
Nanotechnology-Based siRNA Delivery Systems to Overcome Tumor Immune Evasion in Cancer Immunotherapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
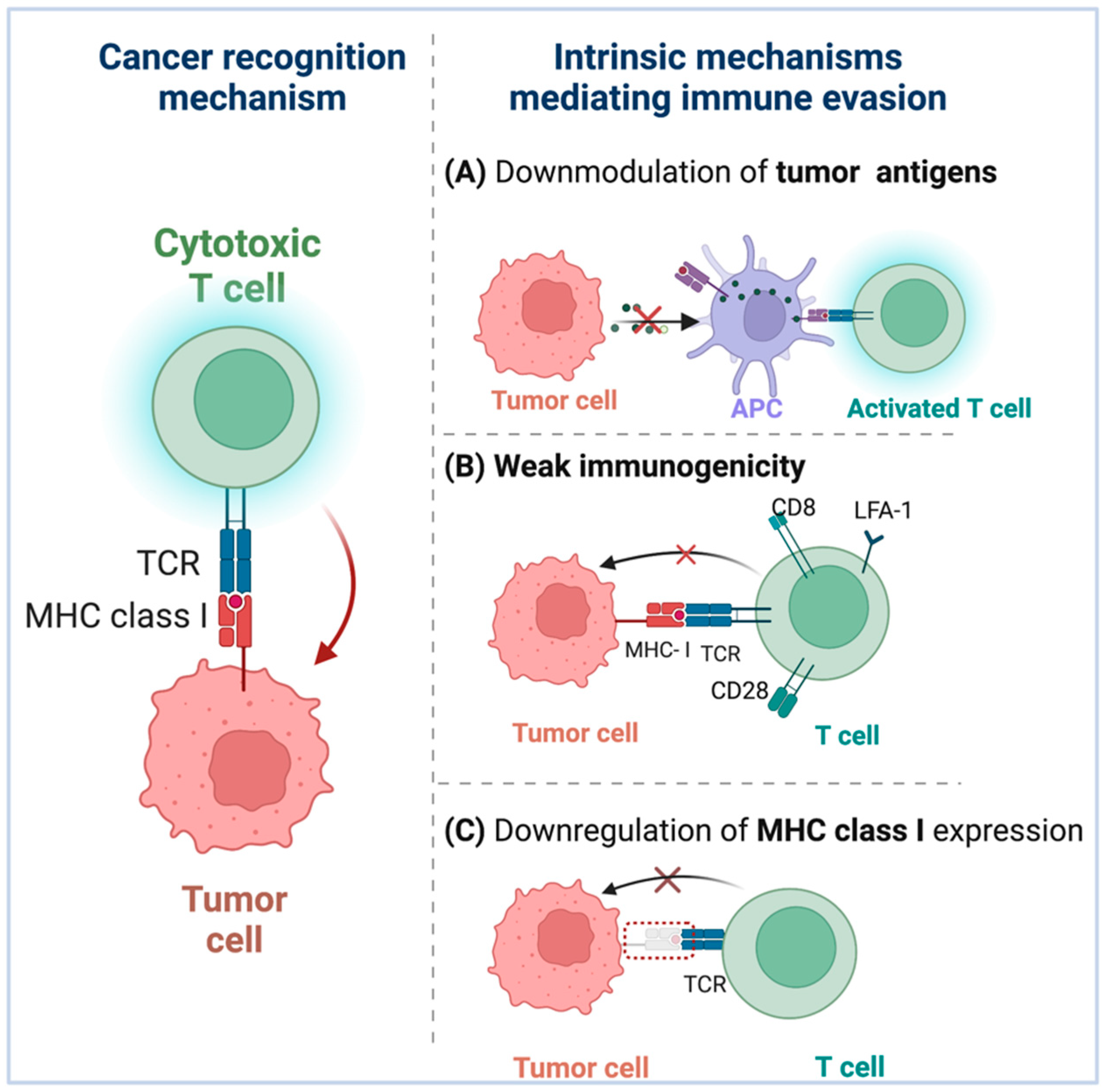
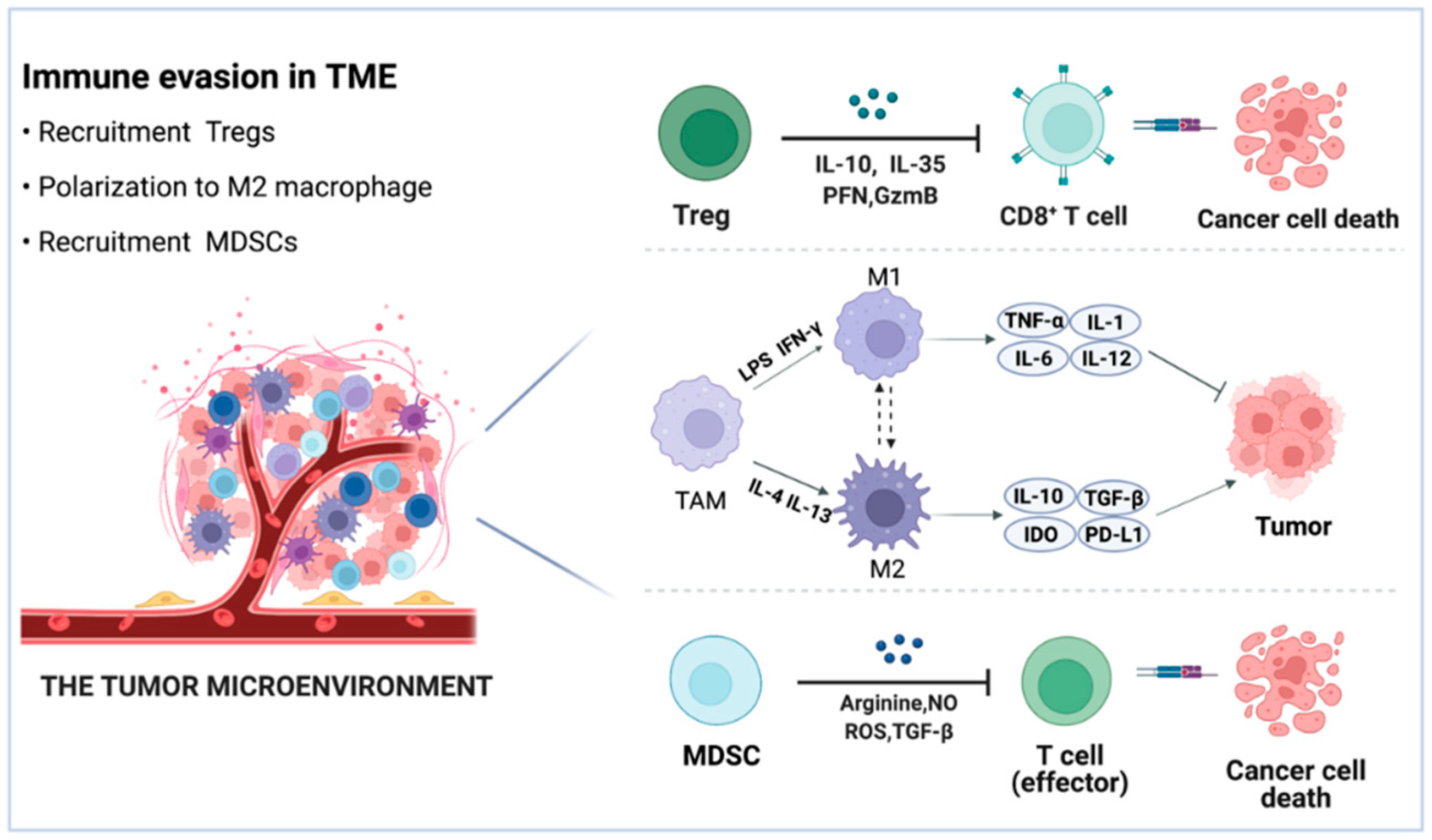
2. Immune Evasion Mechanisms in Tumor
2.1. Intrinsic Mechanisms Mediating Immune Evasion
2.2. Extrinsic Mechanisms Mediating Immune Evasion
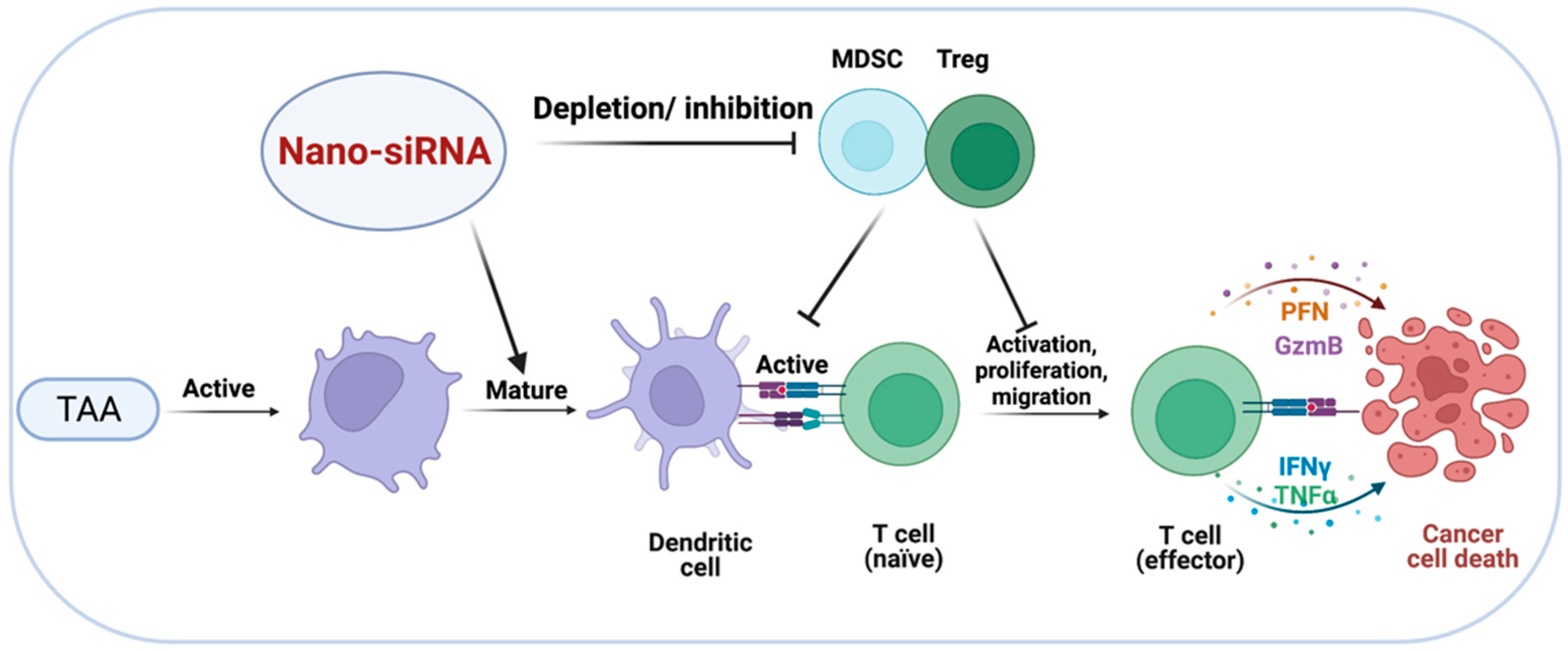
3. Targeting Immunosuppressive Cells in the TME with Nanotechnology-Based siRNA Delivery Systems
3.1. Targeted Delivery of siRNA to Tregs
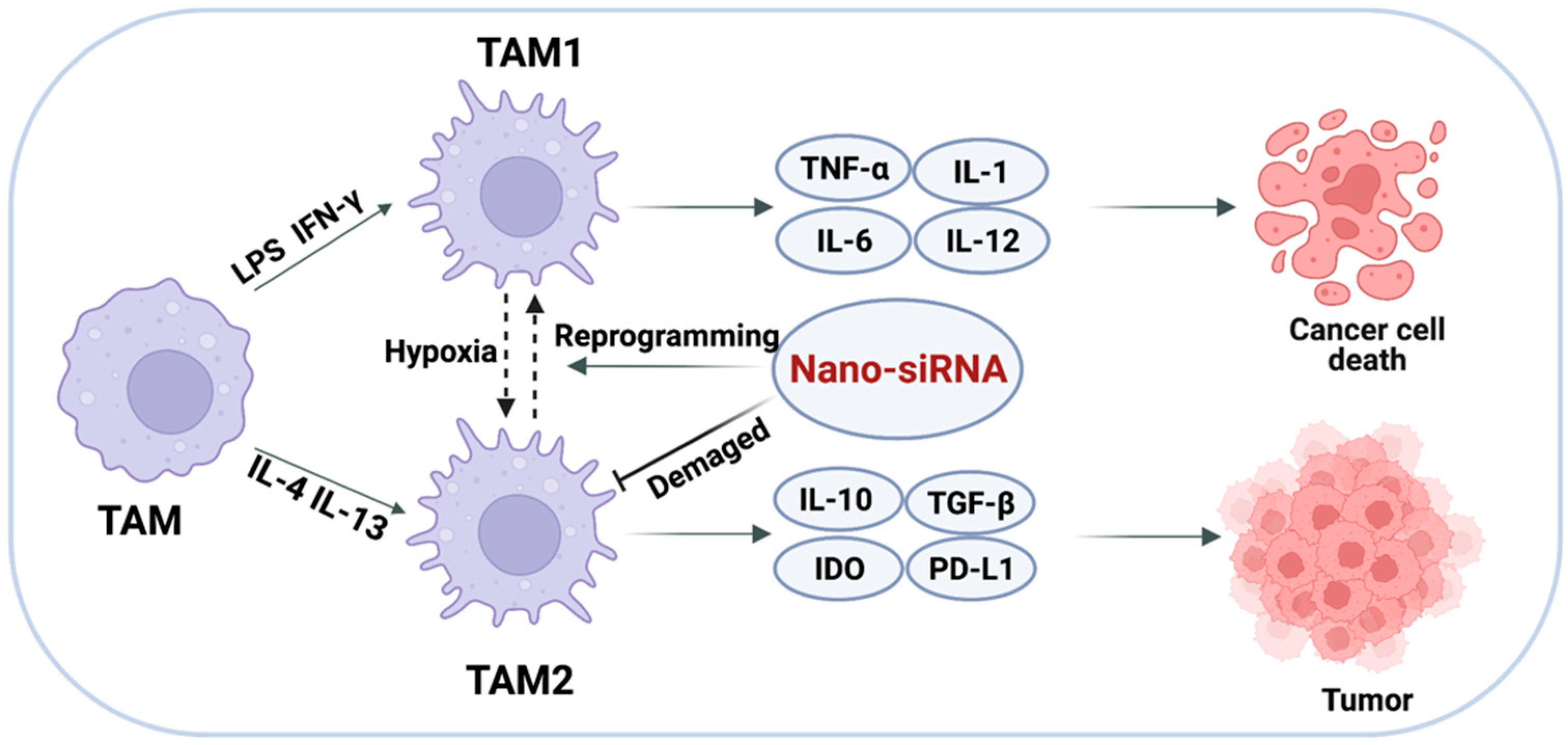
3.2. Targeted Delivery of siRNA to TAMs
3.3. Targeted Delivery of siRNA to MDSCs
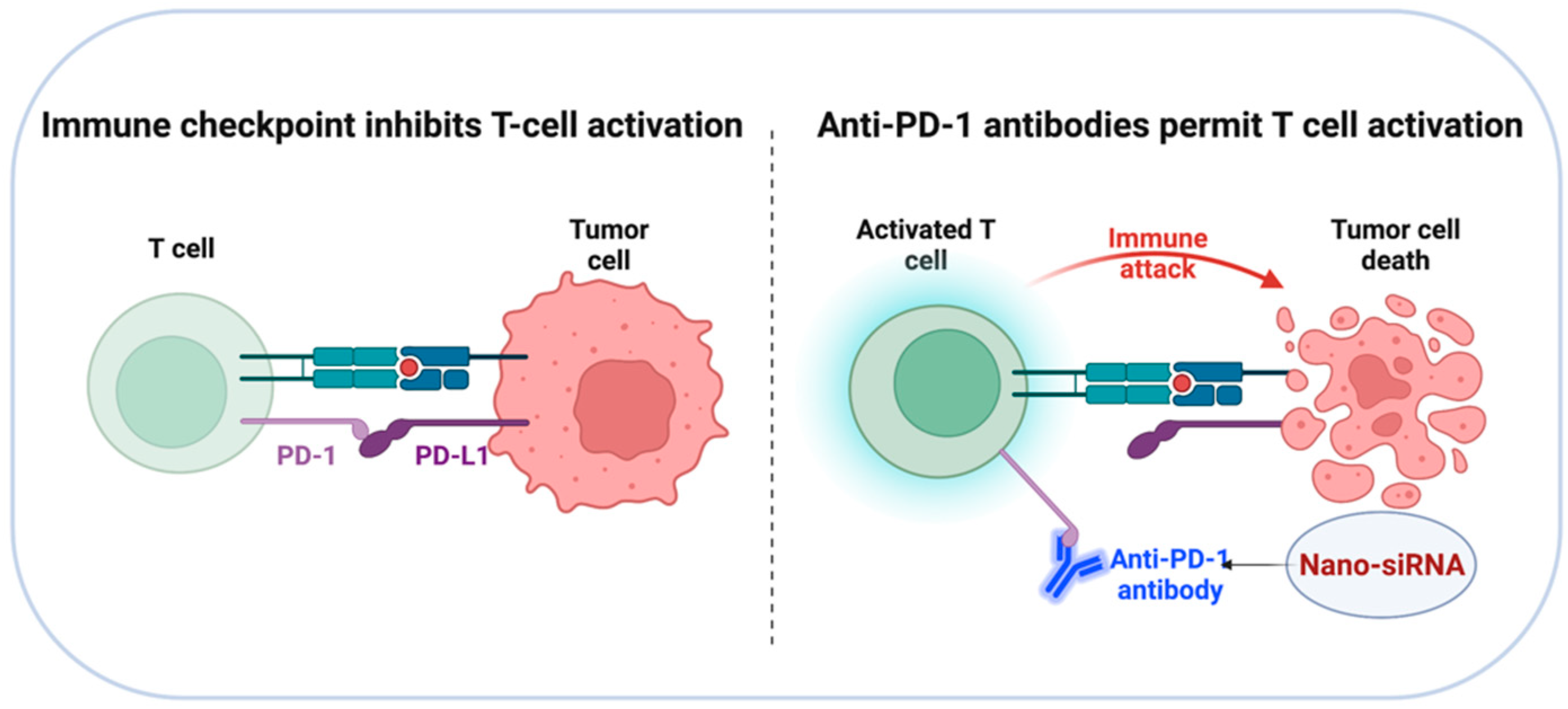
4. Targeted Delivery of siRNA to Checkpoint Inhibitors
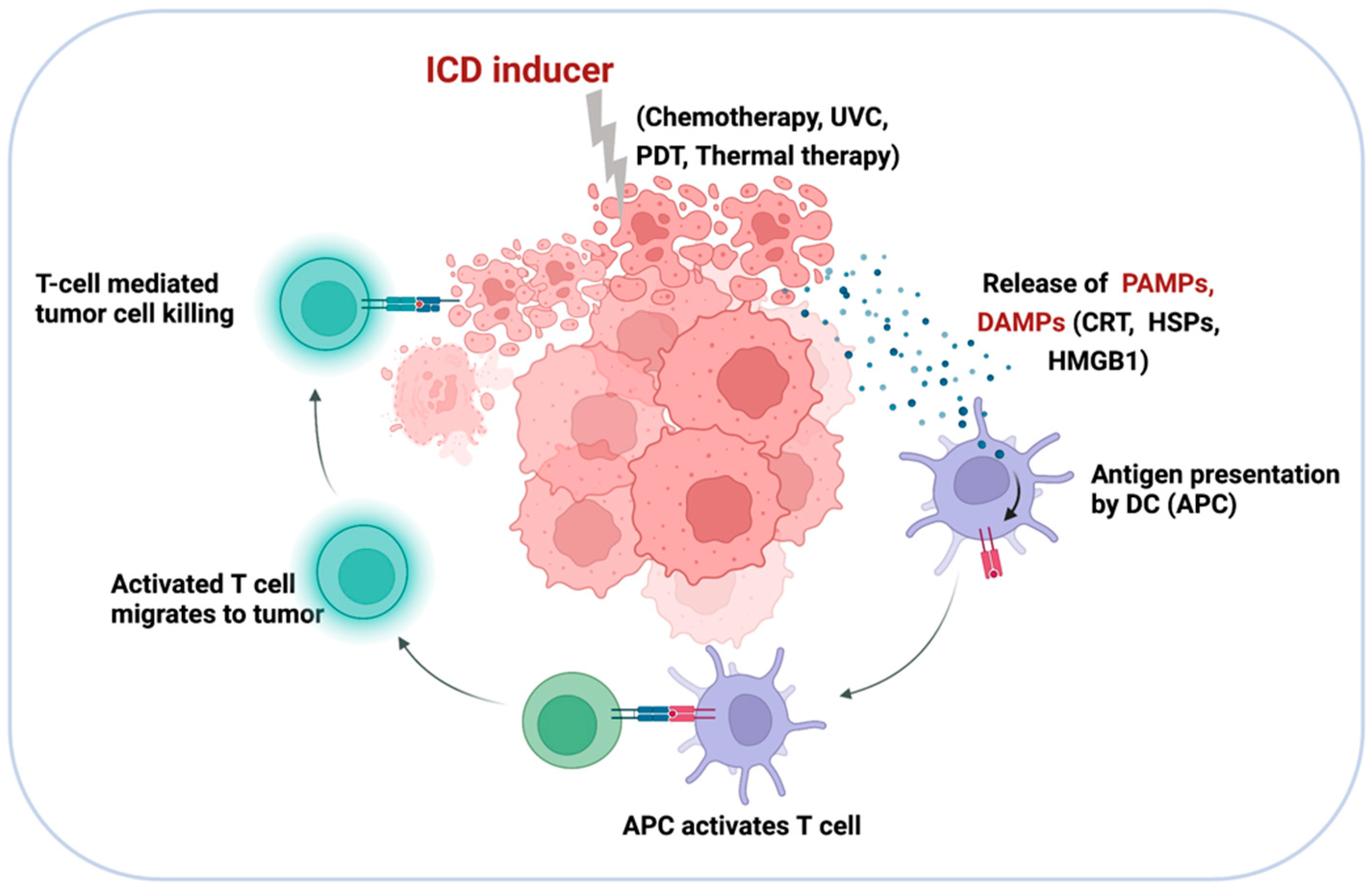
5. Targeting Tumor Cells with Nano-siRNA: ICD-Inducing Strategies
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.; Sadeghi, S.; Tabatabaeian, H. Battling Chemoresistance in Cancer: Root Causes and Strategies to Uproot Them. Int. J. Mol. Sci. 2021, 22, 9451. [Google Scholar] [CrossRef] [PubMed]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shao, C.; Shi, Y.; Han, W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J. Hematol. Oncol. 2018, 11, 31. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune system and melanoma biology: A balance between immunosurveillance and immune escape. Oncotarget 2017, 8, 106132–106142. [Google Scholar] [CrossRef]
- Tuccitto, A.; Shahaj, E.; Vergani, E.; Ferro, S.; Huber, V.; Rodolfo, M.; Castelli, C.; Rivoltini, L.; Vallacchi, V. Immunosuppressive circuits in tumor microenvironment and their influence on cancer treatment efficacy. Virchows Arch. 2019, 474, 407–420. [Google Scholar] [CrossRef]
- Allegrezza, M.J.; Conejo-Garcia, J.R. Targeted Therapy and Immunosuppression in the Tumor Microenvironment. Trends Cancer 2017, 3, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S. Improving cancer immunotherapy through nanotechnology. Nat. Rev. Cancer 2019, 19, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Trivedi, P.; Jain, N.K. Advances in siRNA delivery in cancer therapy. Artif. Cells Nanomed. Biotechnol. 2018, 46, 274–283. [Google Scholar] [CrossRef]
- Ahlquist, P. RNA-dependent RNA polymerases, viruses, and RNA silencing. Science 2002, 296, 1270–1273. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar] [CrossRef]
- Charbe, N.B.; Amnerkar, N.D.; Ramesh, B.; Tambuwala, M.M.; Bakshi, H.A.; Aljabali, A.A.A.; Khadse, S.C.; Satheeshkumar, R.; Satija, S.; Metha, M.; et al. Small interfering RNA for cancer treatment: Overcoming hurdles in delivery. Acta Pharm. Sin. B 2020, 10, 2075–2109. [Google Scholar] [CrossRef]
- Aghamiri, S.; Jafarpour, A.; Malekshahi, Z.V.; Mahmoudi Gomari, M.; Negahdari, B. Targeting siRNA in colorectal cancer therapy: Nanotechnology comes into view. J. Cell. Physiol. 2019, 234, 14818–14827. [Google Scholar] [CrossRef]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Steichen, S.D.; Caldorera-Moore, M.; Peppas, N.A. A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur. J. Pharm. Sci. 2013, 48, 416–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thambi, T.; Park, J.H.; Lee, D.S.J.C.C. Hypoxia-responsive nanocarriers for cancer imaging and therapy: Recent approaches and future perspectives. Chem. Commun. 2016, 52, 8492–8500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Gallis, B.; Taya, M.; Wang, S.; Ho, R.J.Y.; Sasaki, T. pH-responsive artemisinin derivatives and lipid nanoparticle formulations inhibit growth of breast cancer cells in vitro and induce down-regulation of HER family members. PLoS ONE 2013, 8, e59086. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Lin, Y.; Lin, Z.; Wei, Q.; Qian, J.; Ruan, R.; Jiang, X.; Hou, L.; Song, J.; Ding, J.; et al. Stimuli-Responsive Nanoparticles for Controlled Drug Delivery in Synergistic Cancer Immunotherapy. Adv. Sci. 2022, 9, e2103444. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, L.; Jordāo, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Guevara, M.L.; Persano, F.; Persano, S. Nano-immunotherapy: Overcoming tumour immune evasion. Semin. Cancer Biol. 2021, 69, 238–248. [Google Scholar] [CrossRef]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Passardi, A.; Canale, M.; Valgiusti, M.; Ulivi, P. Immune Checkpoints as a Target for Colorectal Cancer Treatment. Int. J. Mol. Sci. 2017, 18, 1324. [Google Scholar] [CrossRef]
- Pandya, P.H.; Murray, M.E.; Pollok, K.E.; Renbarger, J.L. The Immune System in Cancer Pathogenesis: Potential Therapeutic Approaches. J. Immunol. Res. 2016, 2016, 4273943. [Google Scholar] [CrossRef]
- Zhen, Y.; Zhang, H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ploquin, A.; Zhou, Y.; Sullivan, N.J. Ebola Immunity: Gaining a Winning Position in Lightning Chess. J. Immunol. 2018, 201, 833–842. [Google Scholar] [CrossRef]
- Burkholder, B.; Huang, R.-Y.; Burgess, R.; Luo, S.; Jones, V.S.; Zhang, W.; Lv, Z.-Q.; Gao, C.-Y.; Wang, B.-L.; Zhang, Y.-M.; et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta 2014, 1845, 182–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. Immune responses to malignancies. J. Allergy Clin. Immunol. 2010, 125, S272–S283. [Google Scholar] [CrossRef] [Green Version]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Lynn, G.M.; Laga, R.; Jewell, C.M. Induction of anti-cancer T cell immunity by in situ vaccination using systemically administered nanomedicines. Cancer Lett. 2019, 459, 192–203. [Google Scholar] [CrossRef]
- Piepenbrink, K.H.; Borbulevych, O.Y.; Sommese, R.F.; Clemens, J.; Armstrong, K.M.; Desmond, C.; Do, P.; Baker, B.M. Fluorine substitutions in an antigenic peptide selectively modulate T-cell receptor binding in a minimally perturbing manner. Biochem. J. 2009, 423, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, C.G.; Jaffee, E.; Pardoll, D.M. Mechanisms of immune evasion by tumors. Adv. Immunol. 2006, 90, 51–81. [Google Scholar] [PubMed]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Tricks tumors use to escape from immune control. Oral Oncol. 2009, 45, e119–e123. [Google Scholar] [CrossRef]
- Shaaban, M.; Othman, H.; Ibrahim, T.; Ali, M.; Abdelmoaty, M.; Abdel-Kawi, A.-R.; Mostafa, A.; El Nakeeb, A.; Emam, H.; Refaat, A. Immune Checkpoint Regulators: A New Era Toward Promising Cancer Therapy. Curr. Cancer Drug Targets 2020, 20, 429–460. [Google Scholar] [CrossRef]
- Abken, H.; Hombach, A.; Heuser, C.; Kronfeld, K.; Seliger, B. Tuning tumor-specific T-cell activation: A matter of costimulation? Trends Immunol. 2002, 23, 240–245. [Google Scholar] [CrossRef]
- Algarra, I.; García-Lora, A.; Cabrera, T.; Ruiz-Cabello, F.; Garrido, F. The selection of tumor variants with altered expression of classical and nonclassical MHC class I molecules: Implications for tumor immune escape. Cancer Immunol. Immunother. 2004, 53, 904–910. [Google Scholar] [CrossRef]
- Meissner, M.; Reichert, T.E.; Kunkel, M.; Gooding, W.; Whiteside, T.L.; Ferrone, S.; Seliger, B. Defects in the human leukocyte antigen class I antigen processing machinery in head and neck squamous cell carcinoma: Association with clinical outcome. Clin. Cancer Res. 2005, 11, 2552–2560. [Google Scholar] [CrossRef] [Green Version]
- Ogino, T.; Shigyo, H.; Ishii, H.; Katayama, A.; Miyokawa, N.; Harabuchi, Y.; Ferrone, S. HLA class I antigen down-regulation in primary laryngeal squamous cell carcinoma lesions as a poor prognostic marker. Cancer Res. 2006, 66, 9281–9289. [Google Scholar] [CrossRef] [Green Version]
- Jongsma, M.L.M.; Guarda, G.; Spaapen, R.M. The regulatory network behind MHC class I expression. Mol. Immunol. 2019, 113, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Denton, A.E.; Roberts, E.W.; Fearon, D.T. Stromal Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2018, 1060, 99–114. [Google Scholar] [PubMed]
- Musetti, S.; Huang, L. Nanoparticle-Mediated Remodeling of the Tumor Microenvironment to Enhance Immunotherapy. ACS Nano 2018, 12, 11740–11755. [Google Scholar] [CrossRef]
- Thakkar, S.; Sharma, D.; Kalia, K.; Tekade, R.K. Tumor microenvironment targeted nanotherapeutics for cancer therapy and diagnosis: A review. Acta Biomater. 2020, 101, 43–68. [Google Scholar] [CrossRef]
- Sun, X.; Xing, L.; Deng, X.; Hsiao, H.T.; Manami, A.; Koutcher, J.A.; Clifton Ling, C.; Li, G.C. Hypoxia targeted bifunctional suicide gene expression enhances radiotherapy in vitro and in vivo. Radiother. Oncol. 2012, 105, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Rajendrakumar, S.K.; Uthaman, S.; Cho, C.-S.; Park, I.-K. Nanoparticle-Based Phototriggered Cancer Immunotherapy and Its Domino Effect in the Tumor Microenvironment. Biomacromolecules 2018, 19, 1869–1887. [Google Scholar] [CrossRef]
- Larionova, I.; Cherdyntseva, N.; Liu, T.; Patysheva, M.; Rakina, M.; Kzhyshkowska, J. Interaction of tumor-associated macrophages and cancer chemotherapy. Oncoimmunology 2019, 8, 1596004. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-W.; Choi, H.-J.; Ha, S.-J.; Lee, K.-T.; Kwon, Y.-G. Recruitment of monocytes/macrophages in different tumor microenvironments. Biochim. Biophys. Acta 2013, 1835, 170–179. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasievich, E.A.; Huang, L. The suppressive tumor microenvironment: A challenge in cancer immunotherapy. Mol. Pharm. 2011, 8, 635–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer, N.; Kim, E.; Sprang, B.; Leukel, P.; Khafaji, F.; Ringel, F.; Sommer, C.; Tuettenberg, J.; Tuettenberg, A. GARP as an Immune Regulatory Molecule in the Tumor Microenvironment of Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshie, O.; Matsushima, K. CCR4 and its ligands: From bench to bedside. Int. Immunol. 2015, 27, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-L.; Li, J.; Mo, H.-Y.; Qiu, F.; Zheng, L.-M.; Qian, C.-N.; Zeng, Y.-X. Different subsets of tumor infiltrating lymphocytes correlate with NPC progression in different ways. Mol. Cancer 2010, 9, 4. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Cai, S.F.; Fehniger, T.A.; Song, J.; Collins, L.I.; Piwnica-Worms, D.R.; Ley, T.J. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 2007, 27, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.S.K. Treg and CTLA-4: Two intertwining pathways to immune tolerance. J. Autoimmun. 2013, 45, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Fallarino, F.; Grohmann, U.; Hwang, K.W.; Orabona, C.; Vacca, C.; Bianchi, R.; Belladonna, M.L.; Fioretti, M.C.; Alegre, M.-L.; Puccetti, P. Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol. 2003, 4, 1206–1212. [Google Scholar] [CrossRef]
- Wang, H.; Franco, F.; Ho, P.-C. Metabolic Regulation of Tregs in Cancer: Opportunities for Immunotherapy. Trends Cancer 2017, 3, 583–592. [Google Scholar] [CrossRef]
- Yao, S.; Zhu, Y.; Zhu, G.; Augustine, M.; Zheng, L.; Goode, D.J.; Broadwater, M.; Ruff, W.; Flies, S.; Xu, H.; et al. B7-h2 is a costimulatory ligand for CD28 in human. Immunity 2011, 34, 729–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terme, M.; Colussi, O.; Marcheteau, E.; Tanchot, C.; Tartour, E.; Taieb, J. Modulation of immunity by antiangiogenic molecules in cancer. Clin. Dev. Immunol. 2012, 2012, 492920. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Aldawsari, H.M.; Gorain, B.; Alhakamy, N.A.; Md, S. Role of therapeutic agents on repolarisation of tumour-associated macrophage to halt lung cancer progression. J. Drug Target. 2020, 28, 166–175. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Petty, A.J.; Yang, Y. Tumor-associated macrophages: Implications in cancer immunotherapy. Immunotherapy 2017, 9, 289–302. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P. The interaction of anticancer therapies with tumor-associated macrophages. J. Exp. Med. 2015, 212, 435–445. [Google Scholar] [CrossRef]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Pyzer, A.R.; Cole, L.; Rosenblatt, J.; Avigan, D.E. Myeloid-derived suppressor cells as effectors of immune suppression in cancer. Int. J. Cancer 2016, 139, 1915–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condamine, T.; Mastio, J.; Gabrilovich, D.I. Transcriptional regulation of myeloid-derived suppressor cells. J. Leukoc. Biol. 2015, 98, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, K.; Guilliams, M.; Van den Bossche, J.; Van den Bergh, R.; Gysemans, C.; Beschin, A.; De Baetselier, P.; Van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.-I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef]
- Cassim, S.; Pouyssegur, J. Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int. J. Mol. Sci. 2019, 21, 157. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Liu, G.; Liu, S.; Su, H.; Wang, Y.; Li, J.; Luo, C. Remodeling the Tumor Microenvironment with Emerging Nanotherapeutics. Trends Pharmacol. Sci. 2018, 39, 59–74. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [Green Version]
- Phuengkham, H.; Ren, L.; Shin, I.W.; Lim, Y.T. Nanoengineered Immune Niches for Reprogramming the Immunosuppressive Tumor Microenvironment and Enhancing Cancer Immunotherapy. Adv. Mater. 2019, 31, e1803322. [Google Scholar] [CrossRef]
- Li, M.; Zhang, F.; Su, Y.; Zhou, J.; Wang, W. Nanoparticles designed to regulate tumor microenvironment for cancer therapy. Life Sci. 2018, 201, 37–44. [Google Scholar] [CrossRef]
- Overchuk, M.; Zheng, G. Overcoming obstacles in the tumor microenvironment: Recent advancements in nanoparticle delivery for cancer theranostics. Biomaterials 2018, 156, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Pi, F.; Sharma, A.; Rajabi, M.; Haque, F.; Shu, D.; Leggas, M.; Evers, B.M.; Guo, P. Stable RNA nanoparticles as potential new generation drugs for cancer therapy. Adv. Drug Deliv. Rev. 2014, 66, 74–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Yu, T.; Ding, L.; Laurini, E.; Huang, Y.; Zhang, M.; Weng, Y.; Lin, S.; Chen, P.; Marson, D.; et al. A Dual Targeting Dendrimer-Mediated siRNA Delivery System for Effective Gene Silencing in Cancer Therapy. J. Chem. Soc. 2018, 140, 16264–16274. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.J.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, U.B.; Gunnarsson, L.; Géraudie, S.; Scheffler, U.; Griep, R.A.; Reiersen, H.; Duncan, A.R.; Kiprijanov, S.M. Fully human antagonistic antibodies against CCR4 potently inhibit cell signaling and chemotaxis. PLoS ONE 2014, 9, e103776. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L.; Mandapathil, M.; Schuler, P. The role of the adenosinergic pathway in immunosuppression mediated by human regulatory T cells (Treg). Curr. Med.Chem. 2011, 18, 5217–5223. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, A.; Priceman, S.J.; Swiderski, P.; Kujawski, M.; Xin, H.; Cherryholmes, G.A.; Zhang, W.; Zhang, C.; Lahtz, C.; Kowolik, C.; et al. CTLA4 aptamer delivers STAT3 siRNA to tumor-associated and malignant T cells. J. Clin. Investig. 2014, 124, 2977–2987. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-Y.; Liu, Y.; Xu, C.-F.; Shen, S.; Sun, R.; Du, X.-J.; Xia, J.-X.; Zhu, Y.-H.; Wang, J. Restoring anti-tumor functions of T cells via nanoparticle-mediated immune checkpoint modulation. J. Control. Release 2016, 231, 17–28. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, D.; Liu, J.; Han, Y.; Xu, H.; Leng, X.; Kong, D.; Liu, L. Hybrid spherical nucleotide nanoparticles can enhance the synergistic anti-tumor effect of CTLA-4 and PD-1 blockades. Biomater. Sci. 2020, 8, 4757–4766. [Google Scholar] [CrossRef]
- Li, X.; Liu, R.; Su, X.; Pan, Y.; Han, X.; Shao, C.; Shi, Y. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol. Cancer 2019, 18, 177. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Guo, J.; Huang, L. Tackling TAMs for Cancer Immunotherapy: It’s Nano Time. Trends Pharmacol. Sci. 2020, 41, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, J.; Gu, P.; Fan, X. The application of nanoparticles in cancer immunotherapy: Targeting tumor microenvironment. Bioact. Mater. 2021, 6, 1973–1987. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Zeng, Q.; Jewell, C.M. Directing toll-like receptor signaling in macrophages to enhance tumor immunotherapy. Curr. Opin. Biotechnol. 2019, 60, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Klinman, D.M. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat. Rev. Immunol. 2004, 4, 249–258. [Google Scholar] [CrossRef]
- Gao, M.; Ha, T.; Zhang, X.; Wang, X.; Liu, L.; Kalbfleisch, J.; Singh, K.; Williams, D.; Li, C. The Toll-like receptor 9 ligand, CpG oligodeoxynucleotide, attenuates cardiac dysfunction in polymicrobial sepsis, involving activation of both phosphoinositide 3 kinase/Akt and extracellular-signal-related kinase signaling. J. Infect. Dis. 2013, 207, 1471–1479. [Google Scholar] [CrossRef] [Green Version]
- Shan, H.; Dou, W.; Zhang, Y.; Qi, M. Targeted ferritin nanoparticle encapsulating CpG oligodeoxynucleotides induces tumor-associated macrophage M2 phenotype polarization into M1 phenotype and inhibits tumor growth. Nanoscale 2020, 12, 22268–22280. [Google Scholar] [CrossRef]
- Qian, H.; Zhou, T.; Fu, Y.; Guo, M.; Yang, W.; Zhang, D.; Fang, W.; Yao, M.; Shi, H.; Chai, C.; et al. Self-assembled tetrahedral framework nucleic acid mediates tumor-associated macrophage reprogramming and restores antitumor immunity. Mol. Ther. Nucleic Acids 2022, 27, 763–773. [Google Scholar] [CrossRef]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Qian, Y.; Qiao, S.; Dai, Y.; Xu, G.; Dai, B.; Lu, L.; Yu, X.; Luo, Q.; Zhang, Z. Molecular-Targeted Immunotherapeutic Strategy for Melanoma via Dual-Targeting Nanoparticles Delivering Small Interfering RNA to Tumor-Associated Macrophages. ACS Nano 2017, 11, 9536–9549. [Google Scholar] [CrossRef]
- Huang, C.; Jin, H.; Qian, Y.; Qi, S.; Luo, H.; Luo, Q.; Zhang, Z. Hybrid melittin cytolytic Peptide-driven ultrasmall lipid nanoparticles block melanoma growth in vivo. ACS Nano 2013, 7, 5791–5800. [Google Scholar] [CrossRef] [PubMed]
- Pluen, A.; Boucher, Y.; Ramanujan, S.; McKee, T.D.; Gohongi, T.; di Tomaso, E.; Brown, E.B.; Izumi, Y.; Campbell, R.B.; Berk, D.A.; et al. Role of tumor-host interactions in interstitial diffusion of macromolecules: Cranial vs. subcutaneous tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 4628–4633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Messmer, M.N.; Netherby, C.S.; Banik, D.; Abrams, S.I. Tumor-induced myeloid dysfunction and its implications for cancer immunotherapy. Cancer Immunol. Immunother. 2015, 64, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Amoozgar, Z.; Goldberg, M.S. Targeting myeloid cells using nanoparticles to improve cancer immunotherapy. Adv. Drug Deliv. Rev. 2015, 91, 38–51. [Google Scholar] [CrossRef]
- Hossain, D.M.S.; Pal, S.K.; Moreira, D.; Duttagupta, P.; Zhang, Q.; Won, H.; Jones, J.; D’Apuzzo, M.; Forman, S.; Kortylewski, M. TLR9-Targeted STAT3 Silencing Abrogates Immunosuppressive Activity of Myeloid-Derived Suppressor Cells from Prostate Cancer Patients. Clin.Cancer Res. 2015, 21, 3771–3782. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Li, M.; Ran, G.; Wang, X.; Lu, Z.; Li, T.; Tang, X.; Zhang, Z.; He, Q. Redox-responsive nanoassembly restrained myeloid-derived suppressor cells recruitment through autophagy-involved lactate dehydrogenase A silencing for enhanced cancer immunochemotherapy. J. Control. Release 2021, 335, 557–574. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Shipp, M.A.; Ribrag, V.; Michot, J.-M.; Zinzani, P.L.; Kuruvilla, J.; Snyder, E.S.; Ricart, A.D.; Balakumaran, A.; Rose, S.; et al. Programmed Death-1 Blockade with Pembrolizumab in Patients with Classical Hodgkin Lymphoma After Brentuximab Vedotin Failure. J. Clin. Oncol. 2016, 34, 3733–3739. [Google Scholar] [CrossRef] [PubMed]
- Teo, P.Y.; Yang, C.; Whilding, L.M.; Parente-Pereira, A.C.; Maher, J.; George, A.J.T.; Hedrick, J.L.; Yang, Y.Y.; Ghaem-Maghami, S. Ovarian cancer immunotherapy using PD-L1 siRNA targeted delivery from folic acid-functionalized polyethylenimine: Strategies to enhance T cell killing. Adv. Healthc. Mater. 2015, 4, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Roeven, M.W.H.; Hobo, W.; van der Voort, R.; Fredrix, H.; Norde, W.J.; Teijgeler, K.; Ruiters, M.H.J.; Schaap, N.; Dolstra, H. Efficient nontoxic delivery of PD-L1 and PD-L2 siRNA into dendritic cell vaccines using the cationic lipid SAINT-18. J. Immunother. 2015, 38, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Wang, T.; Ma, F.; Zhang, K.; Gao, T.; Pei, R.; Zhang, Y. Aptamer-functionalized targeted siRNA delivery system for tumor immunotherapy. Biomed. Mater. 2022, 17, 14. [Google Scholar] [CrossRef]
- Adkins, I.; Fucikova, J.; Garg, A.D.; Agostinis, P.; Špíšek, R. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology 2014, 3, e968434. [Google Scholar] [CrossRef]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [Green Version]
- Inoue, H.; Tani, K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ. 2014, 21, 39–49. [Google Scholar] [CrossRef] [Green Version]
- De Munck, J.; Binks, A.; McNeish, I.A.; Aerts, J.L. Oncolytic virus-induced cell death and immunity: A match made in heaven? J. Leukoc. Biol. 2017, 102, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krysko, O.; Aaes, T.L.; Kagan, V.E.; D’Herde, K.; Bachert, C.; Leybaert, L.; Vandenabeele, P.; Krysko, D.V. Necroptotic cell death in anti-cancer therapy. Immunol. Rev. 2017, 280, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Chan, C.; Lin, W. Nanoparticle-Mediated Immunogenic Cell Death Enables and Potentiates Cancer Immunotherapy. Angew. Chem. 2019, 58, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, W.; Chen, X.; Wang, Q.; Li, C.; Chen, Q.; Zhang, Y.; Lu, Y.; Ding, X.; Jiang, C. Bone marrow mesenchymal stem cells-derived exosomes for penetrating and targeted chemotherapy of pancreatic cancer. Acta Pharm. Sin. B 2020, 10, 1563–1575. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Liu, X.; Liao, Y.-P.; Salazar, F.; Sun, B.; Jiang, W.; Chang, C.H.; Jiang, J.; Wang, X.; Wu, A.M.; et al. Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression. Nat. Commun. 2017, 8, 1811. [Google Scholar] [CrossRef] [Green Version]
- Niculescu, A.-G.; Grumezescu, A.M. Photodynamic Therapy—An up-to-Date Review. Appl. Sci. 2021, 11, 3626. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Chizenga, E.P.; Abrahamse, H. Nanotechnology in Modern Photodynamic Therapy of Cancer: A Review of Cellular Resistance Patterns Affecting the Therapeutic Response. Pharmaceutics 2020, 12, 632. [Google Scholar] [CrossRef]
- Monge-Fuentes, V.; Muehlmann, L.A.; de Azevedo, R.B. Perspectives on the application of nanotechnology in photodynamic therapy for the treatment of melanoma. Nano Rev. 2014, 5, 24381. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.; Hu, D.; Zheng, M.; Zhao, P.; Liu, H.; Gao, D.; Gong, P.; Gao, G.; Zhang, P.; Ma, Y.; et al. Smart human serum albumin-indocyanine green nanoparticles generated by programmed assembly for dual-modal imaging-guided cancer synergistic phototherapy. ACS Nano 2014, 8, 12310–12322. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, H.; Guo, X.; Wang, Z.; Kong, F.; Luo, L.; Li, Q.; Zhu, C.; Yang, J.; Lou, Y.; et al. Gold Nanospheres-Stabilized Indocyanine Green as a Synchronous Photodynamic-Photothermal Therapy Platform That Inhibits Tumor Growth and Metastasis. ACS Appl. Mater. Interfaces 2017, 9, 3354–3367. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Fan, H.; Zhou, G.; Bai, H.; Liang, H.; Wang, R.; Zhang, X.; Tan, W. Activatable fluorescence/MRI bimodal platform for tumor cell imaging via MnO2 nanosheet-aptamer nanoprobe. J. Chem. Soc. 2014, 136, 11220–11223. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.; Gordijo, C.R.; Abbasi, A.Z.; Maeda, A.; Ip, A.; Rauth, A.M.; DaCosta, R.S.; Wu, X.Y.J. Multifunctional albumin–MnO2 nanoparticles modulate solid tumor microenvironment by attenuating hypoxia, acidosis, vascular endothelial growth factor and enhance radiation response. ACS Nano 2014, 8, 3202–3212. [Google Scholar] [CrossRef]
- Liu, Y.; Pan, Y.; Cao, W.; Xia, F.; Liu, B.; Niu, J.; Alfranca, G.; Sun, X.; Ma, L.; de la Fuente, J.M.; et al. A tumor microenvironment responsive biodegradable CaCO/MnO-based nanoplatform for the enhanced photodynamic therapy and improved PD-L1 immunotherapy. Theranostics 2019, 9, 6867–6884. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, K.; Yang, D.; Zhou, Y. Nanotechnology-Based siRNA Delivery Systems to Overcome Tumor Immune Evasion in Cancer Immunotherapy. Pharmaceutics 2022, 14, 1344. https://doi.org/10.3390/pharmaceutics14071344
Deng K, Yang D, Zhou Y. Nanotechnology-Based siRNA Delivery Systems to Overcome Tumor Immune Evasion in Cancer Immunotherapy. Pharmaceutics. 2022; 14(7):1344. https://doi.org/10.3390/pharmaceutics14071344
Chicago/Turabian StyleDeng, Kaili, Dongxue Yang, and Yuping Zhou. 2022. "Nanotechnology-Based siRNA Delivery Systems to Overcome Tumor Immune Evasion in Cancer Immunotherapy" Pharmaceutics 14, no. 7: 1344. https://doi.org/10.3390/pharmaceutics14071344
APA StyleDeng, K., Yang, D., & Zhou, Y. (2022). Nanotechnology-Based siRNA Delivery Systems to Overcome Tumor Immune Evasion in Cancer Immunotherapy. Pharmaceutics, 14(7), 1344. https://doi.org/10.3390/pharmaceutics14071344