1. Introduction
Paeonia tenuifolia L., also known as the steppe peony, belongs to the
Paeonia L. genus of the Paeonicae family [
1].
Paeonia plants are well known for containing monoterpene glycosides with a “cage-like” pinane skeleton, as well as some other classes of bioactives, such as steroids and polyphenolic compounds [
2]. Specifically, the petals of fern leaf peonies proved to be rich in phenolic acids, anthocyanins and anthocyanidins, flavonoids, and terpene derivatives [
2,
3], which possess antioxidant, antimicrobial, and antibiofilm activities, as well as wound healing potential, and an inhibition effect on the adhesion and invasion of the bacterium
Staphylococcus lugdunensis [
3]. Extracts from the petals of
P. tenuifolia L. were also proposed as topical treatments for bacterial and fungal-induced skin diseases, to inhibit pathogen adhesion and penetration through the skin surface, and accelerate the healing of wounds [
3].
In the past, various plant parts of peonies (seeds, flowers, leaves and roots, as well as whole plants) have been widely used to treat various diseases, pain (head, stomach, and eyes) [
4], problem regarding female genitals (dysmenorrhea and amenorrhea) [
5], aches, neurological diseases (spasm and epilepsy) [
6], infectious diseases (carbuncles) [
4], urinary system diseases, and inflammation (otitis media, appendicitis, and gastritis) [
6], as well as trauma. The petals of herbaceous peonies have been a subject of very few studies, and it has been shown that they possess skin-beneficial biological activities [
3,
7].
Despite the fact that plant extracts are useful for treating a variety of disorders, research demonstrated that their therapeutic value is restricted due to their complicated composition and toxicity when administered to organisms with more complex metabolic systems. Furthermore, organic solvents (e.g., methanol, ethanol, hexane, dichloromethane, ethyl acetate, etc.) are commonly utilized to generate these extracts. As a result, the final vehicle in which the extracts are contained prohibits them from being directly applied in organisms [
8], and their encapsulation is needed. In addition to the limited bioavailability and instability of flavonoids, phenolic acids, and anthocyanins present in
P. tenuifolia L. petal extracts [
9], as well as their limited bioavailability, carriers for the extract ought to be created in order to protect the biologically active components and enable more comfortable dermal applications. To accomplish one or more of the desired effects, plant extracts can be preserved in their health-promoting characteristics, as they are encapsulated in a matrix or membrane, the particulate state. Encapsulation is used to improve the stability of extracted chemicals during processing, storage, or transportation. Furthermore, the primary goal is to convert liquid chemical compounds into solid forms in order to improve active component management. The use of pharmaceutical formulations with encapsulated compounds as delivery systems can be particularly beneficial in situations where the direct consumption of an active ingredient directly disrupts the human skin. Encapsulation can also be used to improve the final product’s quality, separate incompatible chemicals, and administer bioactives in a regulated manner [
10].
Enclosed vesicles known as liposomes are created when lipid components, including phospholipids, are distributed across an aqueous media. The inner water phase is divided from the outer one by one or more produced bilayers with a structure similar to the cell membrane [
11]. Due to their unique structure, liposomes provide a number of significant advantages when used in drug delivery systems. Initially, the capacity of the contained vesicles to differentiate between the inner and exterior phases improves the stability of the encapsulated medicine. Second, when poorly hydrosoluble medicines are integrated into liposomes, their bioavailability improves [
12,
13]. Furthermore, following encapsulation, a regulated or sustained drug release profile may be obtained. Moreover, liposomes have outstanding biodegradability and a strong affinity for cells. Additionally, the target effect of liposomal preparations has the ability to alter the loaded drug’s in vivo distribution, hence increasing the medicinal index of some medications [
14,
15]. Furthermore, the surface of the liposomes may be altered in a number of ways, affecting the ultimate product’s characteristics and biological activity. Liposomes are small, spherical particles composed mainly of lipid mixtures, organized into one or more lipid bilayers. This type of organization allows for their use as a simple approximation to living cells [
16]. Liposomes can be a highly valuable tool for identifying the fundamental interactions of bioactive substances with lipid bilayers since they are fairly simple to make, somewhat stable, and considerably less delicate to handle than human cell lines. To be precise, the encapsulation of bioactive compounds into nanoparticles or liposomes could decrease their toxicity, while increasing bioavailability, and also improve pharmacokinetics, thus leading to a better controlled release profile, and enhanced stability and solubility of the compounds in the organism [
17]. This has led to a large number of areas for their possible medicinal applications, such as a source of treatment for cancer [
18,
19,
20], skin disorders [
21,
22], post ischemia [
23], pulmonary hypertension [
24], etc. The development of liposomes intended for clinical use relied on the development of methods that would allow for the rapid formation of homogeneous small liposomes and efficient loading of drugs or bioactive compounds into them [
25]. This was achieved by employing the extrusion technique and/or changes in the pH value of the mixture.
Biopolymer films and coatings are among the active technologies used to create patches for transdermal administration, providing extra protection of biologically active components in order to retain or increase the overall quality of bioactives from plants and extend their shelf life [
26,
27]. Plant extracts high in polyphenols have been demonstrated to improve the antioxidant activity, UV light barrier capacity, and oxygen barrier ability of biopolymer films, hence enhancing the preservation of biologically active components [
28,
29]. Polyphenol interactions with the biopolymeric matrices may alter the structural and functional properties of the matrix [
29,
30], as well as a decrease the bioactivity of polyphenols; thus, potential interactions and their influence on the biological activities of polyphenols should be thoroughly investigated.
Biologically active compound from plants have been subjected to various encapsulation methods in the past, which could be divided into three larger groups: physical (extrusion, freeze drying, supercritical fluid, pan-coating, electrospinning, etc.), chemical (inclusion complexation, emulsion polymerization, interfacial polymerization), and physicochemical methods (including coacervation, the sol-gel method, solvent evaporation, and similar methods) [
31]. A less explored alternative method for the antioxidant polyphenol encapsulation is the combination of biopolymer films and liposomes. The active ingredients in this combination may be shielded from processing, storage, and environmental factors [
32], preserving their bioactive qualities and allowing for controlled release and prolonged shelf life.
Proliposome technology offers a higher energy input of agitation, resulting in smaller and more homogeneous liposomes [
33,
34]. Using this method might be the easiest way to obtain liposomes [
16]. The main drawback is that although it produces a substantially better encapsulation efficacy, it is not as reproducible when producing smaller amounts of liposomes. During the process of the making of liposomes, the compounds can be combined with ethanol (in the case of lipophilic substances) or in an aqueous solution (in the case of hydrophilic substances). The use of the proliposome method presents a chance for large-scale liposome production [
35].
Wet processing, also known as solvent casting, is based on the drying process of the film-forming solution, which involves the phases of solubilization, casting, and drying [
36]. Firstly, a biopolymer is dissolved in a suitable solvent, organic or inorganic, in order to create a film-forming solution. However, water, ethanol, or their combinations are the only medical-grade solvent systems available for biopolymer films and coatings. The dissolved film-forming solution may be heated or its pH adjusted on occasion to improve film formation or its properties. After that, the solution is dried by being cast onto a flat surface to form a film matrix [
37]. The development of a continuous three-dimensional network among biopolymers is critical for the construction of a cohesive film [
38]. The nature, kind, and amount of the interaction are determined by the polymers involved and film-forming factors such as drying temperature and pace, moisture content, solvent type, plasticizer concentration, and pH.
The rheological, physical, and chemical characteristics of topical drug delivery systems impact drug bioavailability and the production of pharmaceutical and cosmetic formulations. Therefore, it is imperative to investigate the aforementioned characteristics of liposomes and films. The study of the interactions between bioactive substances, medications, vitamins, or hormones with phospholipid liposomal bilayers is frequently carried out using Fourier transform infrared (FT-IR) spectroscopy [
39,
40].
The encapsulation of the petal extracts of Serbian P. tenuifolia into liposomal particles and biopolymer films has not been the focus of any previous studies. As a result, the goal of this study was to create and characterize extract-loaded liposomes, as well as extract- and liposome-loaded films in order to potentially protect the sensitive biologically active components, increase their bioavailability, and enable controlled release, making them suitable for use in a variety of pharmaceutical and cosmetic formulations.
2. Materials and Methods
2.1. Origin of Plant Material
P. tenuifolia L. fresh petals were gathered in May 2023 from plants growing spontaneously in their native habitat in Gulenovci (840 m a.s.l.), Serbia (
Figure 1). The Ministry of Environmental Protection of the Republic of Serbia granted the license for wild-collecting (no. 353-01-121/2023-04, issued on 3 March 2022). The petals were collected by hand from randomly selected full-blooming flowers. A third of the petals per flower were taken from less than one-tenth of the flowering plants found at the locality. Before undergoing the extraction processes, the gathered petals were shade-dried at room temperature.
2.2. Extraction of Plant Material
The biologically active compounds of the petals were extracted using the maceration process, by employing a linear mechanical homogenizer (Roller mixer SRT6, Potsdam, Germany) at room temperature (25 ± 5 °C) for 24 h using methyl alcohol as an extraction medium, with a solid-to-solvent ratio of 1:20. The extracts from the petals were filtered using laboratory filter paper. Before further analysis, the collected extract was evaporated at 30 °C in a drying oven (Sanyo drying oven MOV-212, Eschborn, Germany) to a dry mass, and kept in the dark at 4 °C.
2.3. Preparation of Liposomal Particles
The liposomes containing
P. tenuifolia L. petal extract were prepared utilizing the proliposome technique [
34]. The liposomes were prepared using three mixtures of phospholipids: Phosal SA 75 (containing phosphatidylcholine in ethanol and safflower oil, content ≥ 72.0%), Phosal MCT 53 (phosphatidylcholine in medium-chain triglyceride, content ≥ 53.0%) (from Lipoid, Skopje, Macedonia), and Phospholipon (a commercial lipid mixture, sunflower phosphatidylcholine from non-genetically modified plants, ≥90%; from Lipoid GmbH, Ludwigschafen, Germany). At 50 °C, the phospholipids (4 g), ethanol (15 mL), deionized water (3 mL), and dried
P. tenuifolia L. petal extract (0.4 g) were mixed. After the emulsion had cooled to room temperature, the aqueous phase (20 mL) was added in tiny amounts and the emulsion was agitated at 800 rpm for 1 h. As a control, plain liposomes were also made.
2.4. Preparation of Liposome-Loaded Films
Carboxymethyl cellulose (CMC) (0.48 g) (medium viscosity, 400–800 cP, a molar mass of 250 kDa, approximately, degree of substitution between 0.65–0.9; from Sigma Aldrich, Hamburg, Germany), deionized water (12 mL), and propylene glycol (0.36 mL) were mixed with a magnetic stirrer at 500 rpm for 24 h at room temperature. The resultant mixture was put into Petri plates and dried for 24 h in a laboratory oven (Emmeret UN 160, Emmeret GmbH + Co. KG, Schwalmstadt, Germany). Finally, the biopolymer films were conditioned at room temperature for three days in a desiccator containing magnesium sulfate.
2.5. Preparation of Extract-Loaded Films
CMC (0.96 g), deionized water (24 mL), and propylene glycol (0.36 mL) were added to the extract solution (0.4 g of dried petal extract in 12 mL of deionized water) and mixed for a whole day at room temperature with a magnetic stirrer at 500 rpm. The resulting mixture was put onto Petri plates and dried for twenty-four hours in a laboratory oven (Emmeret UN 160, Emmeret GmbH + Co. KG, Schwalmstadt, Germany). The biopolymers were kept the same way as the liposomes. The film type, CMC-0.2 ex, was created using the same technique, but with the addition of 0.2 g of the dried petal extract.
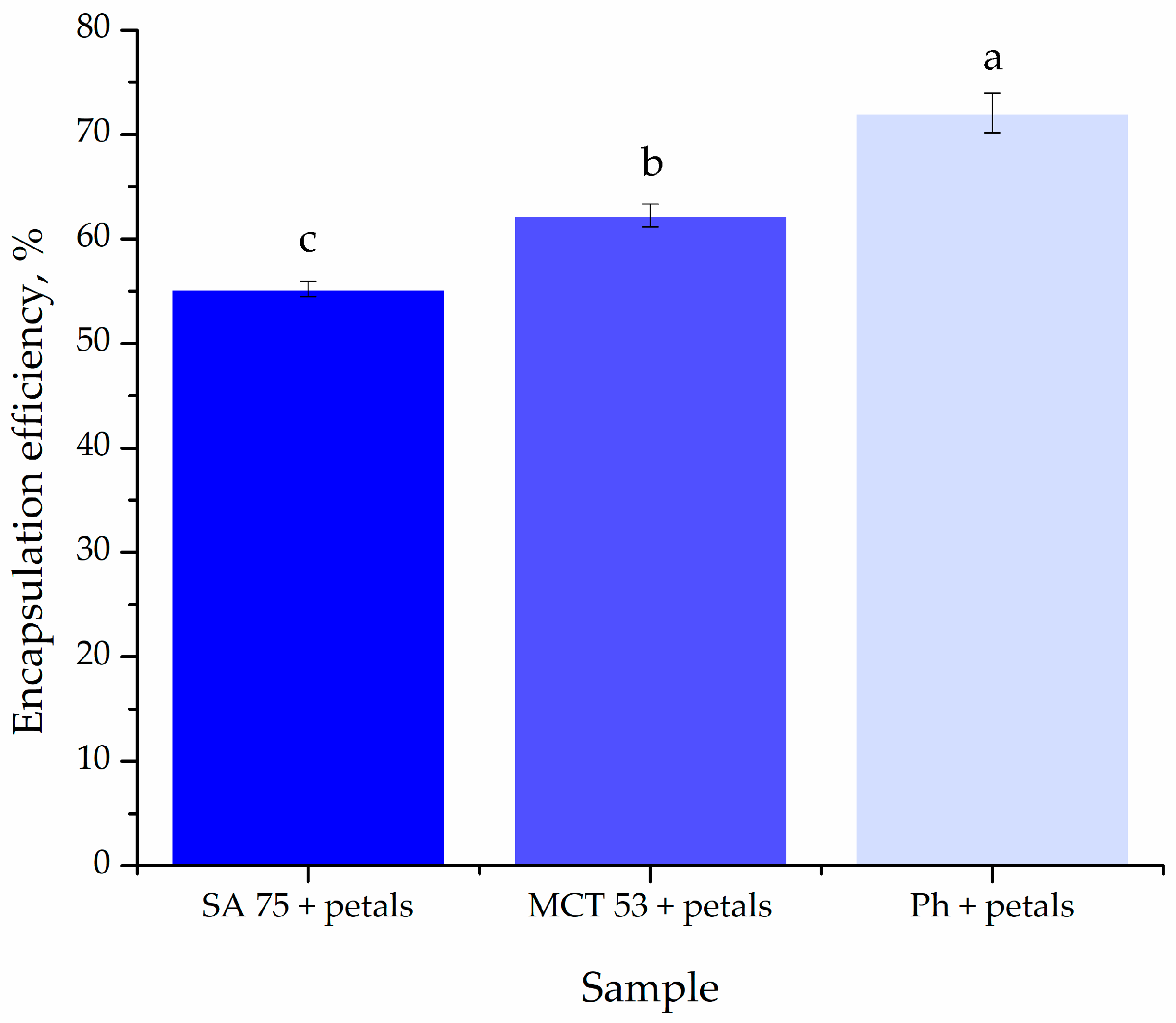
2.6. Encapsulation Efficiency
The liposome–extract particles were separated from the non-encapsulated fraction via centrifugation at 17,500 rpm and 4 °C, for 45 min (Thermo Scientific Sorval WX Ultra series ultracentrifuge, ThermoScientific, Waltham, MA, USA). The encapsulation efficiency (EE) was determined by measuring the total polyphenol content (TPC) in the supernatant using UV-Vis spectrometry (Shimadzu 1800 UV/Vis spectrophotometer, Kyoto, Japan), and the Folin–Ciocalteu method previously described by Čutović et al. [
3]. EE was calculated according to the amount of polyphenols present in the supernatant, obtained after centrifugation as shown in Equation (1):
TPCi denotes the initial total polyphenol content of the P. tenuifolia L. petal extract utilized for liposome synthesis, whereas TPCsup denotes the total polyphenol content found in the supernatant after centrifugation.
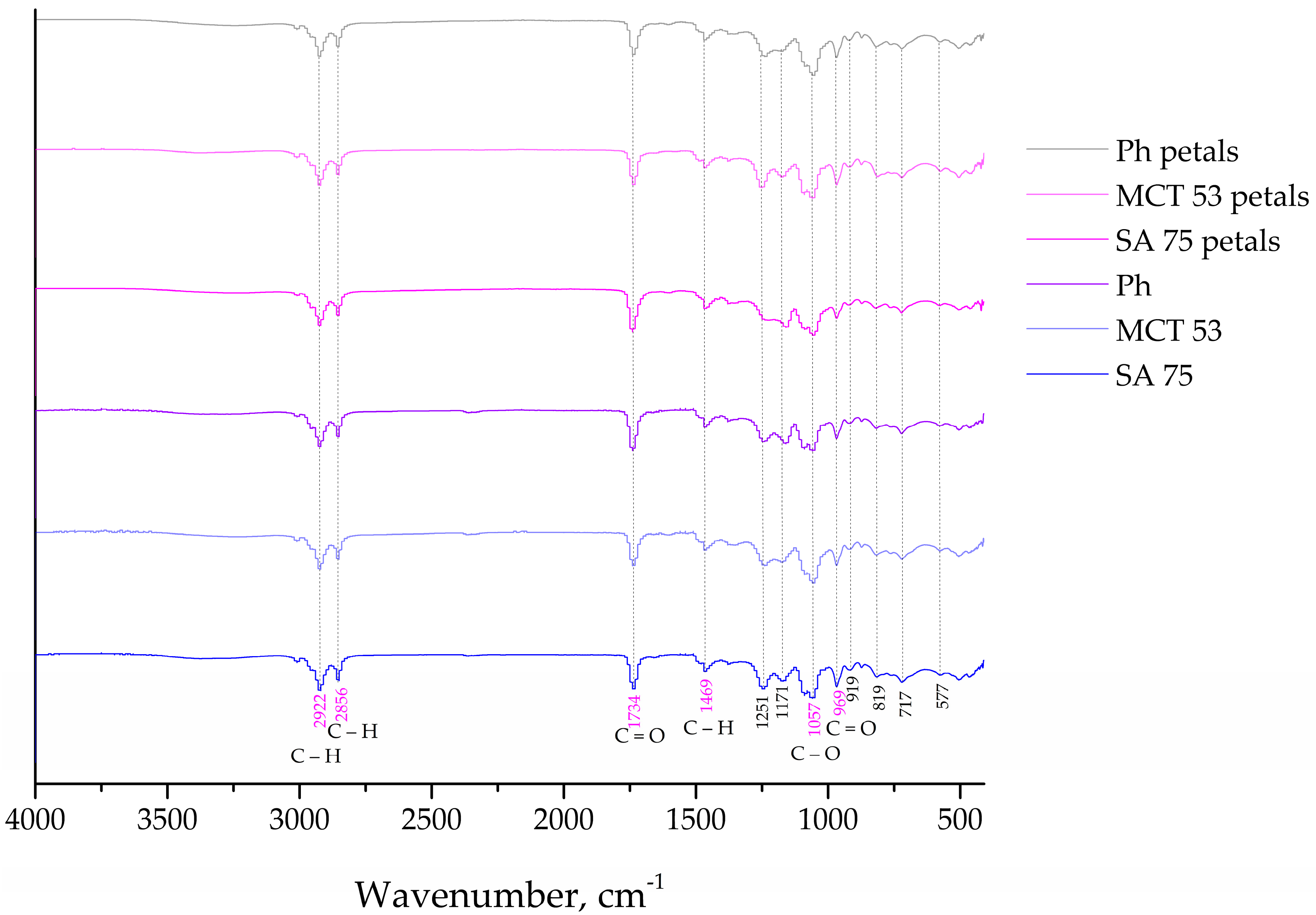
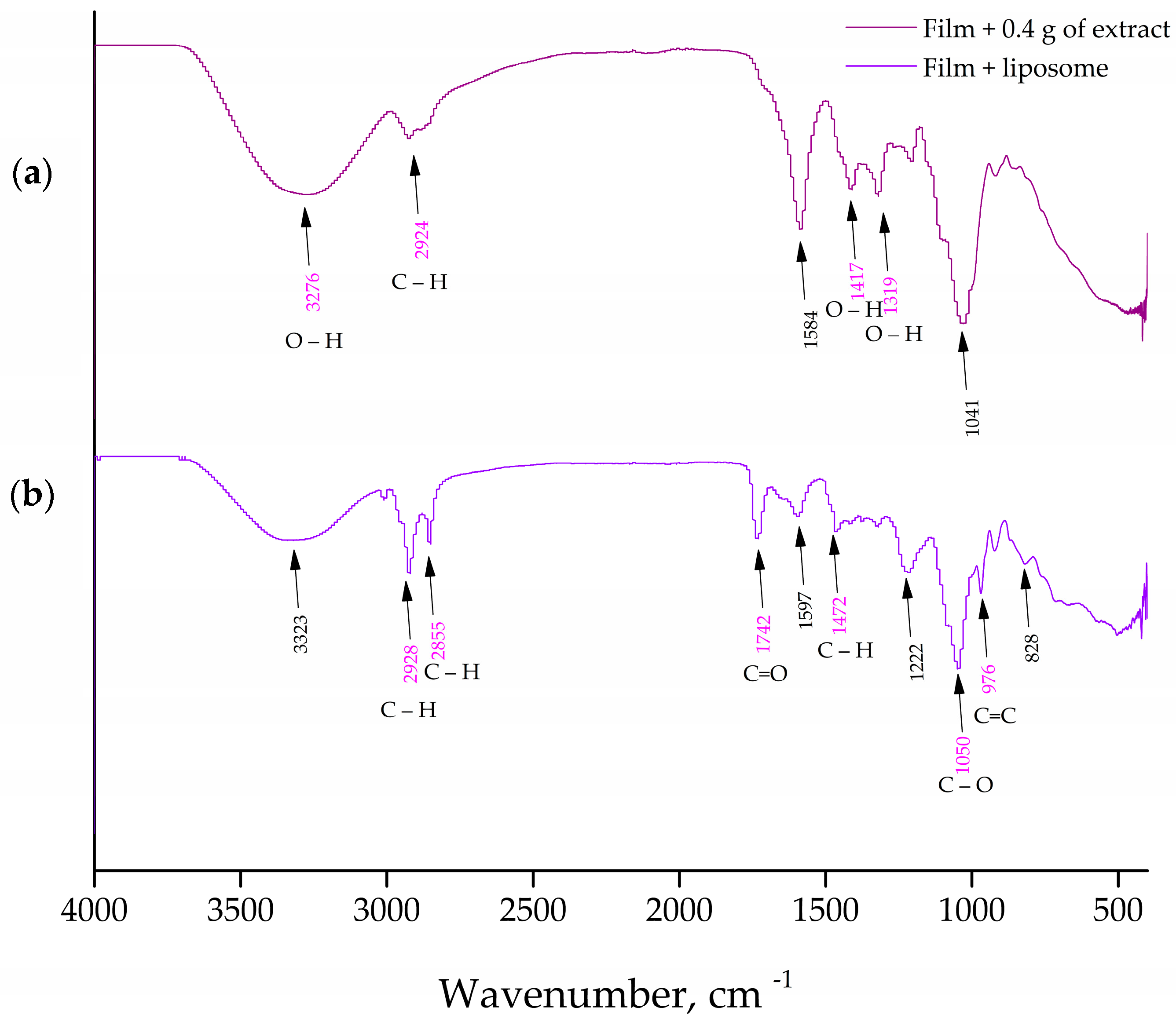
2.7. Fourier Transform Infrared Spectroscopy (FT-IR)
The chemical interactions between the film components, extract compounds, as well as the liposome ones for all prepared liposomes and films were characterized via FTIR spectroscopic analysis. The FTIR spectra were collected using a Nicolet iS10 ATR-IR spectrometer (Thermo Scientific, Stockholm, Sweden) with a scanning resolution of 4000 cm−1. For the FTIR measurements, polymer biofilms were sliced into tiny plates (1 × 1 cm) and mounted on the Diamond chassis. The liposomes were lyophilized (empty and extract-loaded), and the process consisted of centrifugation, after which the liposomes were frozen in the freezer, LAB11/EL19LT (Elcold, Hobro, Denmark), at −80 °C for 1 h and lyophilized in Beta 2–8 LD plus lyophilizator (Christ, Memmingen, Germany) at −75 °C and 0.011 mbar for 24 h.
2.8. Moisture Content
The moisture content was calculated by weighing each film sample after oven drying to a constant weight at 30 ± 0.2 °C. The moisture content was calculated according to Equation (2):
where m
1 stands for the initial weight (g) of the samples, and m
2 and MC
wb are the dry weight (g) and wet basis moisture content of the samples, respectively.
2.9. Stability Study
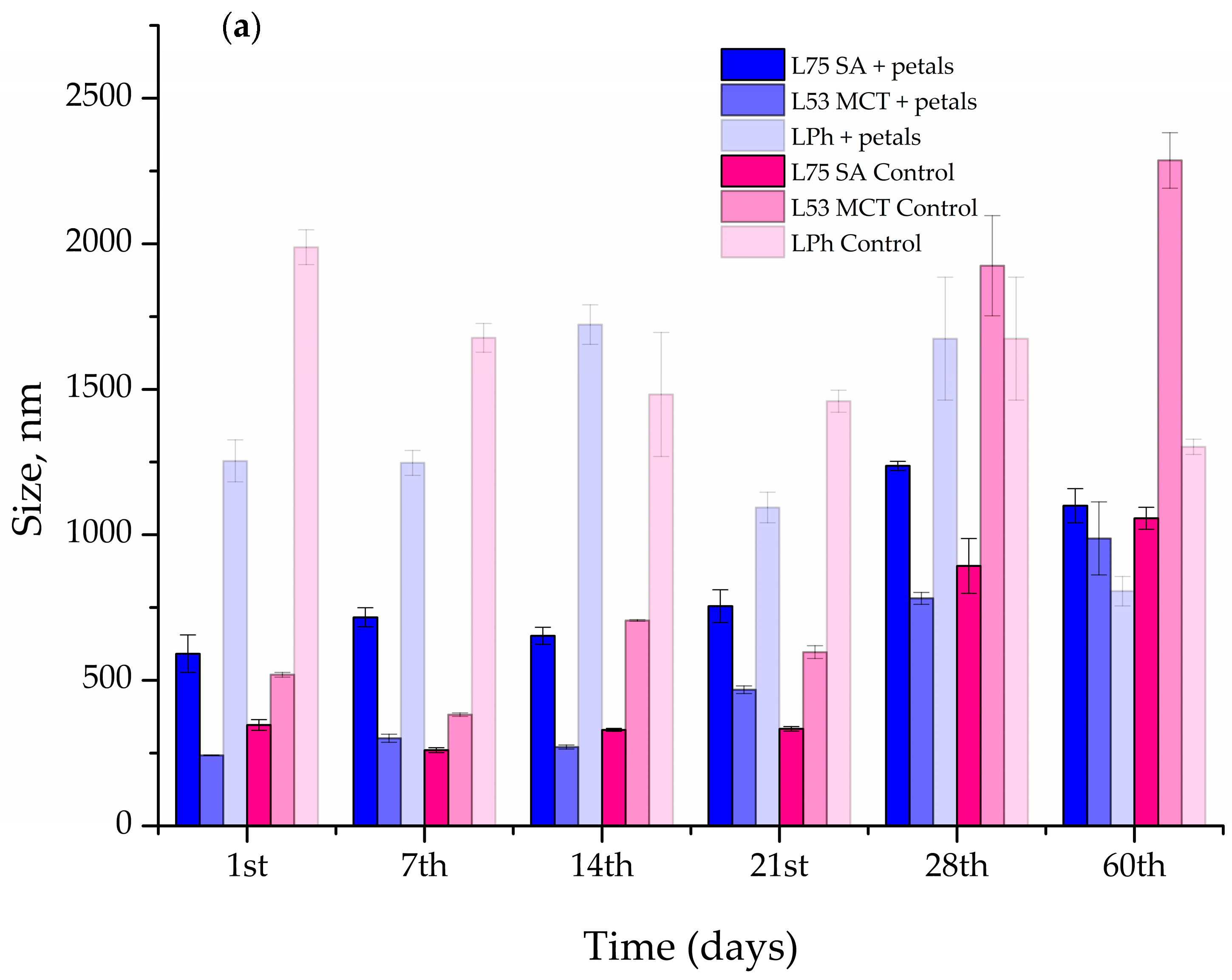
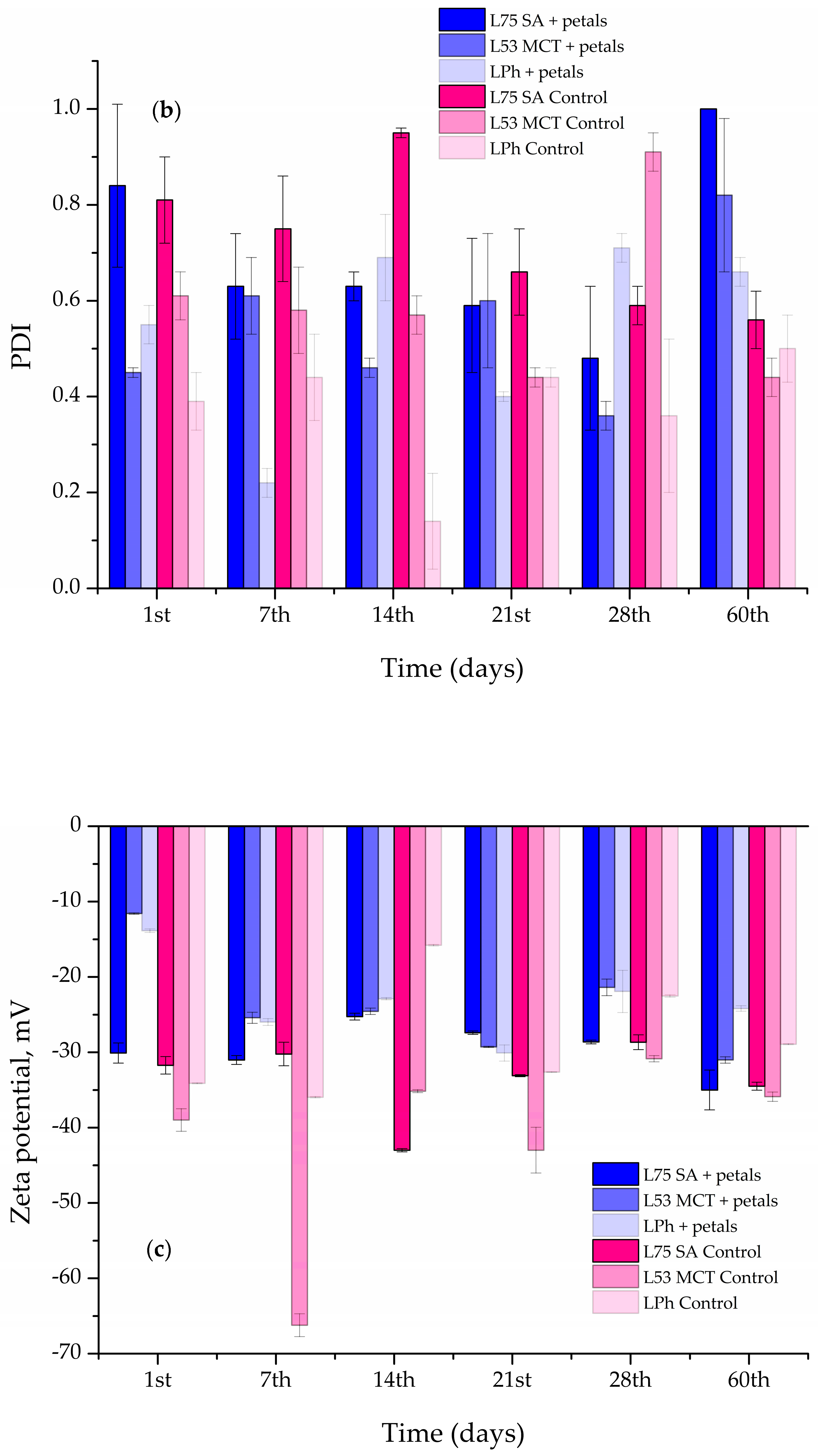
The size of the particles, PDI, and ζ potential of the prepared liposomes were measured every 7th day for the first 28 days, and the on the 60th day, following preparation using the Malvern Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK). During the 60-day stability evaluation, the liposomal system was stored in the refrigerator at 4 °C.
2.10. Rheological Characteristics
The density and surface tension of petal extract-loaded liposomes were measured in a Force Tensiometer K20 (Kruss, Hamburg, Germany) using a silicon crystal as the immersion body and a Wilhelmy plate, respectively. At 25 °C, each sample (20 mL) was analyzed three times.
Using a Rotavisc lo-vi device equipped with a VOL-C-RTD chamber, VOLS-1 adapter, and spindle (IKA, Staufen, Germany), the viscosity of petal extract-loaded liposomes was measured. At 25 °C, each sample (6.7 mL) was analyzed three times.
The density, surface tension, and viscosity measurements were taken on the first and sixtieth day.
2.11. Determination of the Film Mechanical Properties
The tensile strength (TS, MPa), break force (BF, N), and elongation at break (EB,%) of the films were measured with a Universal Testing Machine (Shimadzu Corporation, Kyoto, Japan) outfitted with a 100 N load cell (
Figure 2). The rectangular film strips (25 × 80 mm) were stretched at a crosshead speed of 10 mm/min using stainless steel grips. The gage length was measured as 50 mm. The measurements were obtained three times. The mechanical properties of the films were determined using engineering stress–strain and force–displacement curves at the breaking point [
26]. The film thickness was measured using a digital nonius depth caliper (0-Industrial&Scientific, Pittsburgh, PA, USA, 0–150 mm), and the film weight was determined using an analytical balance (Mettler, Columbus, OH, USA, Type AE 200; 0.0001 g).
2.12. Biological Activities
The assessed biological activities of the encapsulated extracts (in liposomal, film, and liposome–film systems) included their antibacterial and anticandidal, as well as antibiofilm activities, although the antibiofilm activity was performed only for liposomes.
2.12.1. Antibacterial Activity of Liposomal Particles
Gram-positive bacteria (
Staphylococcus aureus ATCC 11632 and
Staphylococcus lugdunensis Ibis 2996) and Gram-negative bacteria (
Proteus vulgaris IBR P004) were examined for antibacterial activity with liposomes. The microdilution technique (96-well microtiter plates) was used to determine the minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC), as reported before [
41]. The samples were placed in a tryptic soy broth (TSB) medium and infected with bacteria at a final concentration of 1 × 10
6 colony-forming units (CFU) per well. Gentamicin (Panfarma, Belgrade, Serbia) was used as the positive control. As the negative control, blank liposomes were utilized. The MIC and MBC values were provided in milligrams per milliliter.
2.12.2. Antibacterial Activity of Liposome- and Extract-Loaded Films
The antibacterial activity of the liposome- and extract-loaded films was determined using the disc diffusion assay [
42]. Inoculums of the test bacteria were produced in the same way that overnight bacterial cultures were. On a Mueller-Hinton agar plate, 300 μL inoculums were used to create uniform bacterial lawns. In the center of the Petri dish, thin film squares (10.0 × 10.0 mm) were inserted. The plates were incubated for 24 h at 37 °C. The zone of inhibition (mm) was used to quantify activity. The net zone of inhibition was calculated by subtracting the square side (i.e., 10.0 mm) from the overall zone of inhibition demonstrated by the test disc in terms of the clear zone surrounding the disc. As the positive control, streptomycin was utilized. As the negative control, blank films were utilized.
2.12.3. Anticandidal Activity of Liposomal Particles
Candida albicans (Y177),
Candida kefyr (Y289), and
Candida krusei (Y454) were used in order to test the extract-loaded liposome antifungal activity. The modified EUCAST protocol (EUCAST, 2002) was used to carry out the anticandidal assay, as previously explained [
43]. The positive control was ketoconazole, while the negative control was blank liposomes. The MIC and MFC (minimum fungicidal concentration) values are presented as mg/mL.
2.12.4. Anticandidal Activity of the Liposome- and Extract-Loaded Films
Anticandidal activity of the liposome- and extract-loaded films was determined using the disc diffusion assay [
42]. Inoculums for testing
Candida strains were produced in the same way that overnight fungal cultures were. On a TSB plate, uniform
Candidal lawns were created using 300 μL inoculums. In the center of the Petri dish, thin film squares measuring 10.0 × 10.0 mm were inserted. The plates were incubated for 24 h at 37 °C. The zone of inhibition (mm) was used to quantify activity. The net zone of inhibition was calculated by subtracting 10.0 mm from the overall zone of inhibition revealed by the test disc in terms of the clear zone around the disc. As the positive control, ketoconazole was utilized. As the negative control, blank films were utilized.
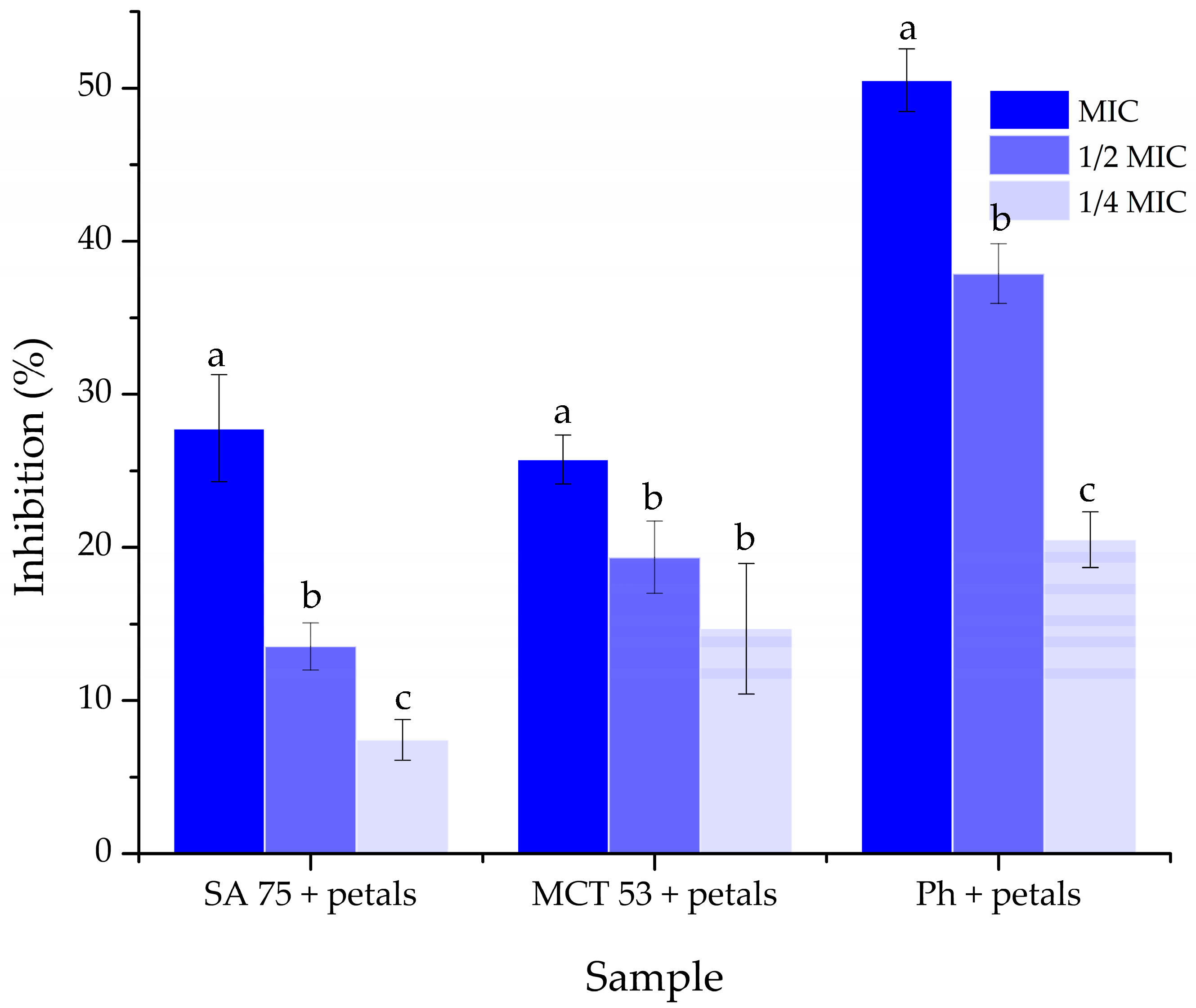
2.12.5. Antibiofilm Activity of Liposomal Particles
The petal extract-loaded liposome effect on the
S. lugdunensis biofilm was assessed with minor alterations, as previously described in Smiljkovic et al. [
44].
S. lugdunensis was grown in TSB with 2% glucose on 96-well microtiter plates with adhesive bottoms (Sigma Aldrich, Taufkirchen, Germany), with MIC, MIC/2, and MIC/4 of the extract-loaded liposomes for 24 h to form a biofilm. After incubation, the wells were washed twice with sterile PBS (phosphate buffer solution). The biofilm was then treated with methyl alcohol and air-dried. For 30 min, the biofilm was stained with crystal violet (Bio-Merieux, Crappone, France). After incubation, the crystal violet was removed, the wells were cleansed with water, air-dried, and then 96% ethanol (Zorka, Šabac, Serbia) was used. Thermo Fisher Scientific’s Multiskan FC Microplate Photometer (Waltham, MA, USA) was used to detect absorbance at 620 nm, and Equation (3) was used to calculate the percentage of biofilm suppression. Gentamicin (Panfarma, Belgrade, Serbia) was employed as the positive control, whereas blank liposomes were used as the negative control.
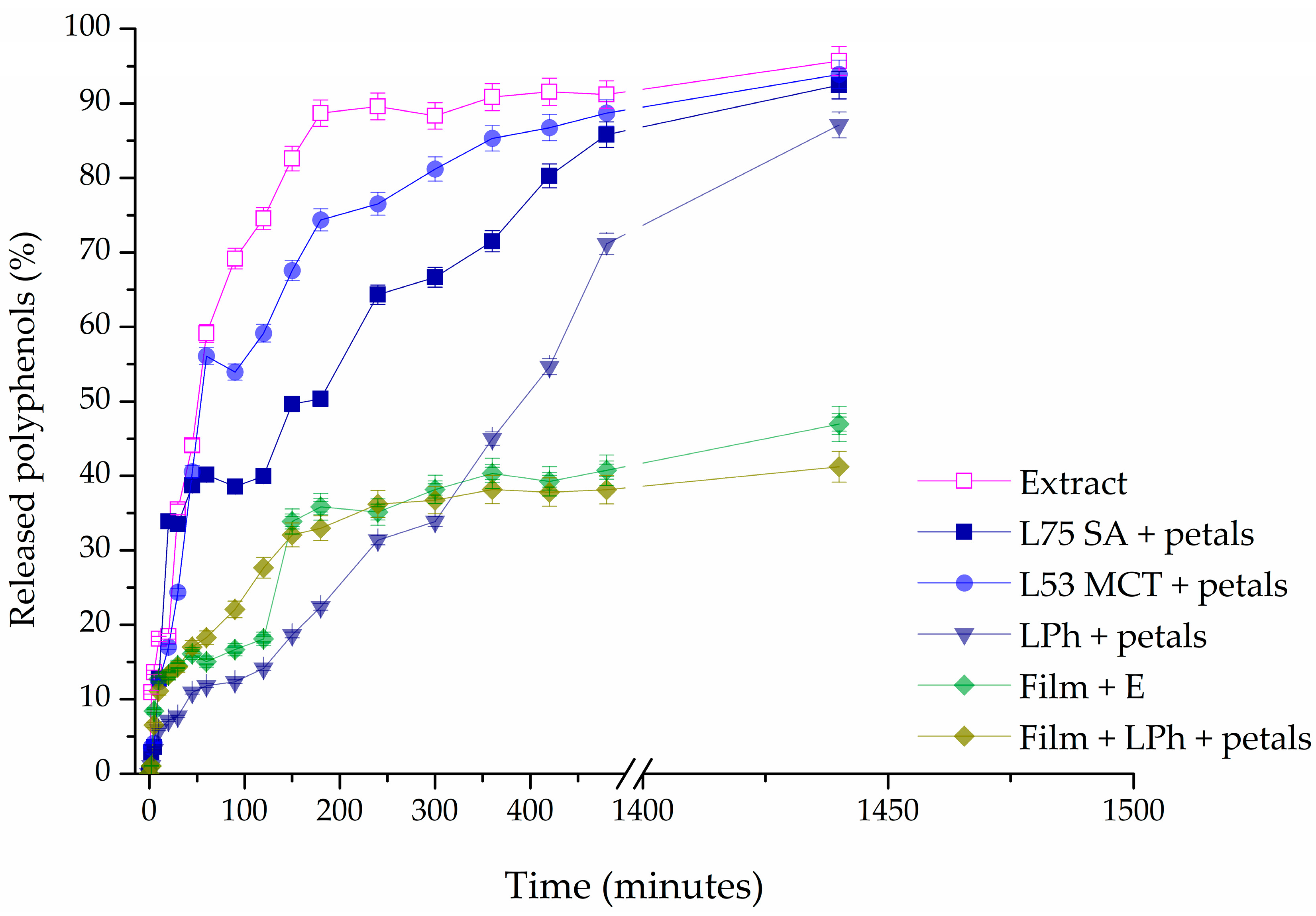
2.13. Release Study
Using the Franz diffusion cell (PermGear, Inc., Hellertown, PA, USA), studies of the controlled release of polyphenols from the extract, liposomes, liposome-loaded films, and pure extract-loaded films were carried out. The donor and acceptor compartments of a Franz cell are divided by a cellulose acetate membrane (Permgear, Hellertown, PA, USA) with a diffusion area of 4.91 cm
2 and a pore size of 0.2 µm. The donor compartment (0.05 g, d = 2.5 cm) received the samples. A magnetic stirrer was used to continuously mix the release media (phosphate buffer, pH = 5.5, c = 0.1 mol/L) at 37 °C and 400 rpm in the receptor compartment [
39]. For 24 h, samples were collected at set times. The Folin–Ciocalteu method, as previously indicated by Čutović et al. [
3], was adjusted for the quantification of the polyphenols in these samples. Briefly, 20 μL of the controlled release sample, 100 μL of the Folin–Ciocalteu reagent, and 1500 μL of deionized water were placed in a 2000 μL flask. Subsequently, 300 μL of sodium carbonate (20%
w/
v) was added, and the mixture was then topped off to a volume of 2000 μL with deionized water. After 120 min of incubation at room temperature and in the dark, the absorbance at 765 nm was measured. The results of each analysis were statistically processed after being run three times.
2.14. Scanning Electron Microscopy (SEM)
The morphology of the biopolymer films containing the P. tenuifolia petal extract or liposomes was evaluated using scanning electron microscopy (Tescan Mira3 XMU, Cranberry Township, PA, USA), operated at 10 keV. The samples were subjected to analysis in their dry forms. Prior to SEM analysis, all three samples of the films were cut on a tile (5 × 5 mm), fixed on a sample holder, and then vacuum-coated with a gold/platinum alloy (15/85) using a Polaron SC502 vacuum sputter coater.
2.15. Statistical Analysis
The statistical analysis in this study was carried out using an analysis of variance (one-way ANOVA) followed by Duncan’s
post hoc test inside the statistical program STATISTICA 7.0 (TIBCO Software Inc., Palo Alto, CA, USA) [
45]. Duncan’s multiple range test is a statistical test known for its use in order to compare the means of multiple groups of results. It is a post hoc test that is performed after the analysis of variance (ANOVA) test reveals a significant difference. The test detects whether group means differ significantly from one another, and divides them into subgroups depending on their similarities. The differences were considered statistically significant at
p < 0.05, n = 3.



 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}