
Mitochondria-Targeted Delivery of Camptothecin Based on HPMA Copolymer for Metastasis Suppression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cells and Animals
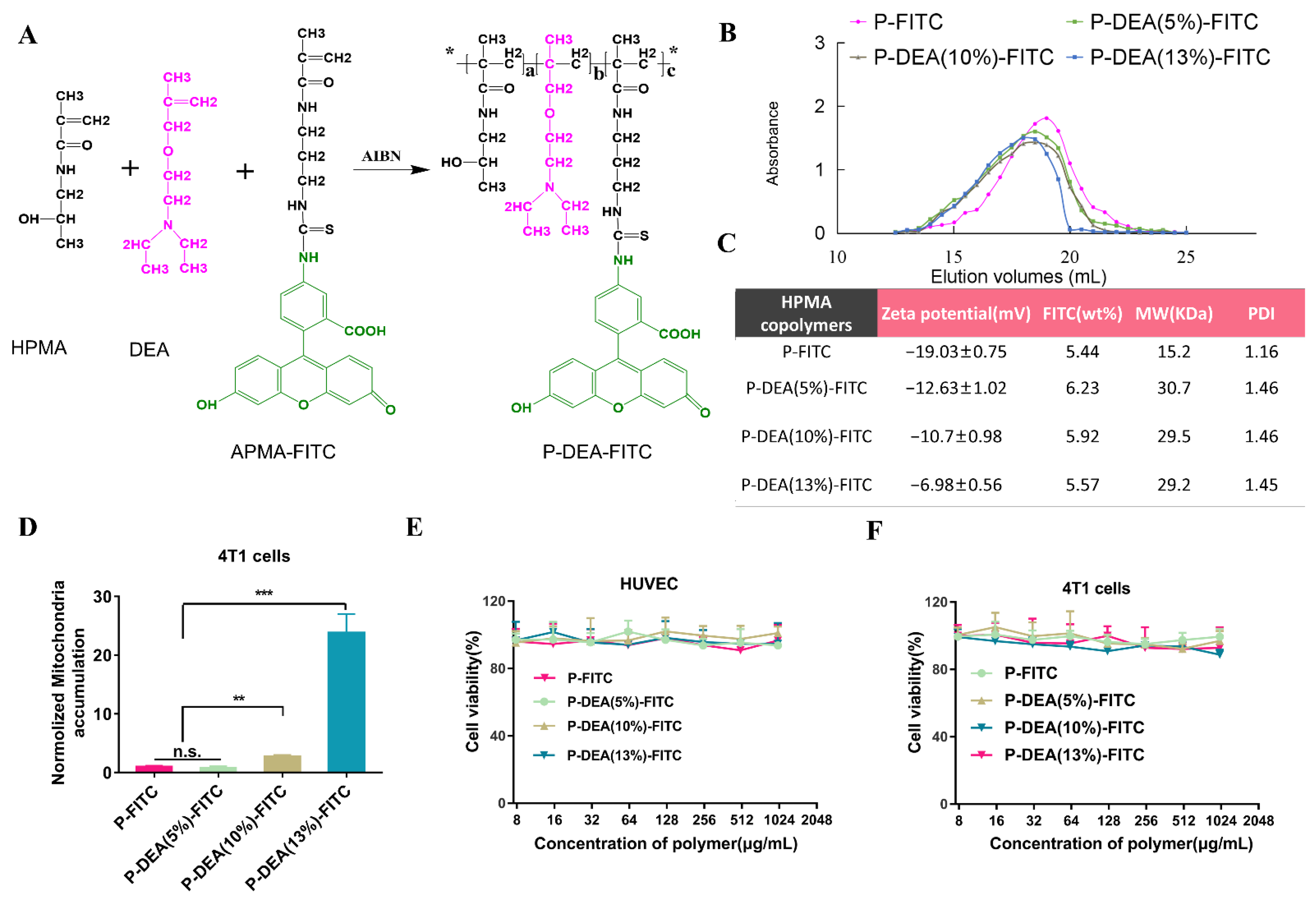
2.3. Synthesis, Characterization, and Mitochondria-Targeting Capacity of HPMA Copolymers with Different Modification Ratios of DEA
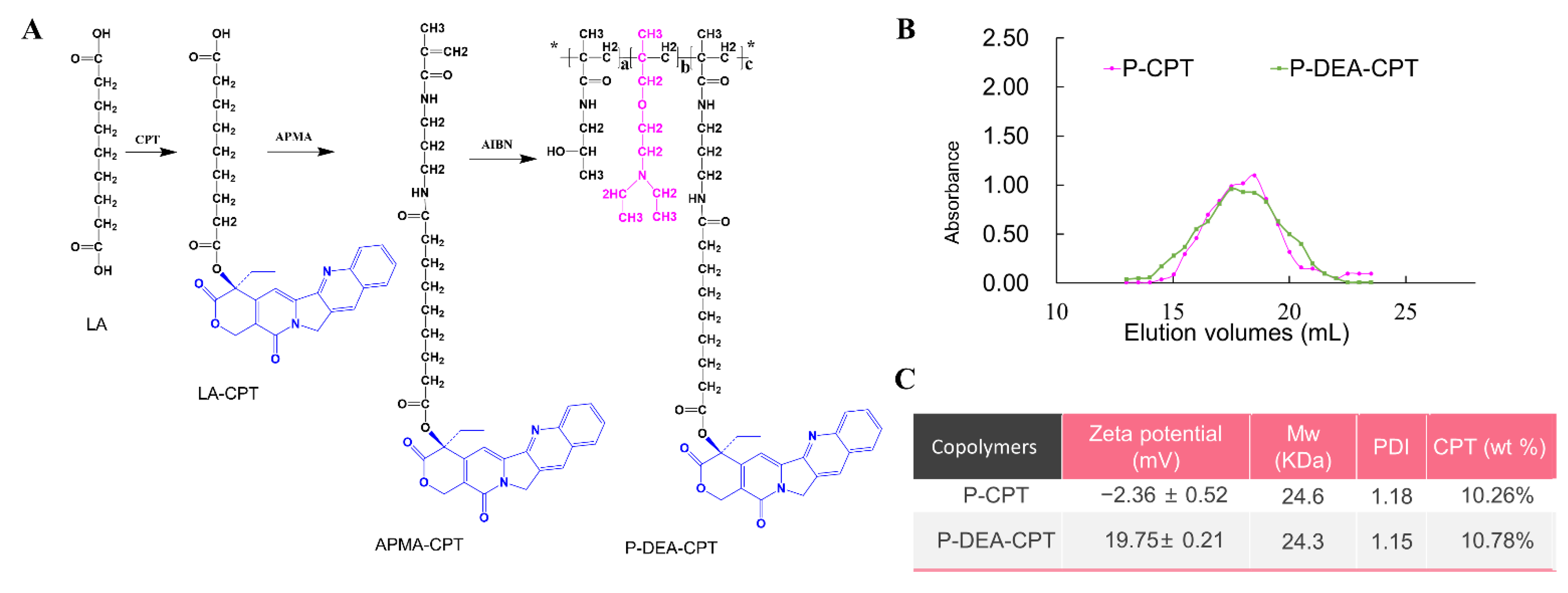
2.4. Synthesis and Characterization of HPMA Copolymer-CPT Conjugates
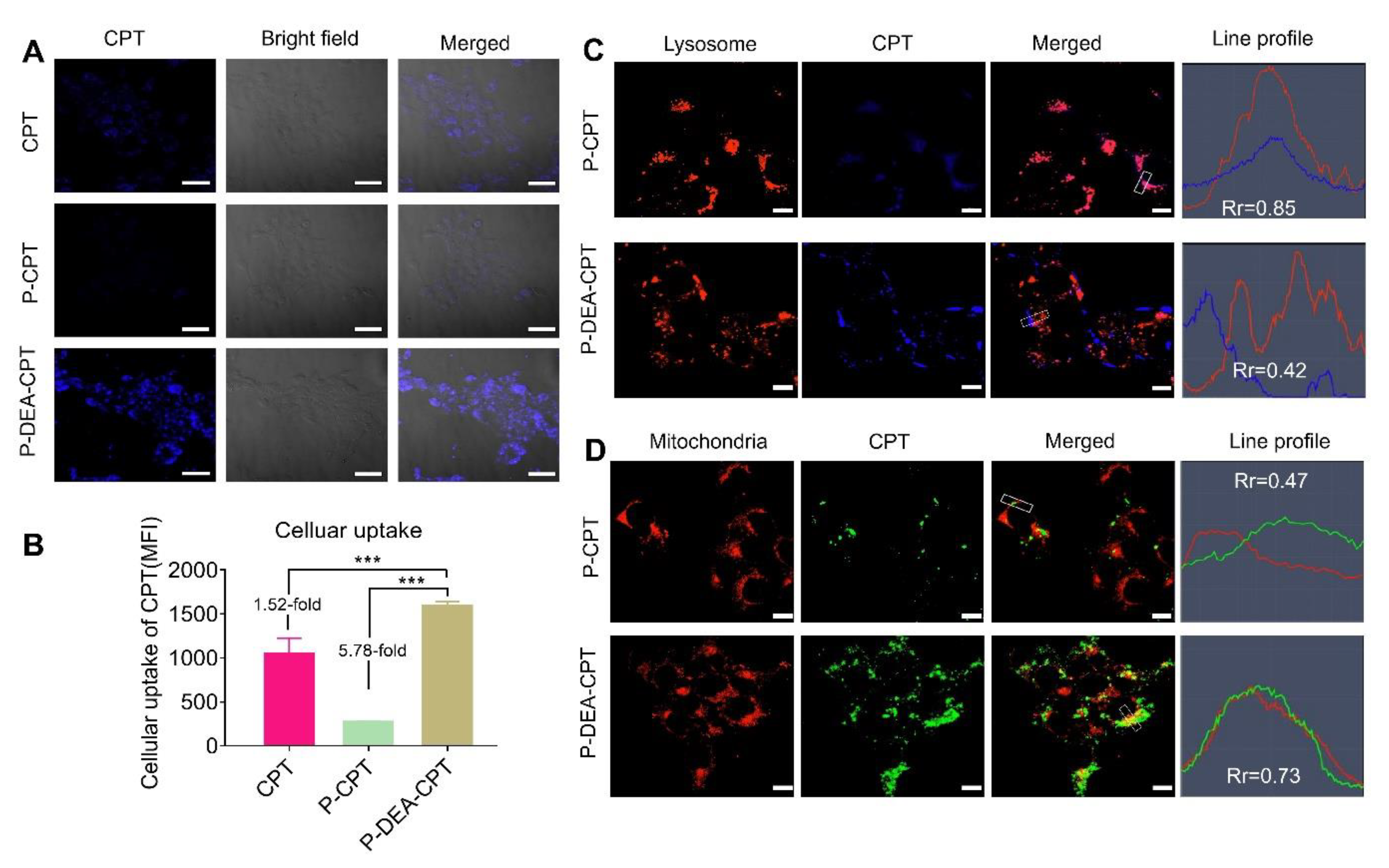
2.5. Cellular Uptake, Lysosome Escape, and Mitochondrial Targeting of HPMA Copolymer–CPT Conjugates
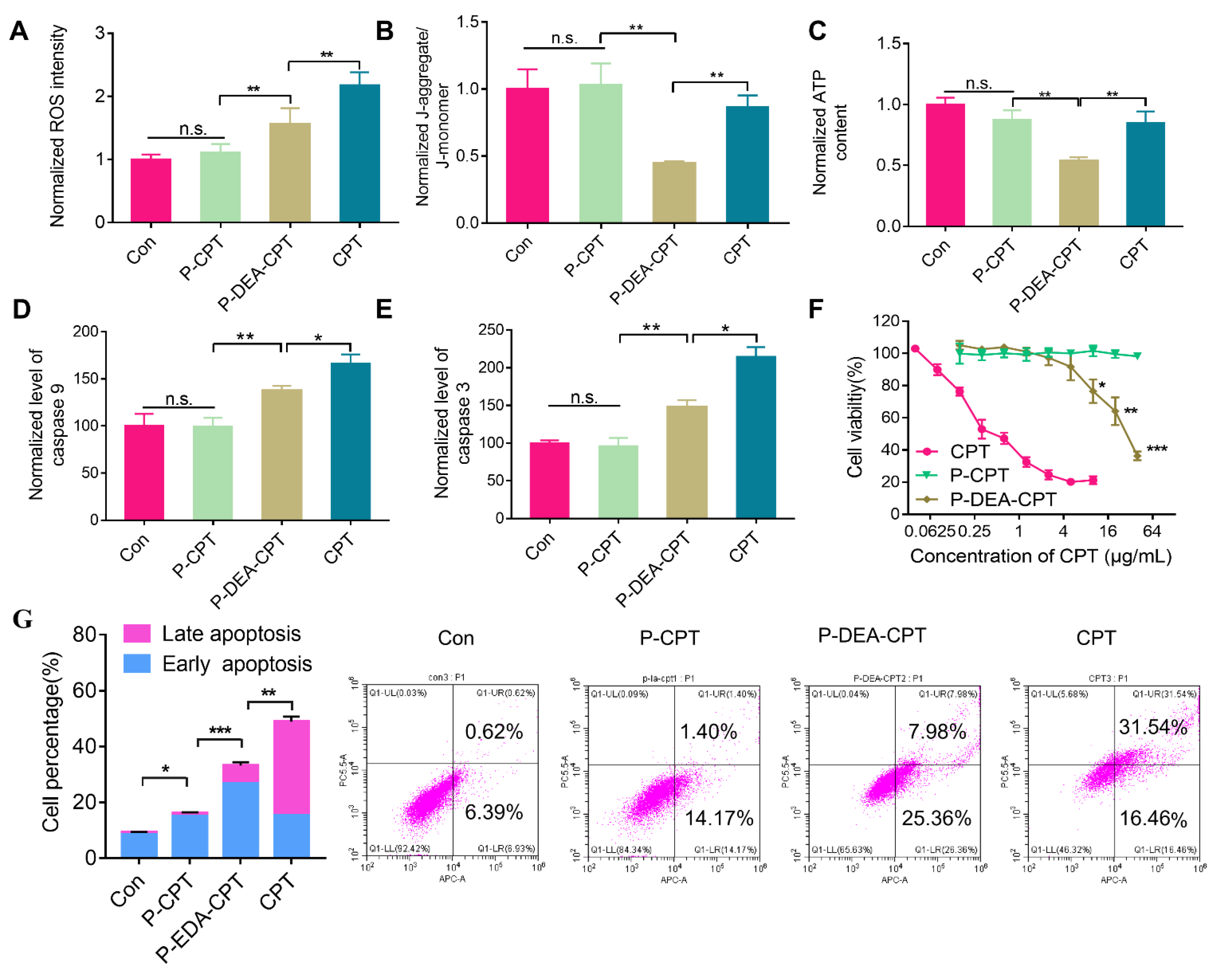
2.6. In Vitro Mitochondrial Damage by HPMA Copolymer–Camptothecin Conjugates
2.6.1. Reactive Oxygen Species Detection
2.6.2. Mitochondrial Membrane Potential Detection
2.6.3. Measurement of ATP Level
2.6.4. Detection of Caspase 9 and Caspase 3
2.7. Cell Proliferation Suppression Efficacy of HPMA Copolymer–Camptothecin Conjugates
2.8. In Vitro Anti-Metastasis Assay
2.9. Intratumoral Mitochondria Targeting Assay
2.10. In Vivo Antitumor and Anti-Metastasis Evaluation
2.11. Statistical Analysis
3. Results and discussion
3.1. DEA Content-Dependent Mitochondrial Targeting Capacity of HPMA Conjugates
3.2. Synthesis and Characterization of HPMA Copolymer–CPT Conjugates
3.3. Cellular Uptake, Lysosome Escape, and Mitochondrial Targeting of P-DEA-CPT
3.4. In Vitro Mitochondrial Damage-Induced Apoptosis and Cytotoxicity of P-DEA-CPT
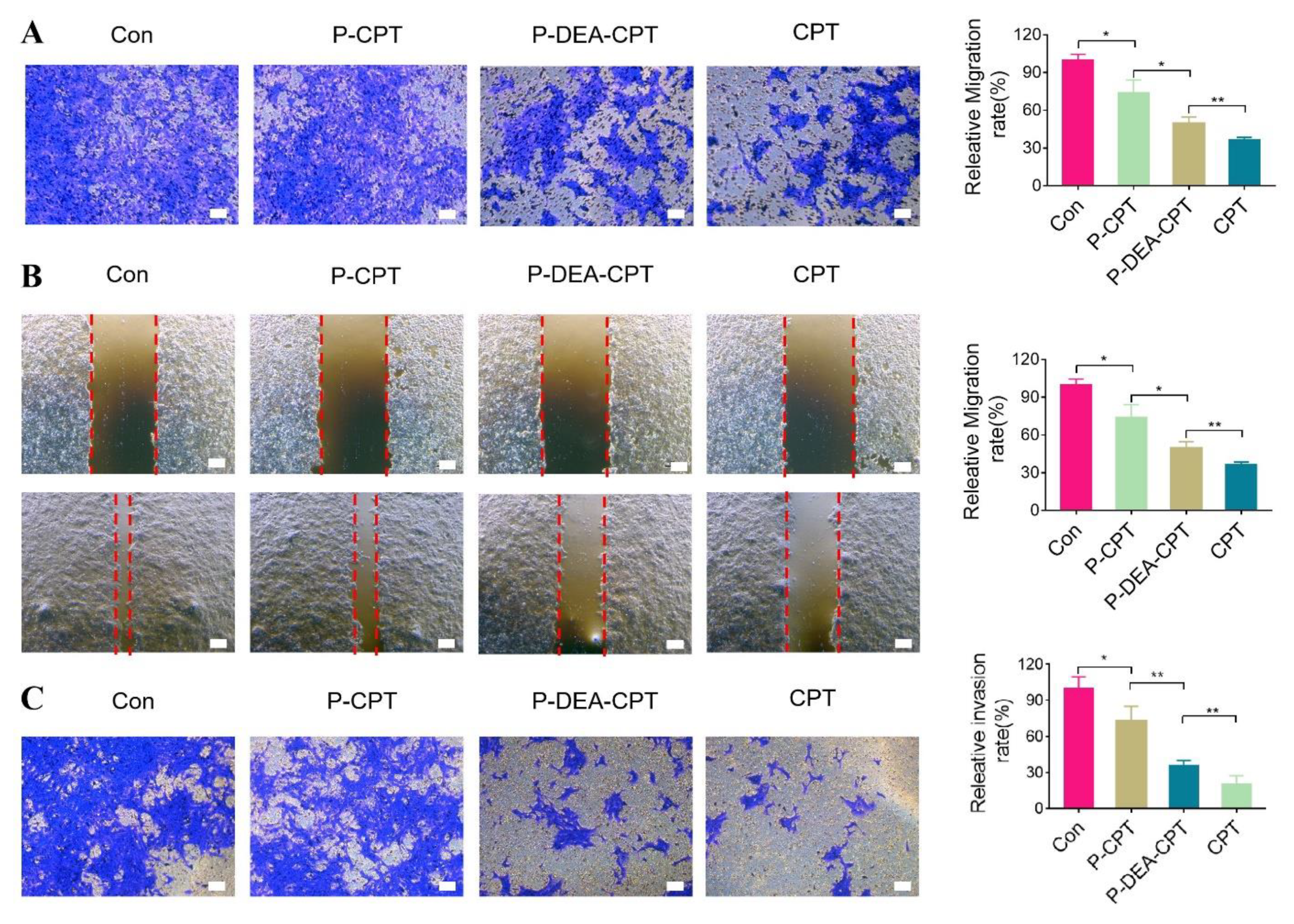
3.5. In Vitro Anti-Metastasis Effect of P-DEA-CPT
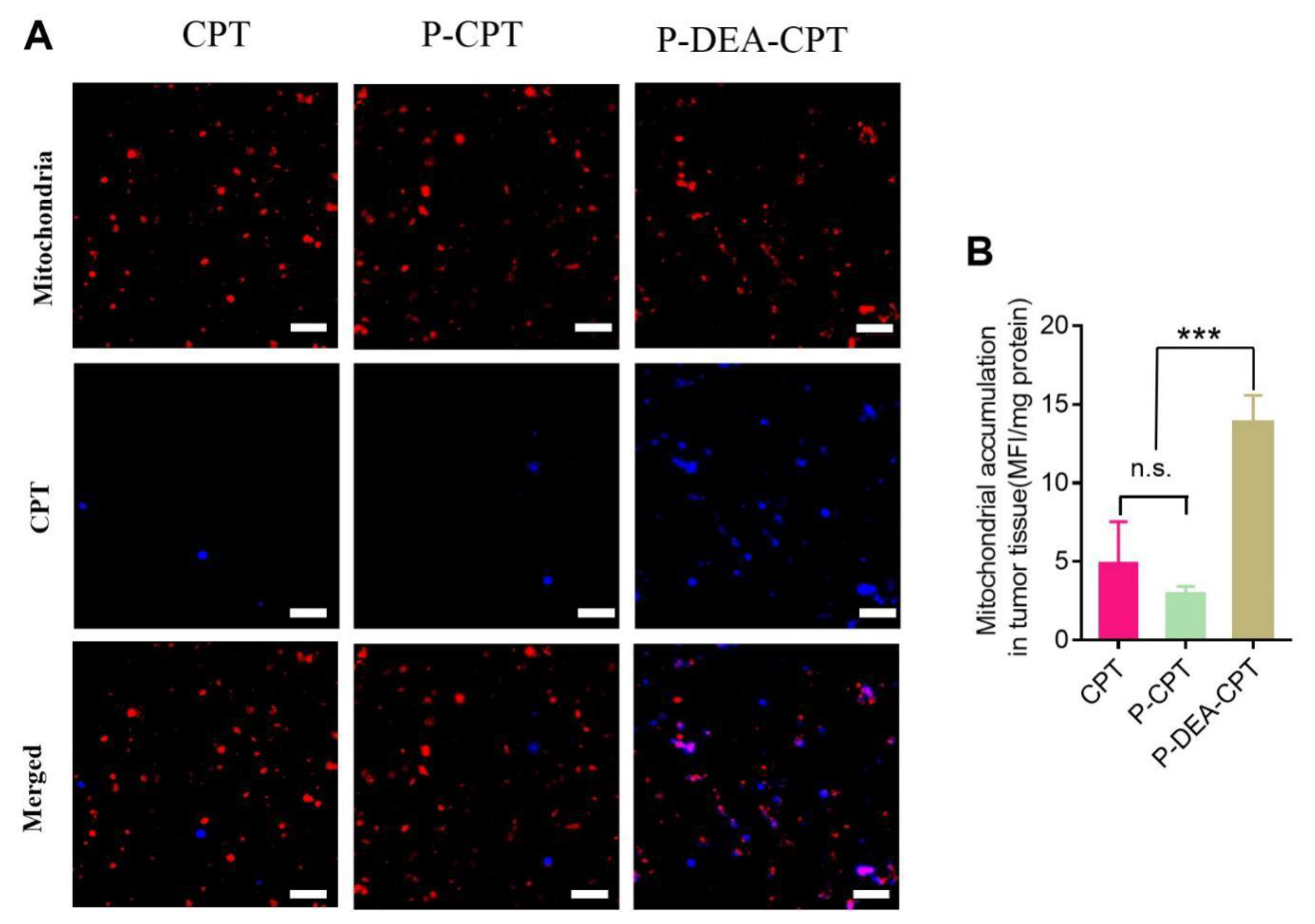
3.6. Intratumoral Mitochondrial Targeting of P-DEA-CPT
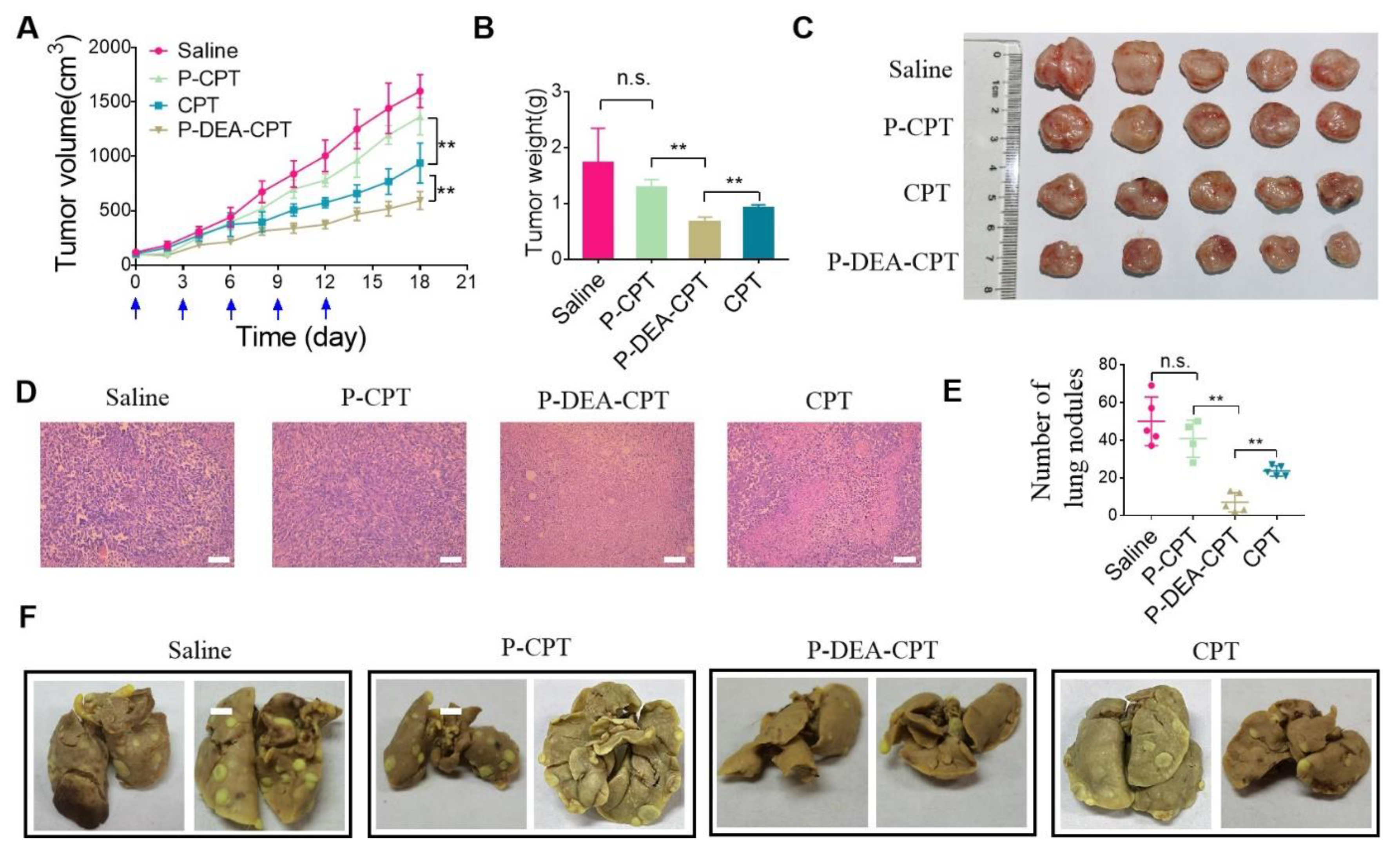
3.7. In Vivo Antitumor and Anti-Metastasis Effect of P-DEA-CPT
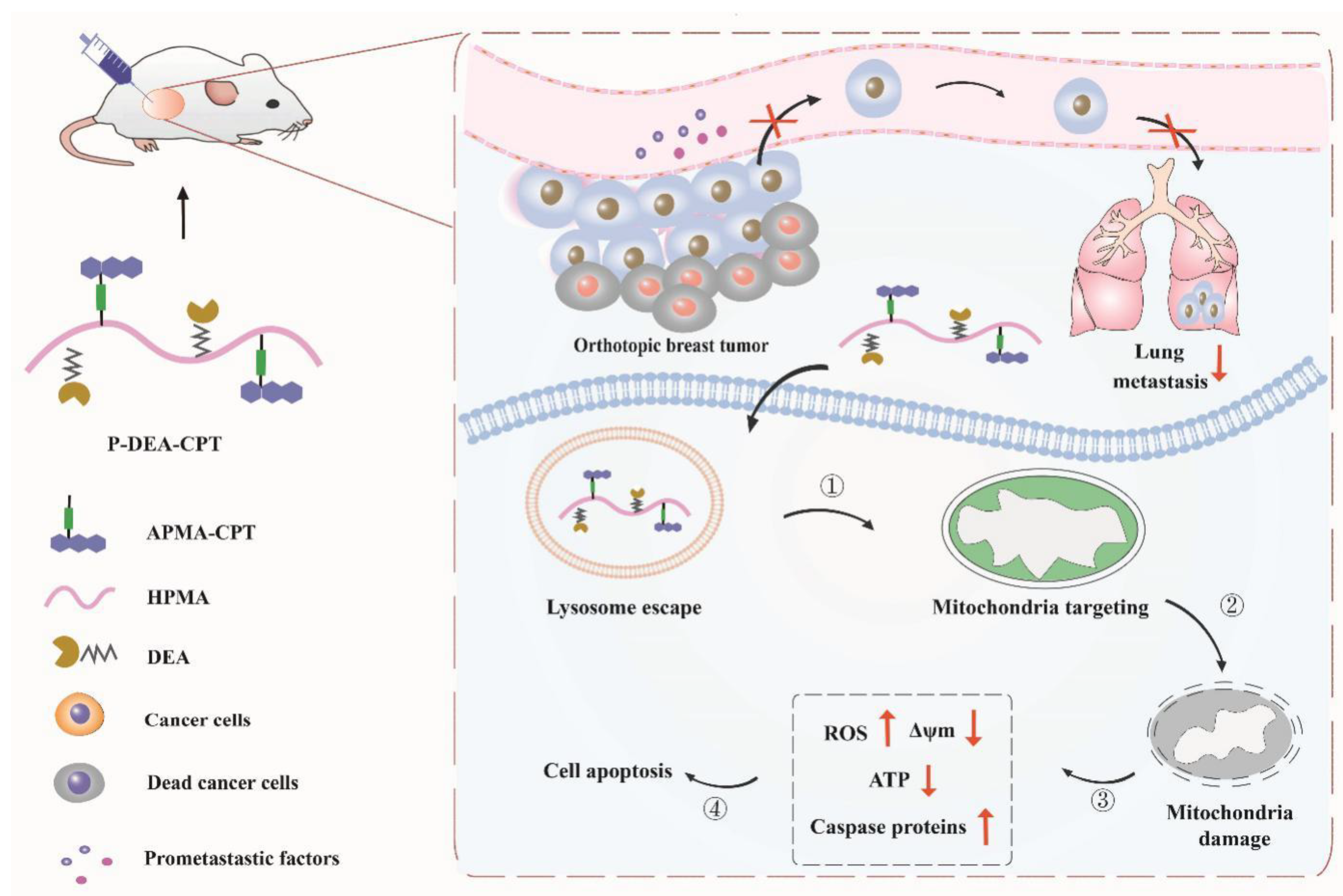
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barua, S.; Mitragotri, S. Synergistic targeting of cell membrane, cytoplasm, and nucleus of cancer cells using rod-shaped nanoparticles. ACS Nano 2013, 7, 9558–9570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Huang, Y.; Shen, Q.; Zhao, Y.; Wang, W.; Yu, J.; Lu, W. A camptothecin-based, albumin-binding prodrug enhances efficacy and safety in vivo. Eur. J. Med. Chem. 2021, 226, 113851. [Google Scholar] [CrossRef]
- Ghanbari-Movahed, M.; Kaceli, T.; Mondal, A.; Farzaei, M.H.; Bishayee, A. Recent advances in improved anticancer efficacies of camptothecin nano-formulations: A systematic review. Biomedicines 2021, 9, 480. [Google Scholar] [CrossRef] [PubMed]
- Sadalage, P.S.; Patil, R.V.; Havaldar, D.V.; Gavade, S.S.; Santos, A.C.; Pawar, K.D. Optimally biosynthesized, PEGylated gold nanoparticles functionalized with quercetin and camptothecin enhance potential anti-inflammatory, anti-cancer and anti-angiogenic activities. J. Nanobiotechnol. 2021, 19, 84. [Google Scholar] [CrossRef] [PubMed]
- Lian, Q.; Xu, J.; Yan, S.; Huang, M.; Ding, H.; Sun, X.; Geng, M. Chemotherapy-induced intestinal inflammatory responses are mediated by exosome secretion of double-strand DNA via AIM2 inflammasome activation. Cell Res. 2017, 27, 784–800. [Google Scholar] [CrossRef] [PubMed]
- Swami, U.; Goel, S.; Mani, S. Therapeutic targeting of CPT-11 induced diarrhea: A case for prophylaxis. Curr. Drug Targets 2013, 14, 777–797. [Google Scholar] [CrossRef] [Green Version]
- Roth, K.G.; Mambetsariev, I.; Kulkarni, P.; Salgia, R. The mitochondrion as an emerging therapeutic target in cancer. Trends Mol. Med. 2020, 26, 119–134. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Reichard, A.; Asosingh, K. The role of mitochondria in angiogenesis. Mol. Biol. Rep. 2019, 46, 1393–1400. [Google Scholar] [CrossRef]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; Kalluri, R. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Li, Q.; Xing, L.; Zhou, M.; Luo, C.; Li, S.; Huang, Y. Co-delivery of mitochondrial targeted lonidamine and PIN1 inhibitor ATRA by nanoparticulate systems for synergistic metastasis suppression. Nano Res. 2022, 15, 3376–3386. [Google Scholar] [CrossRef]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Yan, Y.; Li, L.; Li, Q.; Xiang, Y.; Huang, Y. Sequentially targeting cancer-associated fibroblast and mitochondria alleviates tumor hypoxia and inhibits cancer metastasis by preventing “soil” formation and “seed” dissemination. Adv. Funct. Mater. 2021, 31, 2010283. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, X.; Shen, Q.; Xing, D. Mitochondria-specific drug release and reactive oxygen species burst induced by polyprodrug nanoreactors can enhance chemotherapy. Nat. Commun. 2019, 10, 1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, T.; Hou, C.; Zu, M.; Lu, Y.; Ma, X.; Xu, Z. Mitochondria-specific anticancer drug delivery based on reduction-activated polyprodrug for enhancing the therapeutic effect of breast cancer chemotherapy. ACS Appl. Mater. Interfaces 2019, 11, 29330–29340. [Google Scholar] [CrossRef]
- Ma, X.; Gong, N.; Zhong, L.; Sun, J.; Liang, X.J. Future of nanotherapeutics: Targeting the cellular sub-organelles. Biomaterials 2016, 97, 10–21. [Google Scholar] [CrossRef]
- Li, Q.; Yang, J.; Chen, C.; Lin, X.; Zhou, M.; Zhou, Z.; Huang, Y. A novel mitochondrial targeted hybrid peptide modified HPMA copolymers for breast cancer metastasis suppression. J. Control. Release 2020, 325, 38–51. [Google Scholar] [CrossRef]
- Zhou, M.; Li, L.; Li, L.; Lin, X.; Wang, F.; Li, Q.; Huang, Y. Overcoming chemotherapy resistance via simultaneous drug-efflux circumvention and mitochondrial targeting. Acta Pharm. Sin. B 2019, 9, 615–625. [Google Scholar] [CrossRef]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Kalyanaraman, B. Mitochondria-targeted triphenylphosphonium-based compounds: Syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Qin, J.; Gong, N.; Liao, Z.; Zhang, S.; Timashev, P.; Huo, S.; Liang, X.J. Recent progress in mitochondria-targeting-based nanotechnology for cancer treatment. Nanoscale 2021, 13, 7108–7118. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, J.; Lin, J.; Ye, K.; Wei, P. Mitochondria-targeting and ROS-responsive nanocarriers via amphiphilic TPP-PEG-TK-Ce6 for nanoenabled photodynamic therapy. Adv. Polym. Technol. 2022, 2022, 1178039. [Google Scholar] [CrossRef]
- Yi, X.; Yan, Y.; Li, L.; Zhou, R.; Shen, X.; Huang, Y. Combination of mitochondria impairment and inflammation blockade to combat metastasis. J. Control. Release 2022, 341, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Sun, W.; Li, L.; Li, L.; Liu, Y.; Zhang, Z.R.; Huang, Y. Charge-reversible multifunctional HPMA copolymers for mitochondrial targeting. ACS Appl. Mater. Interfaces 2017, 9, 27563–27574. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, L.; Li, L.J.; Yang, Q.Q.; Zhang, Z.R.; Huang, Y. Two birds, one stone: Dual targeting of the cancer cell surface and subcellular mitochondria by the galectin-3-binding peptide G3-C12. Acta Pharmacologica. Sin. 2017, 38, 806–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [Green Version]
- Lakhani, S.A.; Masud, A.; Kuida, K.; Porter Jr, G.A.; Booth, C.J.; Mehal, W.Z.; Flavell, R.A. Caspases 3 and 7: Key mediators of mitochondrial events of apoptosis. Science 2006, 311, 847. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Snezhkina, A.V. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [Green Version]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Kumar, S.; Eroglu, E.; Stokes III, J.A.; Scissum-Gunn, K.; Saldanha, S.N.; Singh, U.P.; Mishra, M.K. Resveratrol induces mitochondria-mediated, caspase-independent apoptosis in murine prostate cancer cells. Oncotarget 2017, 8, 20895–20908. [Google Scholar] [CrossRef] [PubMed]
- Dilshara, M.G.; Jayasooriya, R.G.P.T.; Karunarathne, W.A.H.M.; Choi, Y.H.; Kim, G.Y. Camptothecin induces mitotic arrest through Mad2-Cdc20 complex by activating the JNK-mediated Sp1 pathway. Food Chem. Toxicol. 2019, 127, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.J.; Xiong, Y.; Wang, Y.; Tong, Q.; Yang, G.S. Sirt1 attenuates camptothecin-induced apoptosis through caspase-3 pathway in porcine preadipocytes. Exp. Cell Res. 2013, 319, 670–683. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Pantel, K.; Kang, Y. Tumor metastasis: Moving new biological insights into the clinic. Nat. Med. 2013, 19, 1450–1464. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [Green Version]
- Valastyan, S.; RWeinberg, A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Jin, Q.; Deng, Y.; Chen, X.; Ji, J. Rational design of cancer nanomedicine for simultaneous stealth surface and enhanced cellular uptake. Acs Nano 2019, 13, 954–977. [Google Scholar] [CrossRef]
- Wang, R.; Yin, C.; Liu, C.; Sun, Y.; Xiao, P.; Li, J.; Jiang, X. Phenylboronic acid modification augments the lysosome escape and antitumor efficacy of a cylindrical polymer brush-based prodrug. J. Am. Chem. Soc. 2021, 143, 20927–20938. [Google Scholar] [CrossRef]
- Liberman, E.A.; Topaly, V.P.; Tsofina, L.M.; Jasaitis, A.A.; Skulachev, V.P. Mechanism of coupling of oxidative phosphorylation and the membrane potential of mitochondria. Nature 1969, 222, 1076–1078. [Google Scholar] [CrossRef]
- Jiang, T.; Chen, L.; Huang, Y.; Wang, J.; Xu, M.; Zhou, S.; Chen, J. Metformin and docosahexaenoic acid hybrid micelles for premetastatic niche modulation and tumor metastasis suppression. Nano Lett. 2019, 19, 3548–3562. [Google Scholar] [CrossRef]
- Arroyo-Crespo, J.J.; Armiñán, A.; Charbonnier, D.; Deladriere, C.; Palomino-Schätzlein, M.; Lamas-Domingo, R.; Vicent, M.J. Characterization of triple-negative breast cancer preclinical models provides functional evidence of metastatic progression. Int. J. Cancer 2019, 145, 2267–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Chen, L.; Li, L.; Huang, Y. Restoration and enhancement of immunogenic cell death of cisplatin by coadministration with digoxin and conjugation to HPMA copolymer. ACS Appl. Mater. Interfaces 2020, 12, 1606–1616. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Pressnall, M.M.; Lu, R.; Huayamares, S.G.; Griffin, J.D.; Groer, C.; Berkland, C.J. Human intratumoral therapy: Linking drug properties and tumor transport of drugs in clinical trials. J. Control. Release 2020, 326, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Mai, X.; Zhang, Y.; Fan, H.; Song, W.; Chang, Y.; Chen, B.; Teng, G. Integration of immunogenic activation and immunosuppressive reversion using mitochondrial-respiration-inhibited platelet-mimicking nanoparticles. Biomaterials 2020, 232, 119699. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, J.; Liu, S.; Li, X.; Miao, L.; Yang, B.; Guan, W. Defeating relapsed and refractory malignancies through a nano-enabled mitochondria-mediated respiratory inhibition and damage pathway. Biomaterials 2020, 229, 119580. [Google Scholar] [CrossRef]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, X.; Yan, Y.; Shen, X.; Li, L.; Huang, Y. Mitochondria-Targeted Delivery of Camptothecin Based on HPMA Copolymer for Metastasis Suppression. Pharmaceutics 2022, 14, 1534. https://doi.org/10.3390/pharmaceutics14081534
Yi X, Yan Y, Shen X, Li L, Huang Y. Mitochondria-Targeted Delivery of Camptothecin Based on HPMA Copolymer for Metastasis Suppression. Pharmaceutics. 2022; 14(8):1534. https://doi.org/10.3390/pharmaceutics14081534
Chicago/Turabian StyleYi, Xiaoli, Yue Yan, Xinran Shen, Lian Li, and Yuan Huang. 2022. "Mitochondria-Targeted Delivery of Camptothecin Based on HPMA Copolymer for Metastasis Suppression" Pharmaceutics 14, no. 8: 1534. https://doi.org/10.3390/pharmaceutics14081534
APA StyleYi, X., Yan, Y., Shen, X., Li, L., & Huang, Y. (2022). Mitochondria-Targeted Delivery of Camptothecin Based on HPMA Copolymer for Metastasis Suppression. Pharmaceutics, 14(8), 1534. https://doi.org/10.3390/pharmaceutics14081534