinetics of Dolutegravir ISFIs
3.3. Deconvolution Analysis of Dolutegravir ISFIs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Orkin, C.; Arasteh, K.; Górgolas Hernández-Mora, M.; Pokrovsky, V.; Overton, E.T.; Girard, P.-M.; Oka, S.; Walmsley, S.; Bettacchi, C.; Brinson, C.; et al. Long-Acting Cabotegravir and Rilpivirine after Oral Induction for HIV-1 Infection. N. Engl. J. Med. 2020, 382, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, K.A.; Vike, N.; Li, X.; Kennedy, J.; Simmons, E.; Rispoli, J.; Solorio, L. Noninvasive characterization of in situ forming implant diffusivity using diffusion-weighted MRI. J. Control Release 2019, 309, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.B.; Solorio, L.; Wu, H.; Krupka, T.; Exner, A.A. Effect of injection site on in situ implant formation and drug release In Vivo. J. Control Release 2010, 147, 350–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Mu, H.; Larsen, S.W.; Jensen, H.; Østergaard, J. Initial Leuprolide Acetate Release from Poly(d,L-lactide-co-glycolide) in Situ Forming Implants as Studied by Ultraviolet-Visible Imaging. Mol. Pharm. 2020, 17, 4522–4532. [Google Scholar] [CrossRef] [PubMed]
- Tipnis, N.; Zou, Y.; Wang, Y.; Choi, S.; Burgess, D.J. Effect of implant formation on drug release kinetics of in situ forming implants. Int. J. Pharm. 2021, 592, 120105. [Google Scholar]
- Zhang, Q.; Fassihi, R. Release rate determination from in situ gel forming PLGA implant: A novel ‘shape-controlled basket in tube’ method. J. Pharm. Pharmacol. 2020, 72, 1038–1048. [Google Scholar] [CrossRef]
- Ravivarapu, H.B.; Moyer, K.L.; Dunn, R.L. Sustained activity and release of leuprolide acetate from an in situ forming polymeric implant. AAPS PharmSciTech 2000, 1, 732–741. [Google Scholar]
- Parent, M.; Nouvel, C.; Koerber, M.; Sapin, A.; Maincent, P.; Boudier, A. PLGA in situ implants formed by phase inversion: Critical physicochemical parameters to modulate drug release. J. Control Release 2013, 172, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Hull, M.W.; Montaner, J.S.G. HIV treatment as prevention: The key to an AIDS-free generation. J. Food Drug Anal. 2013, 21, S95–S101. [Google Scholar] [CrossRef] [Green Version]
- Solorio, L.; Sundarapandiyan, D.; Olear, A.; Exner, A.A. The Effect of Additives on the Behavior of Phase Sensitive In Situ Forming Implants. J. Pharm. Sci. 2015, 104, 3471–3480. [Google Scholar] [CrossRef] [Green Version]
- Astaneh, R.; Erfan, M.; Moghimi, H.; Mobedi, H. Changes in morphology of in situ forming PLGA implant prepared by different polymer molecular weight and its effect on release behavior. J. Pharm. Sci. 2009, 98, 135–145. [Google Scholar] [CrossRef]
- Prabhu, S.; Tran, L.P.; Betageri, G.V. Effect of co-solvents on the controlled release of calcitonin polypeptide from in situ biodegradable polymer implants. Drug Deliv. 2005, 12, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Zare, M.; Mobedi, H.; Barzin, J.; Mivehchi, H.; Jamshidi, A.; Mashayekhi, R. Effect of additives on release profile of leuprolide acetate in an in situ forming controlled-release system: In vitro study. J. Appl. Polym. Sci. 2008, 107, 3781–3787. [Google Scholar] [CrossRef]
- Benhabbour, S.R.; Kovarova, M.; Jones, C.; Copeland, D.J.; Shrivastava, R.; Swanson, M.D.; Sykes, C.; Ho, P.T.; Cottrell, M.L.; Sridharan, A.; et al. Ultra-long-acting tunable biodegradable and removable controlled release implants for drug delivery. Nat. Commun. 2019, 10, 4324. [Google Scholar] [CrossRef] [PubMed]
- Moss, L.; Wagner, D.; Kanaoka, E.; Olson, K.; Yueh, Y.L.; Bowers, G.D. The comparative disposition and metabolism of dolutegravir, a potent HIV-1 integrase inhibitor, in mice, rats, and monkeys. Xenobiotica 2015, 45, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Gouda, R.; Baishya, H.; Qing, Z. Application of Mathematical Models in Drug Release Kinetics of Carbidopa and Levodopa ER Tablets. J. Dev. Drugs 2017, 6, 2. [Google Scholar]
- Kovarova, M.; Benhabbour, S.R.; Massud, I.; Spagnuolo, R.A.; Skinner, B.; Baker, C.E.; Sykes, C.; Mollan, K.R.; Kashuba, A.D.M.; García-Lerma, J.C.; et al. Ultra-long-acting removable drug delivery system for HIV treatment and prevention. Nat. Commun. 2018, 9, 4156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshitake, Y.; Takai, Y.; Kanehisa, H.; Shinohara, M. Muscle shear modulus measured with ultrasound shear-wave elastography across a wide range of contraction intensity. Muscle Nerve 2014, 50, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Diridollou, S.; Berson, M.; Vabre, V.; Black, D.; Karlsson, B.; Auriol, F.; Gregoire, J.M.; Yvon, C.; Vaillant, L.; Gall, Y.; et al. An In Vivo method for measuring the mechanical properties of the skin using ultrasound. Ultrasound Med. Biol. 1998, 24, 215–224. [Google Scholar] [CrossRef]
- Rodger, M.A.; King, L. Drawing up and administering intramuscular injections: A review of the literature. J. Adv. Nurs. 2000, 31, 574–582. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Dose (mg) | Median AUC0–30d (h ∗ ng/mL) | 25% Percentile | 75% Percentile | p-Value | |
|---|---|---|---|---|---|---|
| 100 mg/mL 1 × 40 IM | 4 | 2,367,255.00 | 2,169,626.25 | 3,348,652.50 | 1.000 | |
| 100 mg/mL 2 × 20 IM | 4 | 2,018,574.50 | 1,710,932.00 | 3,779,222.50 | ||
| 100 mg/mL 1 × 40 SQ | 4 | 2,692,192.50 | 2,223,818.25 | 3,814,503.75 | 1.000 | |
| 100 mg/mL 2 × 20 SQ | 4 | 3,477,907.50 | 3,259,230.00 | 3,693,720.00 | ||
| 100 mg/mL 1 × 80 SQ | 8 | 5,302,942.50 | 4,170,672.50 | 6,184,407.50 | 1.000 0.911 | 0.993 |
| 100 mg/mL 2 × 40 SQ | 8 | 4,462,922.50 | 3,750,026.25 | 5,408,896.25 | ||
| 100 mg/mL 4 × 20 SQ | 8 | 5,875,935.00 | 5,296,415.00 | 6,815,062.50 | ||
| 250 mg/mL 1 × 80 SQ | 20 | 7,941,840.00 | 6,143,650.00 | 12,325,440.00 | 1.000 0.923 | 0.945 |
| 250 mg/mL 2 × 40 SQ | 20 | 7,461,075.00 | 6,121,780.00 | 8,639,070.00 | ||
| 250 mg/mL 4 × 20 SQ | 20 | 10,865,925.00 | 9,519,702.50 | 11917185.00 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joiner, J.B.; King, J.L.; Shrivastava, R.; Howard, S.A.; Cottrell, M.L.; Kashuba, A.D.M.; Dayton, P.A.; Benhabbour, S.R. Effects of Injection Volume and Route of Administration on Dolutegravir In Situ Forming Implant Pharmacokinetics. Pharmaceutics 2022, 14, 615. https://doi.org/10.3390/pharmaceutics14030615
Joiner JB, King JL, Shrivastava R, Howard SA, Cottrell ML, Kashuba ADM, Dayton PA, Benhabbour SR. Effects of Injection Volume and Route of Administration on Dolutegravir In Situ Forming Implant Pharmacokinetics. Pharmaceutics. 2022; 14(3):615. https://doi.org/10.3390/pharmaceutics14030615
Chicago/Turabian StyleJoiner, Jordan B., Jasmine L. King, Roopali Shrivastava, Sarah Anne Howard, Mackenzie L. Cottrell, Angela D. M. Kashuba, Paul A. Dayton, and Soumya Rahima Benhabbour. 2022. "Effects of Injection Volume and Route of Administration on Dolutegravir In Situ Forming Implant Pharmacokinetics" Pharmaceutics 14, no. 3: 615. https://doi.org/10.3390/pharmaceutics14030615
APA StyleJoiner, J. B., King, J. L., Shrivastava, R., Howard, S. A., Cottrell, M. L., Kashuba, A. D. M., Dayton, P. A., & Benhabbour, S. R. (2022). Effects of Injection Volume and Route of Administration on Dolutegravir In Situ Forming Implant Pharmacokinetics. Pharmaceutics, 14(3), 615. https://doi.org/10.3390/pharmaceutics14030615