1. Introduction
Tissue regeneration of a hair follicle with complex architecture is not trivial, and requires multiple tissues including epithelium, dermis, muscle, and nerve. In 1954, Dr. Breedis described de novo hair regeneration after large full-thickness (LFT) wounding [
1], and decades later wound-induced hair follicle neogenesis (WIHN) was characterized for the mouse [
2]. The WIHN is a powerful assay for studying de novo skin organ regeneration after full-thickness trauma. Multiple studies have investigated the mechanism of WIHN, namely the requirement for Wnt/β-catenin [
2], derivation of FGF9 from γδT-cells [
3], the activation of TLR3 leading to p-STAT3 induction of Wnt/β-catenin [
4], epithelial competency [
5], and tissue mechanics [
6]. Interestingly, mouse hairs regenerate only in the wound center [
2,
3,
4], and humans fail to regenerate resulting in scar [
7]. Scar tissue does not contain ectodermal organs or rete ridges and is relatively stiff compared to normal tissue [
8]; this stiffness could play a role in regulating tissue regeneration.
Human terminal hair is similar to mouse primary follicles, and human vellus hair is similar to mouse secondary follicles. Humans do not regenerate terminal or vellus hair after LFT wound healing, instead forming scar tissue, and this is a significant clinical issue [
9]. Scar tissue is more stiff than normal tissue and can be reduced by modulating focal adhesion kinase (FAK) signaling [
10]. The laboratory mouse C57BL/6 regenerates hair follicles after LFT wounding in the center of the wound bed but not at the margin during WIHN [
2,
3,
4,
5,
6]. Additionally, these mice only regenerate secondary hair follicles, specifically zigzag [
2,
11]. Secondary hair follicles are less complex than primary follicles [
12]. This raises many questions, for example whether there are inhibitors expressed from the wound edge that inhibit WIHN or if is there another mechanism at work. Is there a minimum distance between the wound center and margin for WIHN and tissue regeneration? Why does WIHN only generate zigzag follicles? Interestingly, African spiny mice can regenerate primary and secondary hair follicles, including all four hair shaft types [
11]. Furthermore, they can regenerate these hair follicles in the wound margin, unlike C57BL/6 mice. This extraordinary regeneration ability is not restricted to the skin organ. Ear cartilage, skeletal muscle, spinal cord, and kidneys also exhibit higher regenerative ability [
13]. A clue to how this ability works is that African spiny mice exhibit softer skin tissue and this associates with greater regenerative ability [
6,
14]. Therefore, we surmised a gradient could be established in the wound bed that modulates tissue regeneration and patterning. Utilizing C57BL/6 mice, we studied how initial wound shape affected tissue regeneration, WIHN, and hair patterning by increasing the distance between the wound margin and center. Our hypothesis is that, when increasing the distance from the wound margin to the wound center during LFT wound healing, both WIHN and tissue regeneration would be increased.
Here we confirm a stiffness gradient in the LFT wound bed, low in the center and high in the margin, and that manipulating wound stiffness physically or chemically regulates regenerative potential. This result partly answers the question of why hair regenerates in the wound center. Previous reports described a role for FAK during scar formation [
8,
10,
15], and here we show a role for the FAK/SMA/MyoII axis during skin regeneration after LFT injury. Finally, these results demonstrate mechanobiological cues are sensed throughout the wound bed to regulate tissue regeneration through p-STAT3 induction. In summary, we were able to control hair follicle neogenesis in LFT wounds by specifically modulating the wound bed stiffness. We studied the wound healing responses of ‘repair’, namely re-epithelialization with scar formation resulting in loss of structure and function, and ‘regeneration’, the restoration of ectodermal organs, tissue integrity, and function to near normal. The need to understand cellular and molecular mechanisms modulating wound healing and tissue regeneration is imperative. Regenerative medicine therapies can be developed targeting the ability to activate regenerative signals that simultaneously inhibit scar formation and enhance regeneration after LFT severe trauma.
2. Materials and Methods
2.1. Animal Models
All animal work was performed according to the approved animal protocol number 109292, approved 12 August 2020, in accordance with the guidelines and regulations for the care and use of laboratory animals at National Cheng Kung University. All mice were housed in climate-controlled indoor facilities. The C56BL/6J mice were purchased from the National Cheng Kung University Animal Center. For live in vivo imaging, Gt(ROSA)26Sortm4(ACTB-tdTomato-EGFP)Luo/J mice purchased from Jackson Laboratory were utilized.
2.2. Wound-Induced Hair Neogenesis Assay
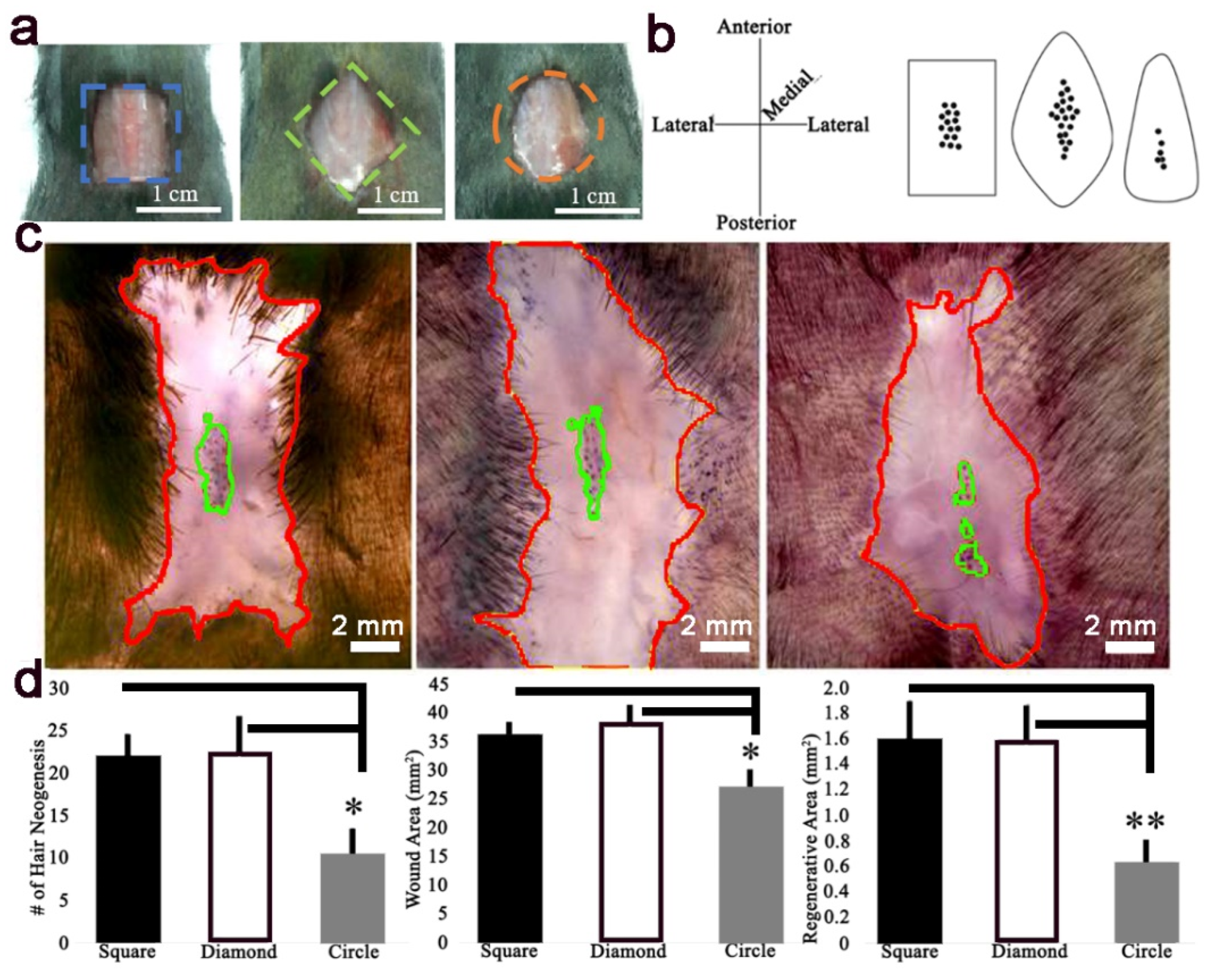
The WHIN assay was previously described [
2]. Briefly, a 1 cm
2 square, 1 cm
2 diamond, or 1 cm
2 circle full-thickness wound was excised on the posterior dorsum of 3-week-old mice and observed for hair neogenesis. Mice were anesthetized using isoflurane, full-thickness skin was excised, and the analgesic ketorolac (12 μg/g b.w.) was given by intraperitoneal injection (IP). To create the diamond wound, a 1 cm
2 square wound was excised at a 45-degree rotation relative to the square wound. To create the circle wound, a circle with radius 0.564 cm was excised. All wound shapes contained equal excised areas of 1 cm
2.
2.3. Inhibitor Treatments
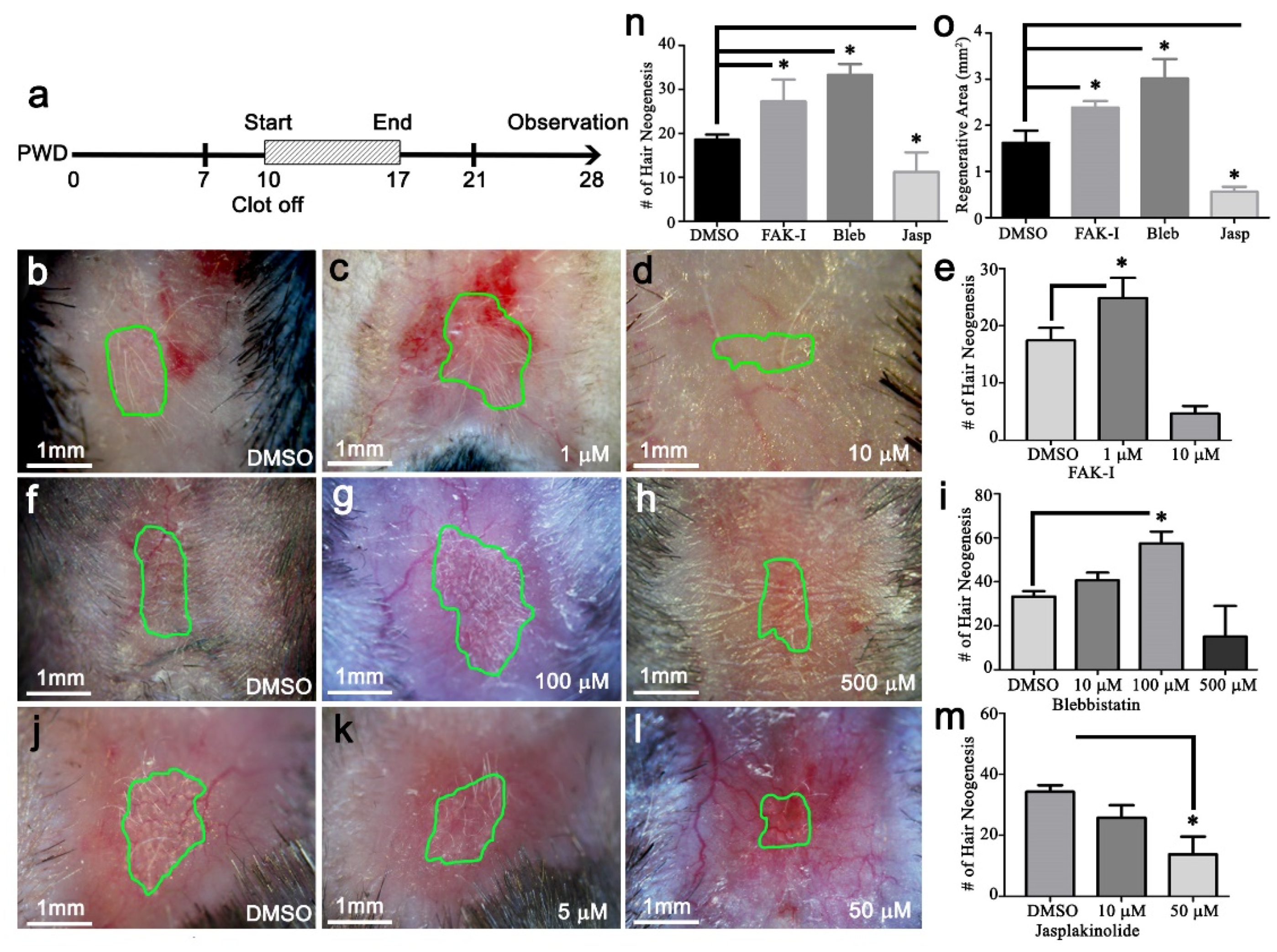
The PF573228, blebbistatin, or jasplakinolide (Cayman, Ann Arbor, MI, USA) were dissolved in DMSO. Then, 20 μL was applied once per day directly to the wound surface, starting at PWD 10 and continuing until PWD 17.
2.4. Separation of Epidermis and Dermis
The epidermis was separated from the dermis of wounds as previously reported [
2]. Briefly, full-thickness wounds were incubated in 20 mm EDTA in phosphate buffered saline (PBS) at 37 °C overnight. The next day, the epithelium was gently peeled away from the dermis with fine watchmaker’s forceps.
2.5. Alkaline Phosphatase (ALP) Stain
To detect newly formed dermal papillae, alkaline phosphatase (ALP) staining was performed as previously reported [
2]. Briefly, full-thickness wounds were excised, the and epidermis was separated from the dermis using 20 mM EDTA. The dermis was fixed in acetone overnight at 4 °C and washed in PBS several times. The dermis was preincubated in ALP buffer (0.1 M Tris-HCl, 0.1 M NaCl, 5 mM MgCl
2, and 0.1% Tween 20) for 30 min, and then incubated with BCIP/NBT color development substrate (Promega) in ALP buffer at 37 °C until color development. The reaction was stopped by washing with pH 8.0 Tris-EDTA, and the tissue was stored in PBS with sodium azide.
2.6. Atomic Force Microscopy (AFM)
In this study, AFM (Nanowizard II, JPK, Berlin, Germany) was set up for contact mode indentation in PBS. The spring constants of all cantilevers were calibrated via the thermal noise method with a correction factor in liquid [
16,
17] prior to each measurement, and were valued at 0.08 N/m. To overcome the unevenness of skin wound tissue, we attached a large polystyrene bead (25 μm in diameter) on tipless cantilevers (CSC12-F, MikroMasch, Wetzlar, Germany) and applied a high indenting force at 5 nN. This value showed the least discrepancy after pre-testing using 1, 3, 5, and 10 nN with the same cantilever on test samples. The approaching and retracting rates of cantilever were set at 1 μm/s. Force–distance curves were collected and calculated with the JPK package software, which was based on the Hertz model with a spherical indentation [
18,
19]. The force on the cantilever F(h) is calculated as follows:
where h is the depth of the indentation, E is the effective modulus of a system tip-sample, v is the Poisson ratio for the sample, and R is the radius of the AFM tip. The unit of Young’s modulus is calculated as N/m
2, and expressed as pascal (Pa) or kilopascal (kPa). The Poisson ratio was set at 0.5, since the spherical tip was incompressible relative to the sample. The temperature of the measurement was controlled at 32 °C to mimic the surface temperate of mouse skin [
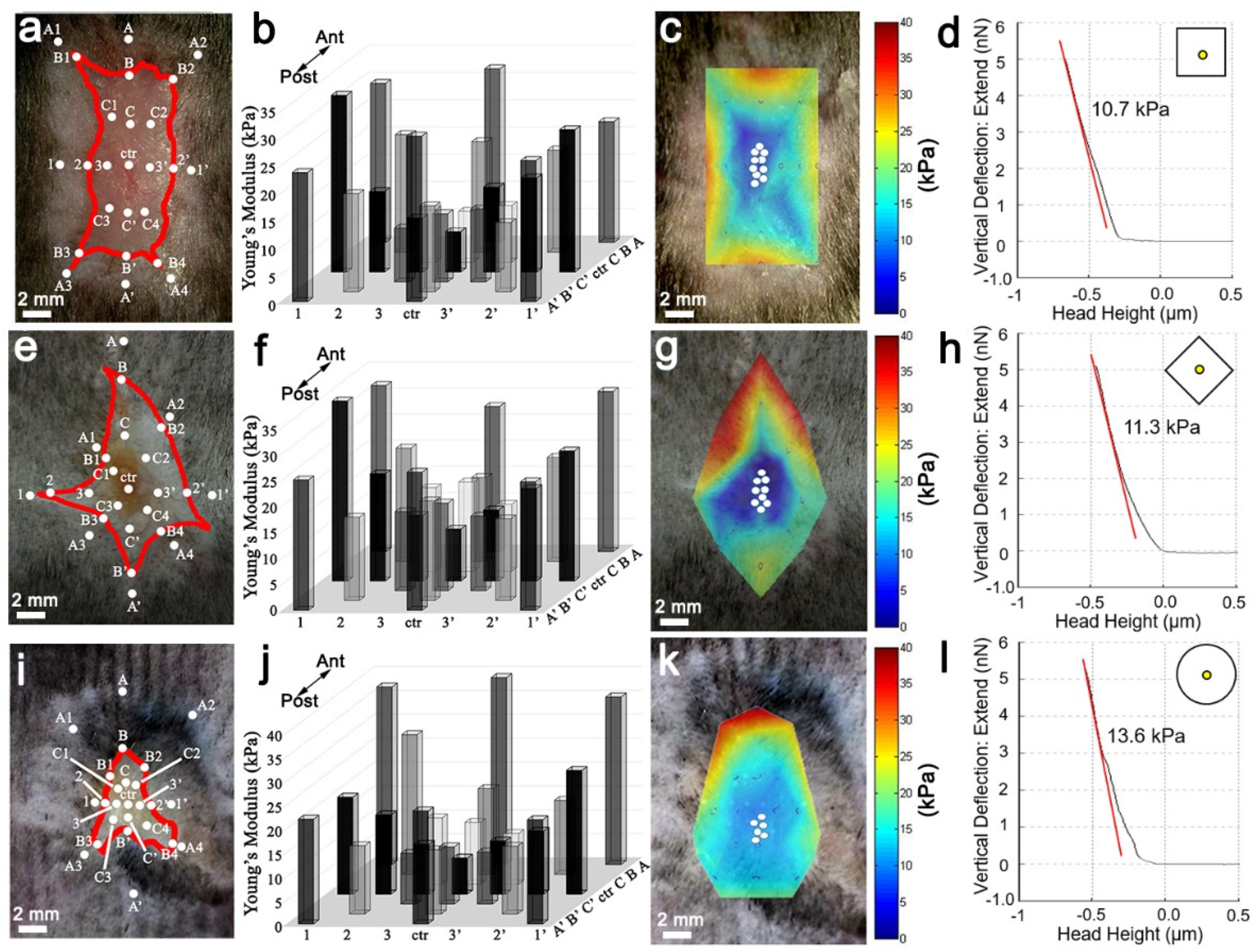
20]. To maintain the biomechanical force integrity of the dorsal wound, the entire mouse skin organ was removed by creating an excision on the ventral side midline, extending from the anterior neck region to the posterior groin region, and then dissecting away the skin organ from the underlying fascia. Normal skin and wound stiffness were immediately measured after skin organ removal to prevent artifacts from tissue decomposition. In order to measure the stiffness of the entire wound, AFM measurements were positioned across the wound starting from unwounded normal skin on one side and progressively traveling through to the opposite side, via the near wound edge, the near wound bed, the wound center, the opposite wound bed, the opposite wound edge, and the opposite normal skin. At least five indentation points were taken for each region of interest.
2.7. Heatmap of Spatial Stiffness of the Wound
The interpolation of Young’s modulus was performed by using the 3D meshgrid function of MATLAB. After obtaining a Young’s modulus (z) at a specific spatial location (x, y) in the wound, a three-dimensional matrix was defined. When the positions and Young’s modulus of all the measured spots were identified, we could interpolate the Young’s modulus of the positions in between to average the Young’s modulus of the nearest parameters using the 3D meshgrid function, by defining (x, y) as the meshgrid and (z) as the griddata. In the end, the heatmap was generated by defining the representative color of Young’s modulus.
2.8. Live in vivo Imaging of the Wound
A 1 cm2 square wound was excised from a 3-week-old (p21) ROSAmTmG mouse. The mouse was anesthetized using inhaled isoflurane and the wound was observed at PWD 17 with an inverted confocal microscope (FV-1000, Olympus) utilizing a 40× or 63× oil lens. The viewing medium between the wound and the cover slide was 50% glycerol in PBS. Then, DMSO with or without FAK-inhibitor (FAK-I) was added for real-time monitoring of cell shape. A Z-stack series of 20 slices was recorded during a time course starting at the cornified layer of the epidermis and ending at a 50 μm depth for each time point.
2.9. Image Quantification and Analysis
The quantification of image area, wound geometries, cell aspect ratio, and orientation were analyzed using ImageJ according to the user guide ImageJ/Fiji 1.46.
2.10. Histology and Immunostaining
The wound tissues were fixed in 4% PFA and dehydrated in a graded alcohol series. The tissue was cleared in xylene and embedded in paraffin wax. Six-micron sections were cut on a microtome. Then, H&E sections were performed according to accepted protocol. Whole-mount tissues were fixed in 4% PFA and then stored at 4 °C in PBS with NaAzide. Paraffin section and whole-mount immunostaining were performed as previously described [
21]. Briefly, fixed tissues were permeabilized with methanol and blocked with 3% H
2O
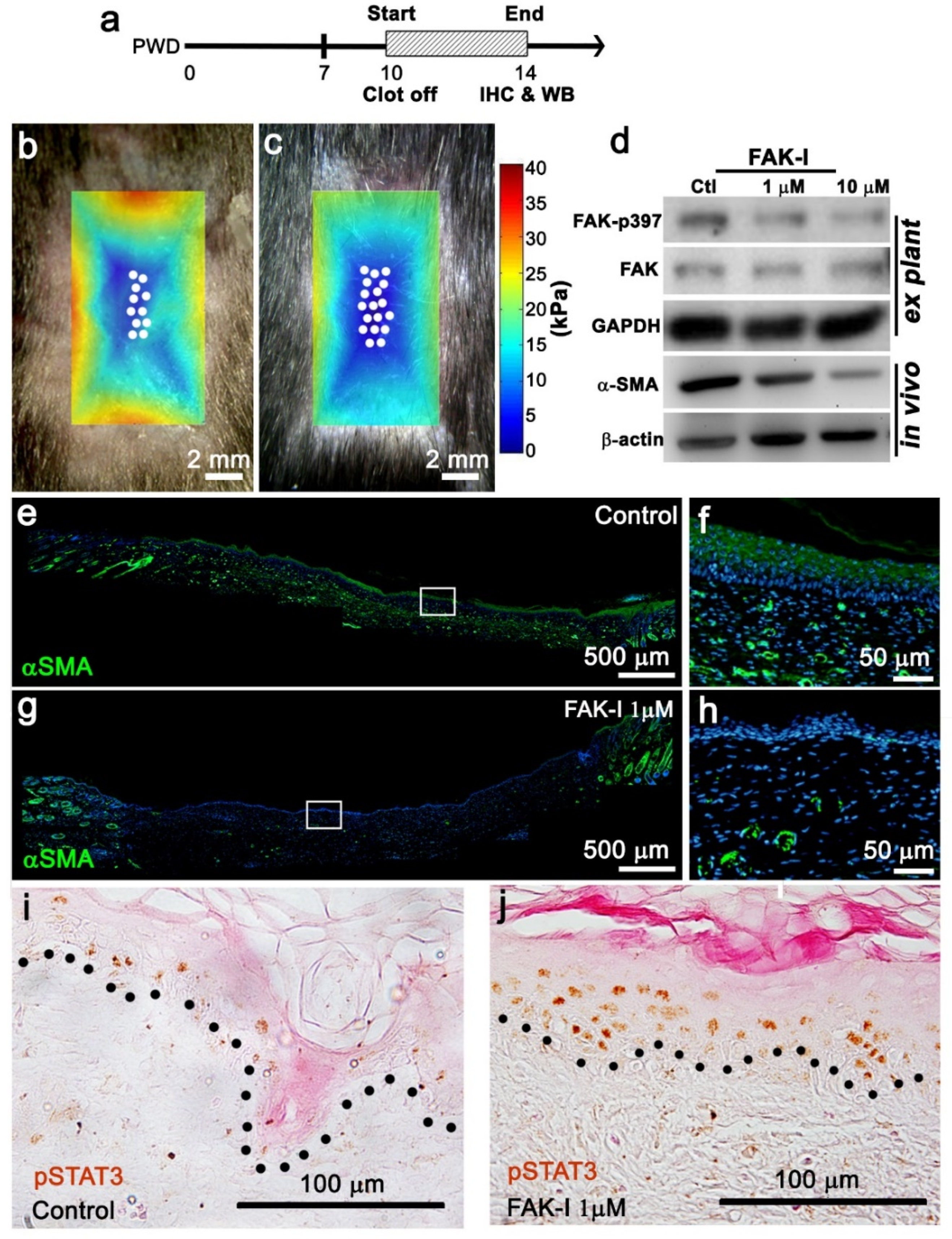
2 for 30 min, and then serum blocked for 1 h. The primary antibody was added and incubated over night at 4 °C with agitation. The tissue was washed with TBST (Tris-buffered saline Tween 20) and the secondary antibody was added for 1 h at room temperature. The tissue was washed with TBST and, if utilized, a tertiary antibody was added for 1 h at room temperature. The tissue was washed, and color was developed using the AEC kit (Vector Laboratories, Burlingame, CA, USA), or fluorescence was visualized with a fluorescence microscope. The whole-mount samples were cleared in a series of glycerol-PBS until 100% glycerol for imaging. The α-SMA antibody is from Thermofisher (Thermofisher, Waltham, MA, USA), the FAK, the pY397-FAK, the EPHA3, and the p-Tyr705-STAT3 antibodies and alexa-488 secondary antibody are from Abcam (Abcam, Waltham, MA, USA). The pSTAT3 sections were counterstained with eosin.
2.11. Tissue Culture, Western Blot, and Immunoprecipitation Blot
To perform tissue culture of the epidermis, the wound was harvested on PWD 14, and the epidermis was isolated under a dissecting microscope and placed in a culture dish with minimal keratinocyte-SFM (Thermofisher, Waltham, MA, USA) plus 10% FBS added for 2 h to allow attachment. FAK-I or DMSO was added to 1 mL of the medium another 6 h. To collect tissue lysate, 250 μL of lysis buffer was added to the dish, and tissue slurry was collected and grinded using a homogenizer in a 1.5 mL microtube. To harvest the tissue directly, the wound was harvested at PWD 14, microdissected, and immediately placed in liquid nitrogen for 30 s. The samples were then grinded using a homogenizer in a 1.5 mL microtube with 500 μL of lysis buffer. The Bio-Rad General Protocol for Western Blotting (Bio-Rad) was followed. The membrane was hybridized with α-SMA (Thermofisher, Waltham, MA, USA), FAK (Abcam, Waltham, MA, USA), pY397-FAK (Abcam, Waltham, MA, USA), GAPDH (Abcam, Waltham, MA, USA) β-actin (Abcam, Waltham, MA, USA), and EPHA3 (Abcam, Waltham, MA, USA) primary antibody at 4 °C overnight, and then with the secondary for 1 h at room temperature. Immunoprecipitation (IP) was performed according to accepted protocol. Briefly, an antibody for the protein of interest was incubated with A/G-couple agarose beads (Millipore, Billerica, MA, USA) at room temperature for 30 min. Target protein was incubated with the antibody–bead complex at 4 °C overnight. The target protein complex was isolated and separated using the aforementioned western blot assay.
2.12. RNA Extraction and RNA-seq
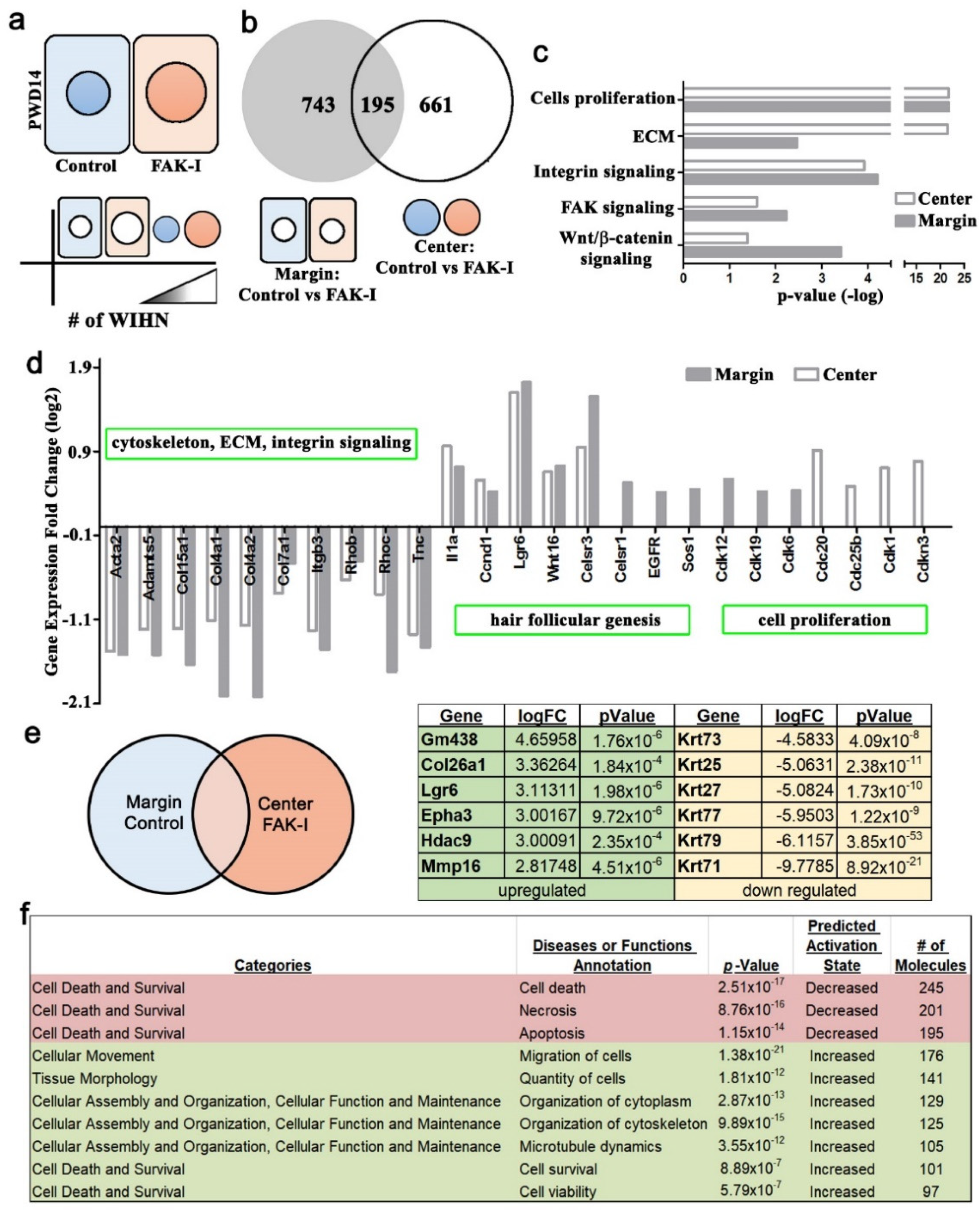
The RNA-seq was performed on replicate samples from treated or control 1 cm2 square PWD 14 wounds, and the samples were further dissected into wound margin and wound center. The wound was harvested, and a 3 mm diameter hole-punch biopsy was taken from the geometric center of the wound to identify the wound center, and the remaining wound tissue was considered the margin. The epidermis and dermis were separated manually under a dissecting microscope. The dissected epidermis tissues were placed in liquid nitrogen, disaggregated individually using a mortar and pestle, and then collected into a 1.5 mL microtube. The RNA was extracted using the RNeasy Mini Kit (QIAGEN, Hilden, Germany). Then, 1 μg of total RNA from each sample was used to construct an RNA-seq library using the TruSeq RNA sample preparation v2 kit (Illumina, San Diego, CA, USA). Sequencing (50 cycles double read) was performed by Welgene (Taipei, Taiwan) using Nextseq (Illumina Solexa, San Diego, CA, USA).
2.13. RNA-seq Analysis
The mouse mm9 reference genome, and RefSeq genome annotation downloaded from the UCSC Genome Browser were used for RNA-Seq analysis [
22]. The alignment, quantification, normalization, and differential expression analysis were performed by TopHat2 [
23], Partek E/M (Partek Inc, Saint Louis, MO, USA), TMM [
24], and GSA [
25], respectively. The false discovery rate < 0.05 was set as a threshold to identify differentially expressed genes. The principal component analysis (PCA), hierarchical clustering, and Venn diagrams were carried out using the Partek Genomics Suite 6.15.1207 (Partek Inc. Saint Louis, MO, USA). The RNAseq data has been uploaded to the Gene Expression Omnibus website. The sample accession numbers are as follows: GSM4850917, GSM4850918, GSM4850919, GSM4850920.
2.14. qPCR
Reverse-transcription was performed using SuperScript II Reverse Transcriptase (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA). In brief, 200 ng of mRNA was added into 500 μg of Oligo(dT), and 10 mM of dNTP Mix, diluted in distilled water to a final volume of 12 μL. The mixture was heated to 65 °C for 5 sec and then quickly chilled on ice. Then, 4 μL of 5X first-strand buffer, 2 μL of 0.1 M DTT, and 1 μL of RNaseOUT (40 units/μL) were added into the tube. After 42 °C incubation for 2 min, 1 μL (200 units) of SuperScript II RT was added, and the mixture was incubated again at 42 °C for 50 min. The reaction was inactivated by heating at 70 °C for 15 min. Then, SYBR Green Master Mix was used to perform qPCR. In brief, SYBR Green Master Mix was mixed with 1 μM of forward and reverse primers and 200 ng of cDNA. All reactions were performed in 20 μL total volume with triplicates (n = 3). The samples were incubated (Mx3000P, Stratagene, San Diego, CA, USA) at 50 °C for 2 min, 95 °C for 2 min, and then cycled 40 times between 95 °C for 15 secs and 60 °C for 1 min. The primers were designed in reference to PrimerBank—MGH-PGA (Boston, MA, USA). The amplification efficiency was calculated using the slope of the regression line of the standard curve. Expressed genes were normalized to the reference gene GAPDH within the same sample to determine dCt. This dCt was then exponentially transformed into dCt expression. After averaging the triplicates, the value was then normalized to the averaged control value ddCt.
2.15. Statistics
The photographs are representative samples of at least four replicates. Hair follicle counts are reported as the average from at least four samples. Each bar on the qPCR graph represents the average and SEM of three independent samples. All data is presented as mean ± SEM unless stated otherwise. Results from a Student’s t-test with p < 0.05 were considered significant. Here,* p < 0.05, ** p < 0.01 and *** p < 0.001.
4. Discussion
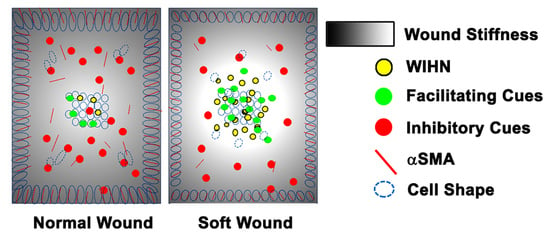
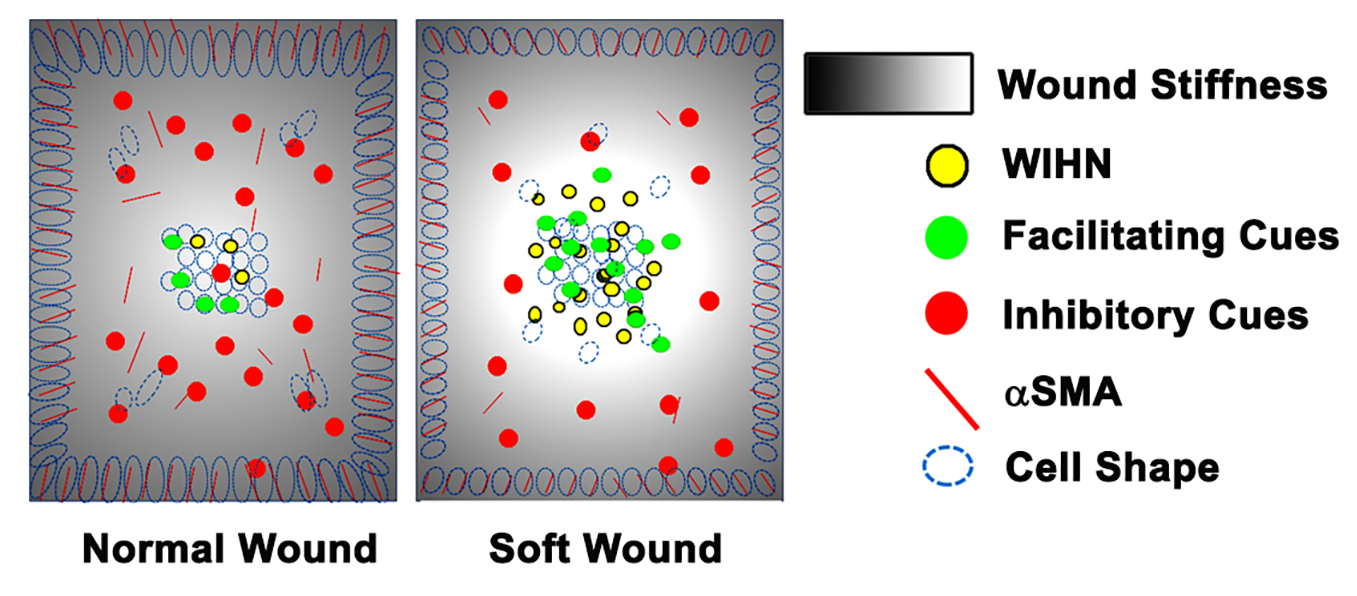
The original hypothesis that increasing the distance between the wound edge and the regenerative wound center would increase WIHN was refuted by the data. Our revised hypothesis is that homeostatic regeneration, wound tissue regeneration, and WIHN requires a relatively soft microenvironment, and this is inhibited by increasing stiffness. In this study, the data show that wound stiffness modulates WIHN and is partially regulated through mechanotransduction pathways. The WIHN presents in the wound center as being associated with the soft microenvironment (
Figure 1 and
Figure 2), lower FAK activity (
Figure 3), lower α-SMA expression (
Figure 3), higher pSTAT3 (
Figure 3), reduced cell aspect ratio (
Figure S2), lower ECM expression (
Figure 4), lower cytoskeletal signaling (
Figure 4), increased cell survival (
Figure 4), and higher EPHA3 (
Figure S3). Furthermore, EPHA3 associates with pSTAT3 (
Figure S3), which leads to increased WHIN signaling (
Figure 4), and targeting of the FAK/SMA/MyoII axis modulates WIHN (
Figure 5). These data support our hypothesis for relatively softer wound microenvironments promoting tissue regeneration both homeostatic and after injury. Furthermore, multiple targets for developing novel regenerative medicine clinical therapies are suggested by the data, including FAK, SMA, MyoII, pSTAT3, and EPHA3.
Regeneration occurs only in the wound center and not at the margin [
2,
3,
4,
5,
6]. The current study shows that WIHN only presented in the wound center, and that it corresponds to the following: softer wound stiffness, lower wound FAK activity, lower epidermal α-SMA expression, higher epidermal pSTAT3 expression, lower epithelial cell aspect ratio, lower ECM expression, lower cytoskeletal signaling, and higher hair neogenesis signaling (
Figure 1,
Figure 2,
Figure 3,
Figure 4 and
Figure 5). These findings concur with previous WIHN studies [
2,
3,
4,
5,
6]. Conversely, higher stiffness at the wound center resulted in less WIHN (
Figure 2,
Figure 3 and
Figure 4). The study by Nelson et al. (2015) shows that WIHN is enhanced via activation of the TLR3 receptor due to increased dsRNA [
4]. This was achieved through a modified WIHN assay where micro-cuts were perpendicularly created along the original 1 cm
2 wound excision border. These micro-cuts can disrupt contraction, resulting in a softer wound bed enhancing WIHN. The current study’s finding of increased regeneration area and WIHN due to softer wounds is similar and delineates a potential mechanism through the cytoskeletal network and adhesion complex signaling. The RNA-seq analysis data of differential gene expression and the EPHA3 IP data between center and margin of PWD 14 wound epidermis support this observation (
Figure 4 and
Figure S3). The FAK inhibitor treatment resulted in downregulation of ECM, integrin, and cell death gene expression, and the upregulation of cell motility, survival, and ephrin gene expression, which fosters a softer environment to enhance cell invagination and promotes WIHN. In addition, cell aspect ratio analysis shows cells in the wound edges are significantly higher than the wound center (
Figure S2) suggesting that cells residing in the wound margin experience a different mechanical stimulation from the wound center, modulating the production of hair neogenesis-related morphogens, for example Wnt or Shh. Taken together, this study proposes that mechanotransduction partly explains why WIHN occurs only in the wound center.
Seifert et al. and Hans et al. show that spiny (Acomys) mice regenerate hair with greater ability than C57Bl/6 mice due in part to softer skin environments [
6,
14]. Stress–strain curves show that spiny mice possess lower tensile skin strength than C57Bl/6 mice [
14]. Atomic force microscopy measurements of healed wounds show that spiny mice hair regenerating wound margins are significantly less stiff than C57Bl/6 mice hairless margins [
6]. This manuscript’s data is in accordance with both Seifert et al. and Hans et al., suggesting that a soft wound microenvironment promotes tissue regeneration. Interestingly, spiny mice can regenerate primary and secondary hair follicles, including all four hair follicle types of guard, awl, auchene, and zigzag [
11]. Even though C57BL6 mouse wound center stiffness is very similar to spiny mice [
6] and this regenerative area can be increased for WIHN (
Figure 3 and
Figure 5), these mice cannot regenerate all hair types and only regenerate secondary follicles [
2,
5,
11]. This is a significant clinical issue, because human terminal hair is more similar to mouse primary follicles, and regional specific hair follicle regeneration requires further study.
Wong et al. demonstrate wound stretching increases tension and scarring, a pro-inflammatory process [
8]. The high skin tension induces FAK activity by recruiting immune cells, thus, mediating scarring. The current study shows that WIHN favors a physical environment resembling the relatively soft skin stiffness of anagen (
Figure 2 and
Figure S1). Furthermore, our data show increasing stiffness via jasplakinolide significantly reduces regenerative total area and WIHN (
Figure 5). The relative stiffness of hair cycling skin changes from ~27 kPa during telogen to ~7 kPa during anagen (
Figure S1). Interestingly, the square wound stiffness from the non-regenerating margin is ~40 kPa, while the regenerative wound center is ~13 kPa (
Figure 2). Accordingly, FAK inhibitor reduced the stiffness of the entire wound bed, resulting in enhanced regeneration and WIHN (
Figure 3). Conversely, actin cytoskeleton stabilizer treatment and circle wounds exhibited relatively stiffer wounds, resulting in WIHN reduction (
Figure 5). This data is in agreement with Wong et al., and suggests relatively high skin stiffness during telogen and during full-thickness wound healing inhibits tissue regeneration and hair neogenesis.
An intriguing aspect is that WIHN favors a physical environment resembling anagen stiffness (
Figure 2 and
Figure S1). In order for hair follicular neogenesis to occur, activated epidermal cells proliferate, condense, form hair placodes, and invaginate into the dermis (30–32). Greco et al. showed that cell proliferation is an important step during wound closure, and hair follicle growth requires cell division and stem cell motility [
26,
36,
37]. For activating epidermal cells to invaginate, these cells overcome wound stiffness (Harn 21). Thus, downregulation of ECM gene expression upon FAK inhibitor treatment and the sequential downregulation of integrin signaling fosters a relatively softer environment to enhance cell invagination for WIHN (
Figure 4;
Figure 5). Consequently, the FAK inhibitor treatment downregulation of ECM, integrin, and cell death gene expression and upregulation of cell motility, survival, and ephrin gene expression fosters a softer environment to enhance cell invagination and promotes WIHN. Regeneration is an intricate interaction of biochemical signals and mechanical stimulations. These cues work in concert in a spatiotemporally dynamic manner to form a favorable environment for the desired result of structure and function. This study shows how the topological distribution of mechanical cues and molecular signaling pathways work synergistically to modulate tissue response during regeneration. As a result, the production and distribution of morphogens, for example Wnt or Shh, could be affected in the wound. Whether mechanical force plays a role in regulating the distribution of these biochemical molecules and morphogens begets further study. Taken together, this study proposes a mechanism that partly explains why WIHN occurs at the wound center, and that modulation of mechanobiological cues can enhance tissue regeneration (
Figure 2,
Figure 4,
Figure 5 and
Figure S3).
Finally, the RNAseq data exhibited an increase in
Epha3 in the wound center during WIHN (
Figure 4). Additional follow-up experiments showed an increase in EPHA3 in the wound center and an association of EPHA3 with pSTAT3 (
Figure S3). Of note, pSTAT3 has been shown to bind the -catenin promoter to promote WIHN [
4]. This manuscript confirmed pSTAT3 presence in the wound center epithelium where hair regeneration occurs during WIHN (
Figure 3j). Furthermore, the data show that a reduction in wound tissue stiffness (FAK inhibitor PF573228 treatment of the entire wound) increases pSTAT3 in the wound center (
Figure 3b–d,j). Taken together, these data suggest STAT3 is phosphorylated in the wound center by an additional kinase. The RNAseq data identified EphA3 as a potential candidate kinase for STAT3 in the wound center epithelium (
Figure 4e). It is known that ephrin receptors are protein–tyrosine kinases and phosphorylate STAT3 [
33]. The immunoprecipitation blot data showed that EPHA3 binds to pSTAT3 in the wound center epithelium (
Figure S3f). Taken together, this data suggests that EPHA3 phosphorylates STAT3 in the wound center epithelium during WIHN, and this is downstream and modulated by wound stiffness. Furthermore, EphA3 levels increase in the wound center epithelium during WIHN after FAK inhibitor treatment (
Figure 4e). This is one reason why pSTAT3 is increased only in the wound center when FAK is inhibited. Ephrin receptors and ligands are located on adjacent cell surfaces. Ephrin receptors contain extracellular, transmembrane, and intracellular domains. including a tyrosine kinase domain that phosphorylates STAT3 (Bong 2007). A pertinent question is how the mechanical stiffness gradient signal is transmitted cell to cell during full-thickness wound regeneration and WIHN. Ephrin signaling is an interesting candidate and will be the focus of future studies.



 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}