
Development of Nafamostat Mesylate Immediate-Release Tablet by Drug Repositioning Using Quality-by-Design Approach


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Solubility Studies
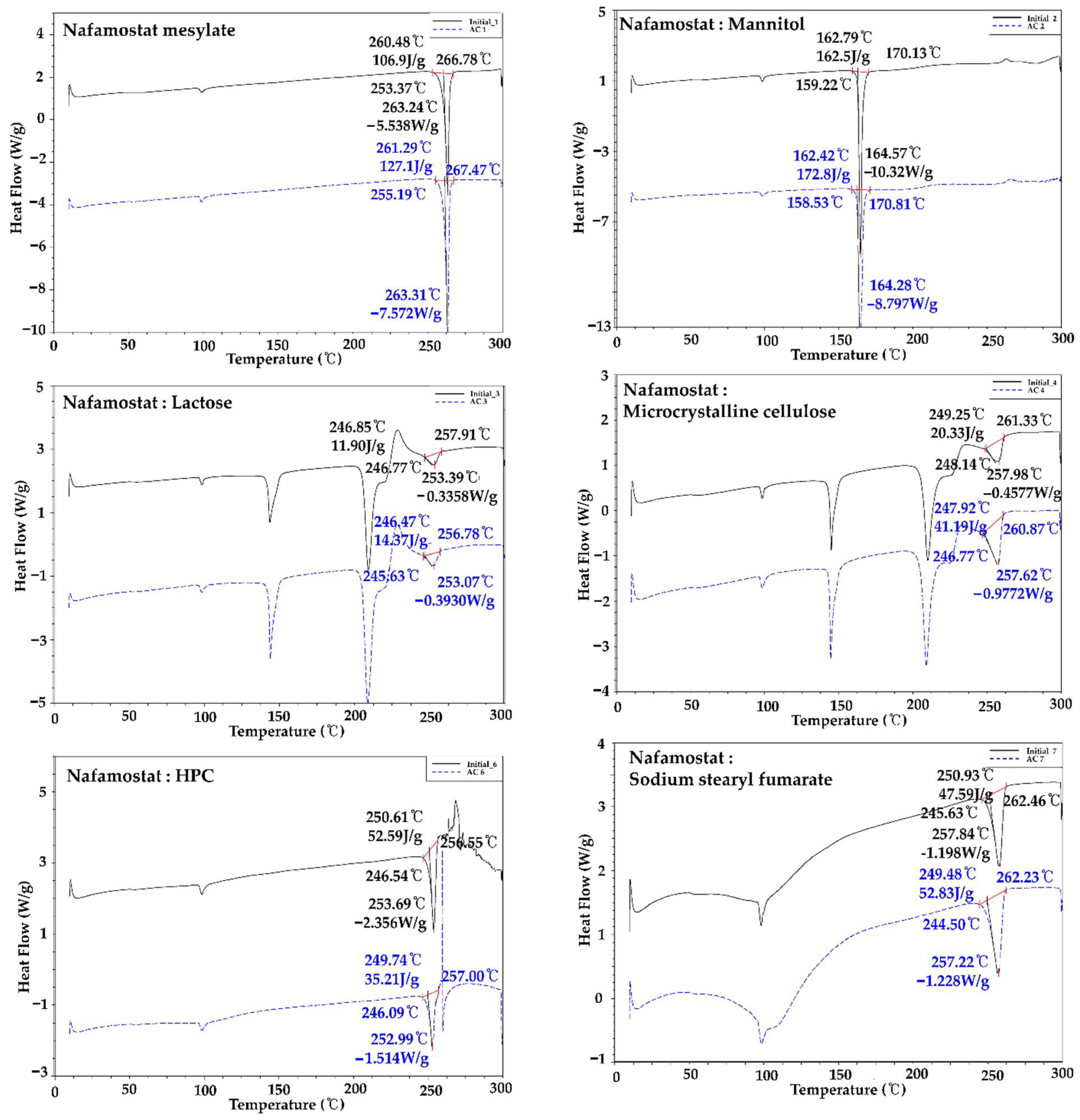
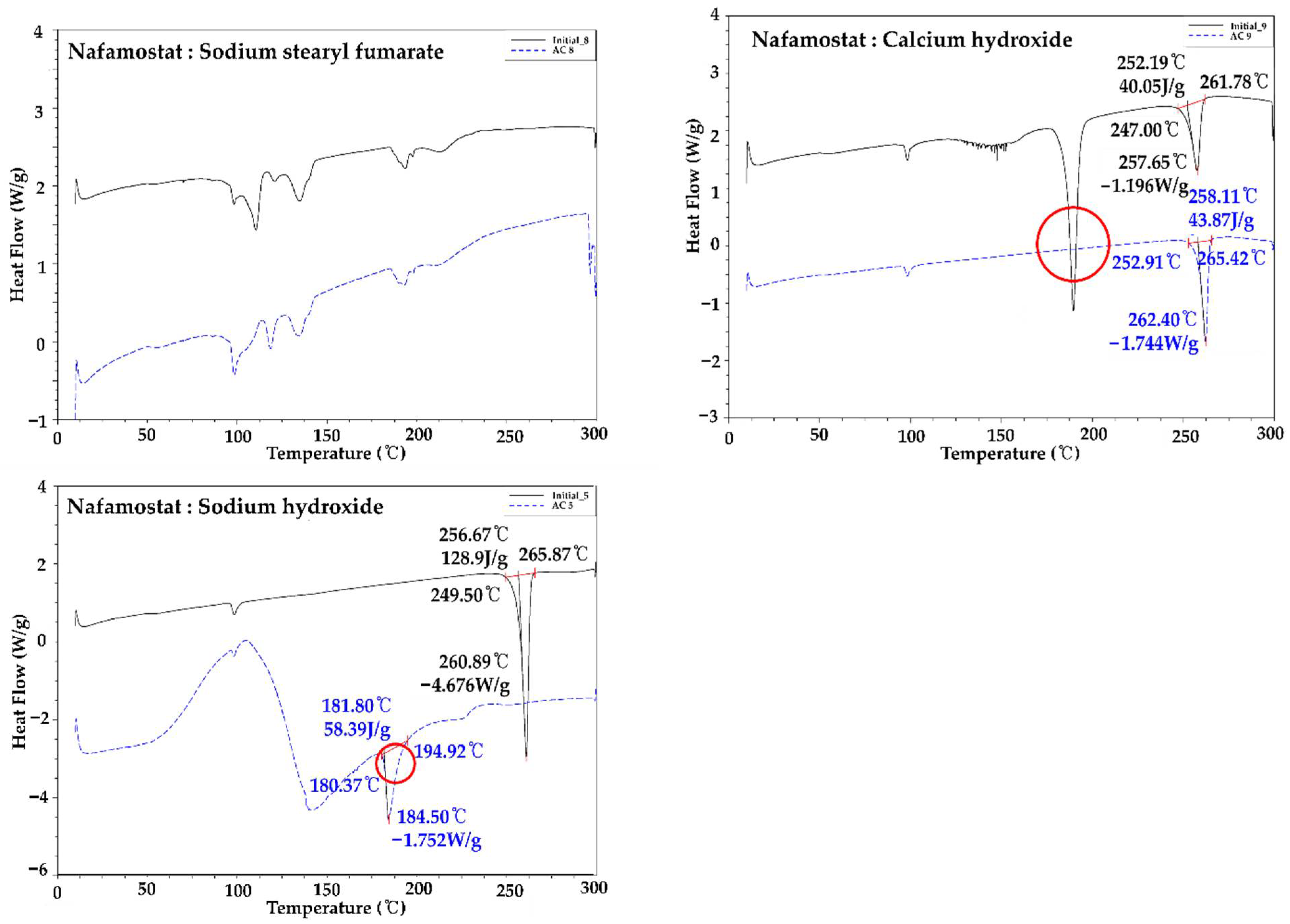
2.3. Compatibility Studies between API and Excipients
2.4. Formulation of Nafamostat Mesylate
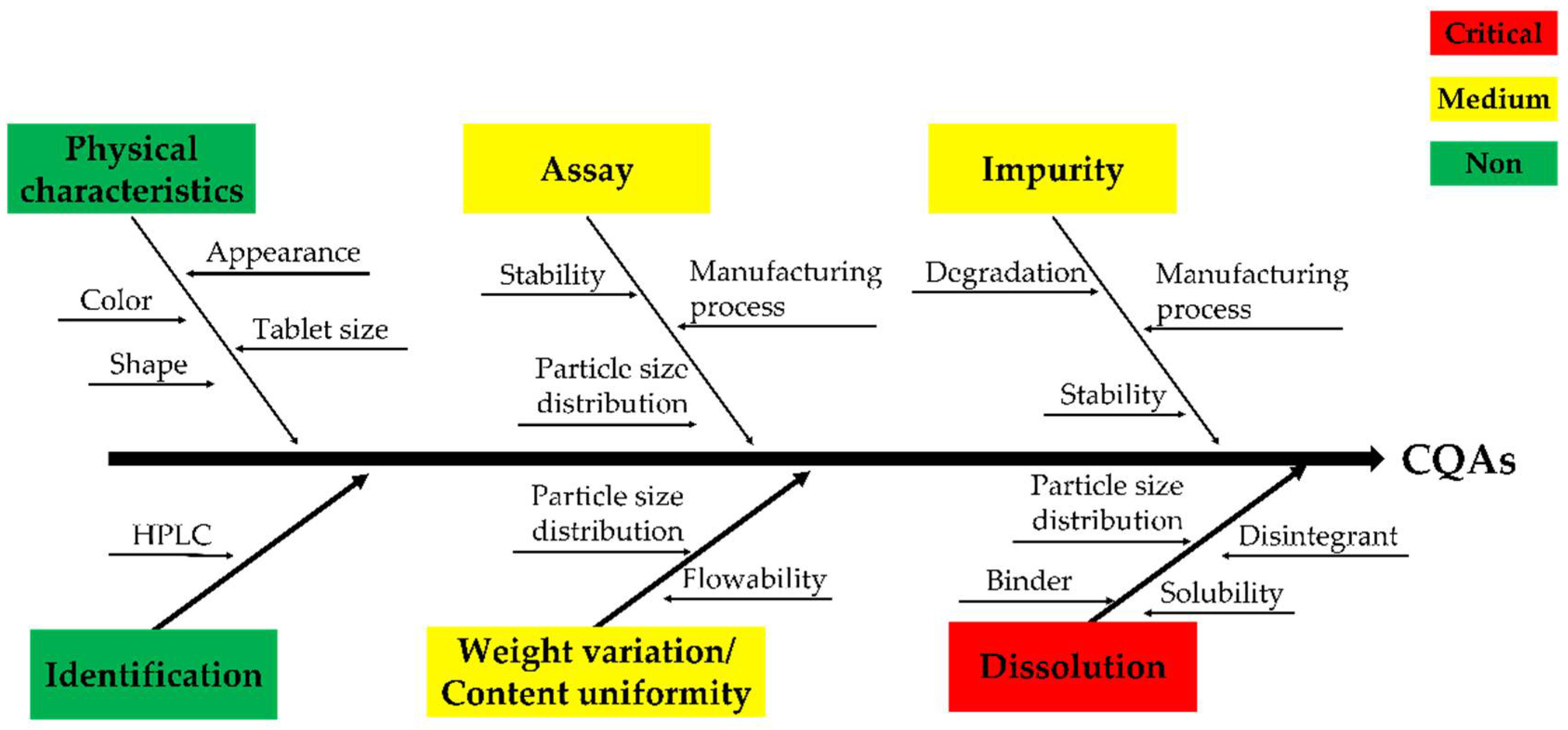
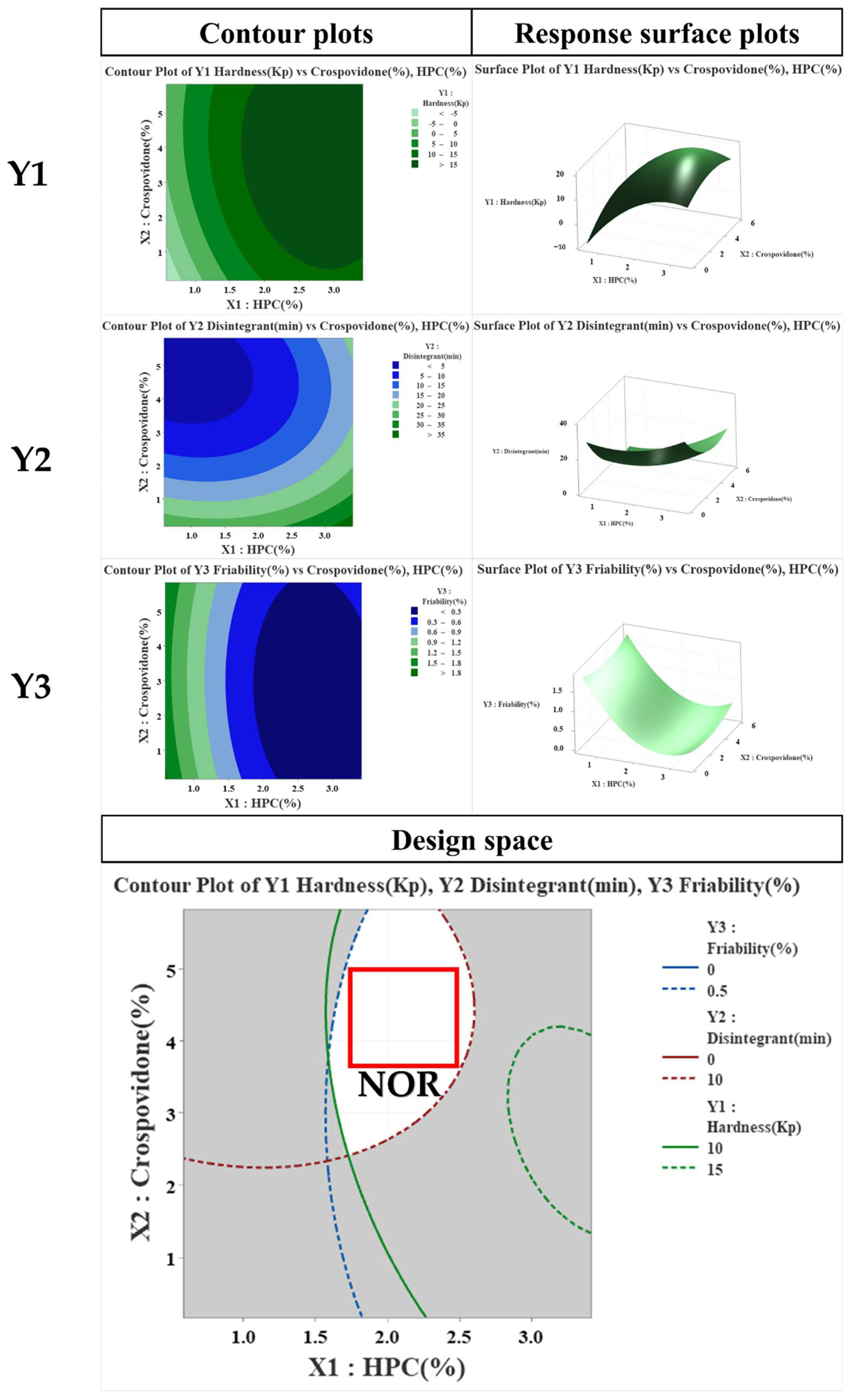
2.5. Critical Quality Attributes (CQA)/Risk Assessment (RA) of Critical Material Attributes (CMA) and Critical Process Parameters (CPP)
2.6. HPLC Analysis
2.6.1. Assay
2.6.2. Impurity
2.7. Stability Studies
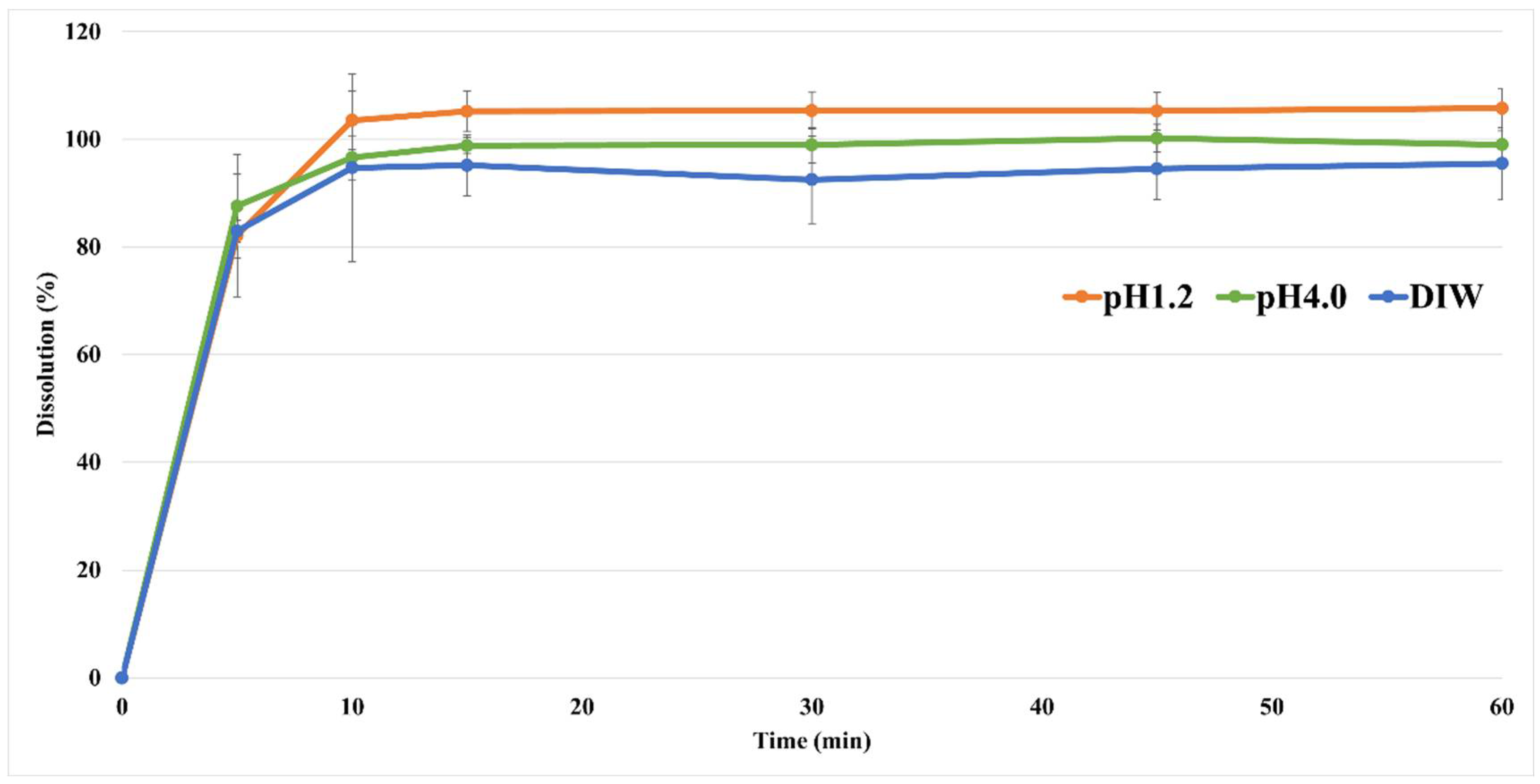
2.8. In Vitro Dissolution Studies
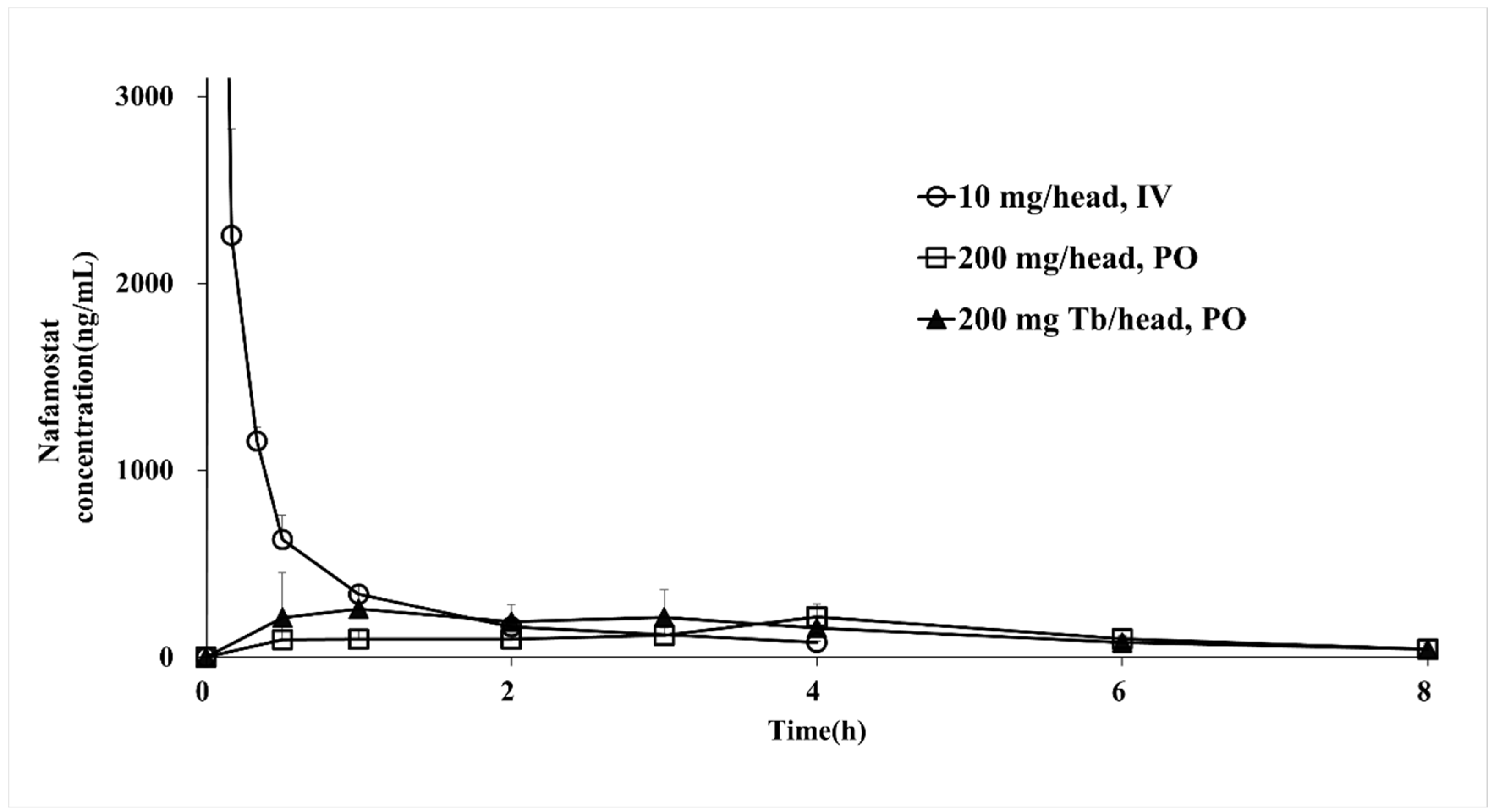
2.9. Pharmacokinetics Studies
2.10. Statistical Analysis
3. Results and Discussion
3.1. Physicochemical Properties of Nafamostat Mesylate
3.2. Solubility Studies
3.3. Compatibility Studies between API and Excipients
3.4. Stability Studies
3.5. CQA/RA of CMAs and CPPs
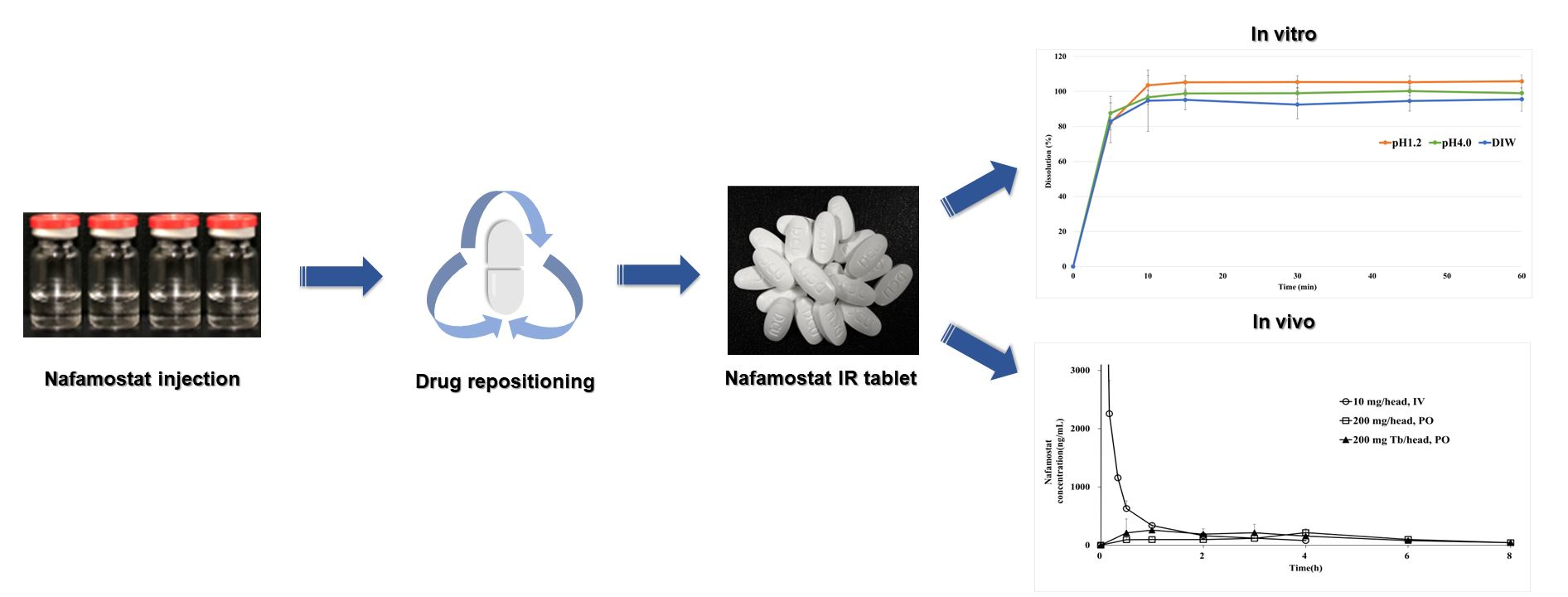
3.6. In Vitro Dissolution Studies
3.7. Pharmacokinetic Study in Monkeys
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Daniel, S.J. Education and the COVID-19 pandemic. Prospects 2020, 49, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shereen, M.A.; Khan, S.; Kazmi, A.; Bashir, N.; Siddique, R. COVID-19 infection: Emergence, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020, 24, 91–98. [Google Scholar] [CrossRef]
- Tian, S.; Hu, N.; Lou, J.; Chen, K.; Kang, X.; Xiang, Z.; Chen, H.; Wang, D.; Liu, N.; Liu, D. Characteristics of COVID-19 infection in Beijing. J. Infect. 2020, 80, 401–406. [Google Scholar] [CrossRef] [Green Version]
- Zietz, M.; Zucker, J.; Tatonetti, N.P. Associations between blood type and COVID-19 infection, intubation, and death. Nat. Commun. 2020, 11, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J. Preventing a COVID-19 pandemic. Bmj 2020, 368, m810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stopsack, K.H.; Mucci, L.A.; Antonarakis, E.S.; Nelson, P.S.; Kantoff, P.W. TMPRSS2 and COVID-19: Serendipity or opportunity for intervention? Cancer Discov. 2020, 10, 779–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strope, J.D.; Chau, C.H. TMPRSS2: Potential biomarker for COVID-19 outcomes. J. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Krüger, N.; Müller, M.; Drosten, C.; Pöhlmann, S. The novel coronavirus 2019 (2019-nCoV) uses the SARS-coronavirus receptor ACE2 and the cellular protease TMPRSS2 for entry into target cells. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.W.; Mao, H.J.; Wu, Y.L.; Tanaka, Y.; Zhang, W. TMPRSS2: A potential target for treatment of influenza virus and coronavirus infections. Biochimie 2017, 142, 1–10. [Google Scholar] [CrossRef]
- Azimi, A. TMPRSS2 Inhibitors, Bromhexine, Aprotinin, Camostat and Nafamostat as Potential Treatments for COVID-19. 2020. Available online: https://osf.io/preprints/frenxiv/a3rvm/ (accessed on 19 May 2020).
- Hoffmann, M.; Schroeder, S.; Kleine-Weber, H.; Müller, M.A.; Drosten, C.; Pöhlmann, S. Nafamostat mesylate blocks activation of SARS-CoV-2: New treatment option for COVID-19. Antimicrob. Agents Chemother. 2020, 64, e00754-20. [Google Scholar] [CrossRef] [Green Version]
- Ellinger, B.; Bojkova, D.; Zaliani, A.; Cinatl, J.; Claussen, C.; Westhaus, S.; Reinshagen, J.; Kuzikov, M.; Wolf, M.; Geisslinger, G. Identification of inhibitors of SARS-CoV-2 in-vitro cellular toxicity in human (Caco-2) cells using a large scale drug repurposing collection. Res. Sq. 2020. [Google Scholar] [CrossRef] [Green Version]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Jin, G.; Wong, S.T. Toward better drug repositioning: Prioritizing and integrating existing methods into efficient pipelines. Drug Discov. Today 2014, 19, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of drug repositioning approaches and resources. Int. J. Biol. Sci. 2018, 14, 1232. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.; Jeon, S.; Ryu, W.-S.; Kim, S. Comparative analysis of antiviral efficacy of FDA-approved drugs against SARS-CoV-2 in human lung cells: Nafamostat is the most potent antiviral drug candidate. bioRxiv 2020. [Google Scholar] [CrossRef]
- Savosina, P.; Druzhilovskii, D.; Poroikov, V. COVID-19: Analysis of Drug Repositioning Practice. Pharm. Chem. J. 2021, 54, 989–996. [Google Scholar] [CrossRef]
- Ko, M.; Jeon, S.; Ryu, W.S.; Kim, S. Comparative analysis of antiviral efficacy of FDA-approved drugs against SARS-CoV-2 in human lung cells. J. Med. Virol. 2021, 93, 1403–1408. [Google Scholar] [CrossRef]
- Parisi, D.; Adasme, M.F.; Sveshnikova, A.; Bolz, S.N.; Moreau, Y.; Schroeder, M. Drug repositioning or target repositioning: A structural perspective of drug-target-indication relationship for available repurposed drugs. Comput. Struct. Biotechnol. J. 2020, 18, 1043–1055. [Google Scholar] [CrossRef]
- Park, K.; Kim, D. Drug-drug relationship based on target information: Application to drug target identification. BMC Syst. Biol. 2011, 5, S12. [Google Scholar] [CrossRef] [Green Version]
- Rutherford, K.D.; Mazandu, G.K.; Mulder, N.J. A systems-level analysis of drug–target–disease associations for drug repositioning. Brief. Funct. Genom. 2018, 17, 34–41. [Google Scholar] [CrossRef]
- Low, Z.Y.; Farouk, I.A.; Lal, S.K. Drug repositioning: New approaches and future prospects for life-debilitating diseases and the COVID-19 pandemic outbreak. Viruses 2020, 12, 1058. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Decroly, E.; Khatib, A.-M.; Villoutreix, B.O. Structure-based drug repositioning over the human TMPRSS2 protease domain: Search for chemical probes able to repress SARS-CoV-2 Spike protein cleavages. Eur. J. Pharm. Sci. 2020, 153, 105495. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, K.; Barale, S.S.; Dhanavade, M.J.; Waghmare, S.R.; Nadaf, N.H.; Kamble, S.A.; Mohammed, A.A.; Makandar, A.M.; Fandilolu, P.M.; Dound, A.S. Homology modeling and docking studies of TMPRSS2 with experimentally known inhibitors Camostat mesylate, Nafamostat and Bromhexine hydrochloride to control SARS-Coronavirus-2. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Burgstaller-Muehlbacher, S.; Pache, L.; De Jesus, P.P.; Hull, M.V.; Chang, M. A large-scale drug repositioning survey for SARS-CoV-2 antivirals. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Shin, B.; Kang, K.; Park, S.; Beck, B.R. Target-centered drug repurposing predictions of human angiotensin-converting enzyme 2 (ACE2) and transmembrane protease serine subtype 2 (TMPRSS2) interacting approved drugs for coronavirus disease 2019 (COVID-19) treatment through a drug-target interaction deep learning model. Viruses 2020, 12, 1325. [Google Scholar]
- Lee, J.; Kim, J.-E. Application of Open Source Based DoE R Program for the Development of QbD. Yakhak Hoeji 2019, 63, 274–281. [Google Scholar] [CrossRef]
- Oh, G.-H.; Park, J.-H.; Shin, H.-W.; Kim, J.-E.; Park, Y.-J. Quality-by-design approach for the development of telmisartan potassium tablets. Drug Dev. Ind. Pharm. 2018, 44, 837–848. [Google Scholar] [CrossRef]
- Lee, S.-H.; Kim, J.-E. Quality by Design Applied Development of Immediate-Release Rabeprazole Sodium Dry-Coated Tablet. Pharmaceutics 2021, 13, 259. [Google Scholar] [CrossRef]
- Kim, J.-E.; Park, Y.-J. QbD Consideration for Developing a Double-Layered Tablet into a Single-Layered Tablet with Telmisartan and Amlodipine. Pharmaceutics 2022, 14, 377. [Google Scholar] [CrossRef]
- ICHHT Guideline. Pharmaceutical Development. Q8 (R2). 2009. Available online: https://www.google.com.hk/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&ved=2ahUKEwiB2tPfoJr4AhXxwIsBHUk0DtwQFnoECAQQAQ&url=https%3A%2F%2Fdatabase.ich.org%2Fsites%2Fdefault%2Ffiles%2FQ8_R2_Guideline.pdf&usg=AOvVaw1Y33pbg2S6x-qt_zdeeUx2 (accessed on 16 August 2020).
- Salazar, J.; Heinzerling, O.; Müller, R.H.; Möschwitzer, J.P. Process optimization of a novel production method for nanosuspensions using design of experiments (DoE). Int. J. Pharm. 2011, 420, 395–403. [Google Scholar] [CrossRef]
- Visser, J.C.; Dohmen, W.M.; Hinrichs, W.L.; Breitkreutz, J.; Frijlink, H.W.; Woerdenbag, H.J. Quality by design approach for optimizing the formulation and physical properties of extemporaneously prepared orodispersible films. Int. J. Pharm. 2015, 485, 70–76. [Google Scholar] [CrossRef]
- Kaljević, O.; Djuriš, J.; Djurić, Z.; Ibrić, S. Application of failure mode and effects analysis in quality by design approach for formulation of carvedilol compression coated tablets. J. Drug Deliv. Sci. Technol. 2016, 32, 56–63. [Google Scholar] [CrossRef]
- Mishra, S.M.; Rohera, B.D. An integrated, quality by design (QbD) approach for design, development and optimization of orally disintegrating tablet formulation of carbamazepine. Pharm. Dev. Technol. 2017, 22, 889–903. [Google Scholar] [CrossRef]
- Carlson, C. Effective FMEAs: Achieving Safe, Reliable, and Economical Products and Processes Using Failure Mode and Effects Analysis; John Wiley & Sons: Hoboken, NJ, USA, 2012; Volume 1. [Google Scholar]
- Montgomery, T.A.; Marko, K.A. Quantitative FMEA automation. In Proceedings of the Annual Reliability and Maintainability Symposium, Philadelphia, PA, USA, 13–16 January 1997; pp. 226–228. [Google Scholar]
- Teng, S.H.G.; Ho, S.Y.M. Failure mode and effects analysis: An integrated approach for product design and process control. Int. J. Qual. Reliab. Manag. 1996, 13, 8–26. [Google Scholar] [CrossRef]
- Jordan, W.E. Failure modes, effects and criticality analyses. In Annual Reliability and Maintainability Symposium; Institute of Electrical and Electronics Engineers: New York, NY, USA, 1972. [Google Scholar]
- The Ministry of Health, Labour and Welfare. The Japanese Pharmacopoeia, 17th ed.; Pmda: Tokyo, Japan, 2016; pp. 1303–1304.
- Guideline, I.H.T. Impurities in new drug products. Q3B (R2) Curr. Step 2006, 4, 1–5. [Google Scholar]
- SAT RS. 711 Dissolution; The United States Pharmacopeial Convention: Rockville, MD, USA, 2011. [Google Scholar]
- Elder, D.; Teasdale, A. ICH Q9 quality risk management. In ICH Quality Guidelines: An Implementation Guide; Wiley Online Library: Hoboken, NJ, USA, 2017; pp. 579–610. [Google Scholar]
- Maruyama, Y.; Yoshida, H.; Uchino, S.; Yokoyama, K.; Yamamoto, H.; Takinami, M.; Hosoya, T. Nafamostat mesilate as an anticoagulant during continuous veno-venous hemodialysis: A three-year retrospective cohort study. Int. J. Artif. Organs 2011, 34, 571–576. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nafamostat Mesylate | |||
|---|---|---|---|
| Chemical structure |  | CAS No. | 82956-11-4 |
| Chemical name | Nafamostat mesylate | ||
| Formula | C19H17N5O2∙2CH4O3S | ||
| Mol. mass | 539.6 g/mol | Description | White powder |
| Melting point | 637.2 °C | Solubility | 0.0341 mg/mL (in water) |
| Boiling point | Log p | 1.91, 2.52 | |
| pKa | 11.32 | BCS class | |
| Storage condition | A light-shielding airtight container, stored at room temperature (1–30 °C) | ||
| Mechanism of action | TMPRSS2 activity inhibitor. Nafamostat exhibits inhibitory action on trypsin; it also inhibits clotting factors such as thrombin, Xa, XIIa, VIIa, kallikrein, and complement, and inhibits platelet aggregation | ||
| Pharmacokinetics | Half-life: 8 min | ||
| Solvent | Apparent | Equilibrium (mg/mL) | |
|---|---|---|---|
| 1 h | 4 h | ||
| Water | ++++ | 18.918 ± 0.047 | 18.825 ± 0.019 |
| Ethanol | + | 2.022 ± 0.046 | 2.201 ± 0.027 |
| pH 1.2 | +++ | 9.979 ± 0.003 | 8.558 ± 0.209 |
| pH 2.0 | +++ | 14.786 ± 0.040 | 16.463 ± 0.066 |
| pH 3.0 | + | 0.002 ± 0.004 | 0.001 ± 0.002 |
| pH 4.0 | +++ | 15.470 ± 0.160 | 16.016 ± 0.226 |
| pH 5.0 | + | 0.001 ± 0.034 | 0.000 ± 0.008 |
| pH 6.0 | + | 0.025 ± 0.003 | 0.019 ± 0.004 |
| pH 6.8 | + | 0.010 ± 0.003 | 0.009 ± 0.001 |
| pH 7.0 | + | 0.008 ± 0.001 | 0.006 ± 0.002 |
| pH 8.0 | + | 17.500 ± 0.014 | 15.371 ± 0.019 |
| pH 9.0 | + | 0.096 ± 0.003 | 0.078 ± 0.005 |
| pH 10.0 | + | 0.000 ± 0.001 | 0.000 ± 0.001 |
| pH 11.0 | + | 0.004 ± 0.002 | 0.008 ± 0.004 |
| pH 12.0 | + | 0.000 ± 0.002 | 0.042 ± 0.009 |
| Items | Total Impurities | ||
|---|---|---|---|
| Initial (%) | 40 °C/60% RH 4 Weeks (%) | 60 °C/75% RH 4 Weeks (%) | |
| Nafamostat mesylate | 0.04 ± 0.01 | 0.10 ± 0.03 | 0.13 ± 0.01 |
| Mannitol | 0.04 ± 0.01 | 0.09 ± 0.03 | 0.14 ± 0.02 |
| Lactose | 0.07 ± 0.03 | 0.10 ± 0.01 | 0.13 ± 0.01 |
| Dicalcium phosphate dihydrate | 0.04 ± 0.02 | 3.20 ± 0.01 | 1.26 ± 0.03 |
| Microcrystalline cellulose | 0.05 ± 0.01 | 0.09 ± 0.01 | 0.12 ± 0.01 |
| Pregelatinized starch | 0.05 ± 0.01 | 0.09 ± 0.01 | 0.13 ± 0.02 |
| Precipitated calcium carbonate | 0.07 ± 0.01 | 0.16 ± 0.01 | 0.28 ± 0.02 |
| Sodium starch glyconate | 0.06 ± 0.01 | 0.15 ± 0.02 | 0.23 ± 0.01 |
| Crospovidone | 0.05 ± 0.01 | 0.10 ± 0.02 | 0.12 ± 0.03 |
| Sodium croscamellose | 0.03 ± 0.01 | 0.11 ± 0.04 | 0.12 ± 0.01 |
| Sodium stearyl fumarate | 0.03 ± 0.05 | 0.12 ± 0.03 | 0.15 ± 0.02 |
| Magnesium stearate | 0.03 ± 0.02 | 0.13 ± 0.01 | 0.20 ± 0.01 |
| Povidone K | 0.05 ± 0.01 | 0.10 ± 0.01 | 0.14 ± 0.03 |
| Hydroxypropyl methylcellulose | 0.05 ± 0.01 | 0.05 ± 0.03 | 0.16 ± 0.05 |
| Hydroxypropyl cellulose | 0.05 ± 0.03 | 0.08 ± 0.08 | 0.14 ± 0.04 |
| Calcium hydroxide | 1.08 ± 0.01 | 0.64 ± 0.01 | 9.00 ± 0.01 |
| Sodium hydroxide | 0.32 ± 0.04 | 3.82 ± 0.01 | 9.05 ± 0.01 |
| (a) | |||||||
| Process Method | Initial (%) | 1 M | 2 M | ||||
| RT (%) | AC (%) | HC (%) | RT (%) | AC (%) | HC (%) | ||
| Wet granulation | 0.05 ± 0.02 | 0.25 ± 0.02 | 1.53 ± 0.01 | 6.16 ± 0.05 | 0.25 ± 0.02 | 2.10 ± 0.01 | 9.35 ± 0.01 |
| Direct compression | 0.06 ± 0.01 | 0.05 ± 0.01 | 0.11 ± 0.01 | 0.95 ± 0.04 | 0.11 ± 0.02 | 0.33 ± 0.01 | 3.04 ± 0.01 |
| (b) | |||||||
| Direct Compression | Initial (%) | 1 M | 2 M | ||||
| RT (%) | AC (%) | HC (%) | RT (%) | AC (%) | HC (%) | ||
| 100/300 mg | 0.06 ± 0.02 | 0.06 ± 0.02 | 0.07 ± 0.02 | 0.19 ± 0.02 | 0.11 ± 0.02 | 0.23 ± 0.02 | 0.34 ± 0.02 |
| 100/500 mg | 0.05 ± 0.02 | 0.05 ± 0.02 | 0.11 ± 0.02 | 0.95 ± 0.02 | 0.11 ± 0.02 | 0.33 ± 0.02 | 3.04 ± 0.02 |
| (a) | |||||||||
| CQAs1 | Nafamostat Mesylate | Diluent | Binder | Disintegrant | Anti-Adherent | Glidant | Lubricant | ||
| Physical | Low | Low | Low | Low | Low | Low | Low | ||
| Assay | Low | Low | Low | Low | Low | Low | Low | ||
| Uniformity | Low | Low | Medium | Low | Low | Low | Medium | ||
| Impurities | Low | Low | Low | Low | Low | Low | Low | ||
| Dissolution | Low | Low | High | High | Low | Low | Medium | ||
| (b) | |||||||||
| Functions | CMAs2 | Failure Mode (Critical Event) | Effect on CQAs with Respect to QTPP 3 (Justification of Failure Mode) | P4 | S5 | D6 | RPN7 | ||
| Physical property of API 8 | Solid state form | Different crystal form/Different form | The solubility of active pharmaceutical ingredient (API) may be affected, and the dissolution of the drug product is affected, thus causing damage to bioavailability and efficacy | 1 | 2 | 2 | 4 | ||
| Chemical property of API | Solubility | Different Salt/ Different form | May affect the dissolution of tablets; thus, bioavailability and efficacy may be compromised | 1 | 1 | 2 | 2 | ||
| Impurity in the manufacturing process | Low purity | May affect the assay and impurity of tablets; thus, quality and safety may be compromised | 1 | 2 | 2 | 4 | |||
| Chemical stability | Unstable | Decomposition products may be affected by dry hear/oxidation/hydrolysis/UV light, thus causing quality and safety damage | 1 | 1 | 2 | 2 | |||
| Diluent | PSD 9 | Uneven | It can affect the flow properties of blending and can affect the content uniformity; thus, quality/safety may be compromised | 1 | 1 | 2 | 2 | ||
| Moisture content | High | May affect the impurity profile, thus causing damage to safety | 3 | 2 | 2 | 12 | |||
| Binder | Volume of binder | Higher than optimum | Produces hard mixtures, which can affect disintegration and dissolution time; thus, bioavailability and efficacy may be compromised | 4 | 3 | 3 | 36 | ||
| Lower than optimum | Loose, fragile mixtures can produce tablets of weaker hardness (fast disintegration); thus, bioavailability and efficacy may be compromised | 4 | 3 | 3 | 36 | ||||
| Disintegrant | Concentration of disintegrant | Higher than optimum | The desired dissolution pattern cannot be obtained, and the hardness of the tablet may be affected; thus, bioavailability and efficacy may be compromised | 4 | 4 | 3 | 48 | ||
| Lower than optimum | The desired dissolution pattern cannot be obtained; thus, bioavailability and efficacy may be compromised | 4 | 3 | 3 | 36 | ||||
| Anti-adherent | Concentration of anti-adherent | Lower than optimum | It may be difficult to discharge tablets from tooling; the excipient can be stuck on the surface of the filing die; thus, product quality may be compromised | 3 | 3 | 2 | 18 | ||
| Glidant | Concentration of glidant | Lower than optimum | By reducing the friction in the particles, it may affect the flowability of granules or powders such as die friction; may affect content uniformity; thus, content uniformity and product quality may be compromised | 2 | 2 | 2 | 8 | ||
| Lubricant | Concentration of lubricant | Higher than optimum | Hydrophobic lubricants can be coated on the surface of drug particles, which can delay dissolution; thus, efficacy may be compromised | 3 | 3 | 3 | 27 | ||
| Lower than optimum | The powder can stick to the surface of tooling/punch and cause picking; thus, product quality may be compromised | 3 | 3 | 3 | 27 | ||||
| Run | CMAs | CQAs | |||
|---|---|---|---|---|---|
| X1 HPC (%) | X2 Crospovidone (%) | Y1 Hardness (%) | Y2 Disintegration (%) | Y3 Friability (%) | |
| 1 | 1.0 | 1.0 | 1.8 | 19.3 | 1.3 |
| 2 | 3.0 | 1.0 | 18.4 | 28.3 | 0.2 |
| 3 | 1.0 | 5.0 | 5.9 | 4.0 | 1.3 |
| 4 | 3.0 | 5.0 | 18.6 | 18.0 | 0.3 |
| 5 | 0.6 | 3.0 | 0.9 | 6. | 1.5 |
| 6 | 3.4 | 3.0 | 16.3 | 18.0 | 0.1 |
| 7 | 2.0 | 0.2 | 8.8 | 28.3 | 0.3 |
| 8 | 2.0 | 3.0 | 16.8 | 4.0 | 0.3 |
| 9 | 2.0 | 3.0 | 17.1 | 8.5 | 0.2 |
| 10 | 2.0 | 3.0 | 16.5 | 7.9 | 0.1 |
| 11 | 2.0 | 3.0 | 16.9 | 8.0 | 0.2 |
| 12 | 2.0 | 3.0 | 17.6 | 9.1 | 0.3 |
| 13 | 2.0 | 3.0 | 17.0 | 8.5 | 0.3 |
| Parameter | Reference Drug | Test Drug | |
|---|---|---|---|
| Injection (10 mg/Head, IV) | Solution (200 mg/Head, PO) | IR Tablet (200 mg Tb/Head, PO) | |
| AUClast (ng·h/mL) | 2182.1 ± 32.7 | 898.6 ± 76.2 | 1140.5 ± 291.7 |
| Cmax (ng/mL) | 11,108.0 ± 1340.7 | 216.1 ± 69.2 | 327.3 ± 144.3 |
| Tmax (h) | 0.02 ± 0.0 | 4.0 ± 0.0 | 1.00 ± 1.3 |
| t1/2 (h) | 1.5 ± 0.0 | 1.9 ± 0.9 | 2.2 ± 1.1 |
| Clinf/F | 4.2 ± 0.02 | 197.2 ± 34.7 | 160.1 ± 45.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.-A.; Kim, J.-E. Development of Nafamostat Mesylate Immediate-Release Tablet by Drug Repositioning Using Quality-by-Design Approach. Pharmaceutics 2022, 14, 1219. https://doi.org/10.3390/pharmaceutics14061219
Kim H-A, Kim J-E. Development of Nafamostat Mesylate Immediate-Release Tablet by Drug Repositioning Using Quality-by-Design Approach. Pharmaceutics. 2022; 14(6):1219. https://doi.org/10.3390/pharmaceutics14061219
Chicago/Turabian StyleKim, Hyeon-A, and Joo-Eun Kim. 2022. "Development of Nafamostat Mesylate Immediate-Release Tablet by Drug Repositioning Using Quality-by-Design Approach" Pharmaceutics 14, no. 6: 1219. https://doi.org/10.3390/pharmaceutics14061219
APA StyleKim, H. -A., & Kim, J. -E. (2022). Development of Nafamostat Mesylate Immediate-Release Tablet by Drug Repositioning Using Quality-by-Design Approach. Pharmaceutics, 14(6), 1219. https://doi.org/10.3390/pharmaceutics14061219