

New Biocompatible Nanohydrogels of Predefined Sizes for Complexing Nucleic Acids



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
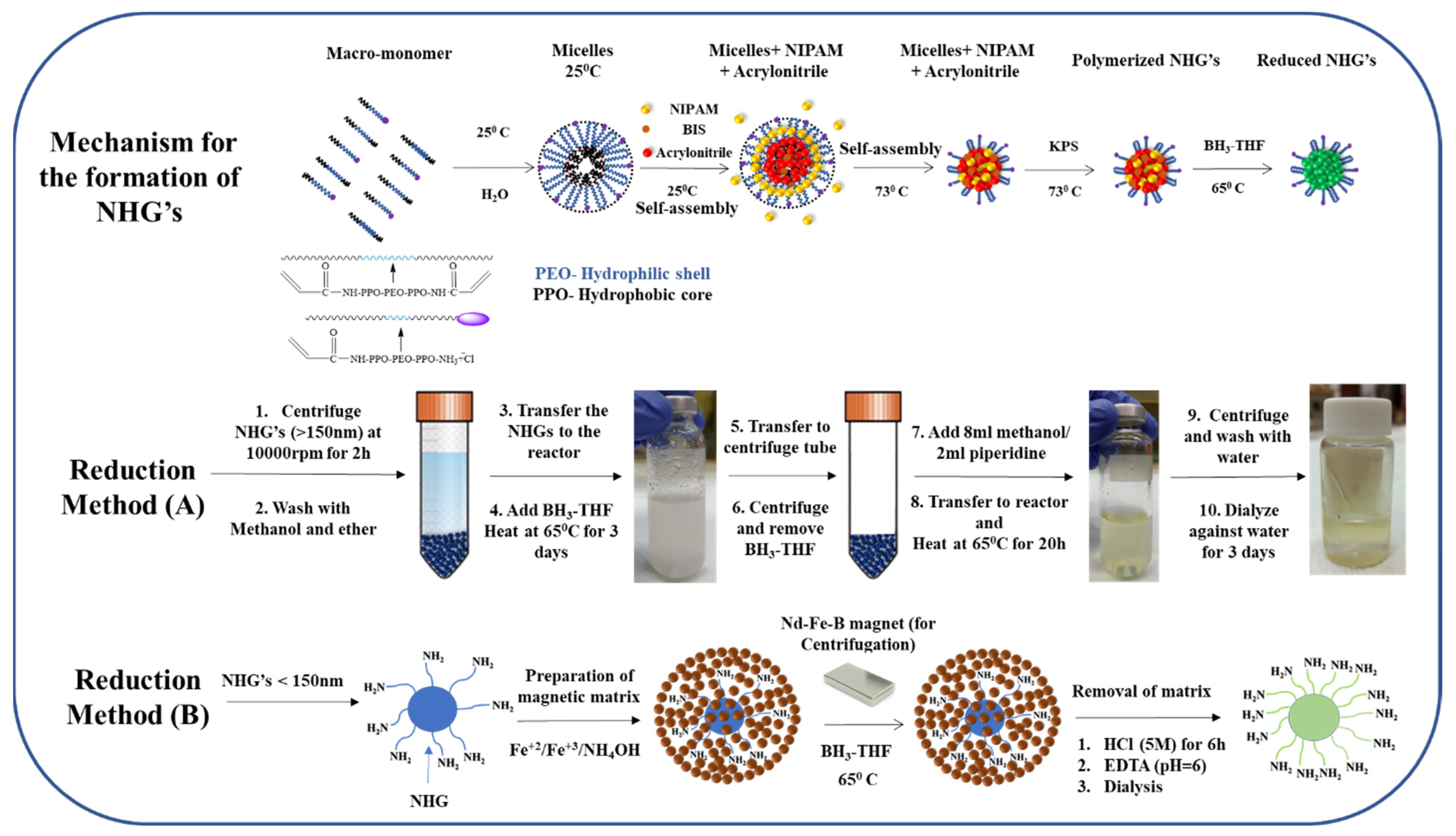
2.2. Synthesis of NHG’s Containing Highly Reducible Groups
2.3. Reduction of NHG’s (Method A)
2.4. Reduction of NHG’s (Method B)
Preparation of the Magnetic Embedded NHG’s Matrix
2.5. Reduction of Magnetic Embedded NHG’s
2.6. Dissolving the Magnetic Matrix for Releasing the NHG’s
2.7. DLS and Zeta Potential
2.8. Plasmid Preparation
2.9. Preparation of Polyplexes for Size and Zeta Potential Measurement
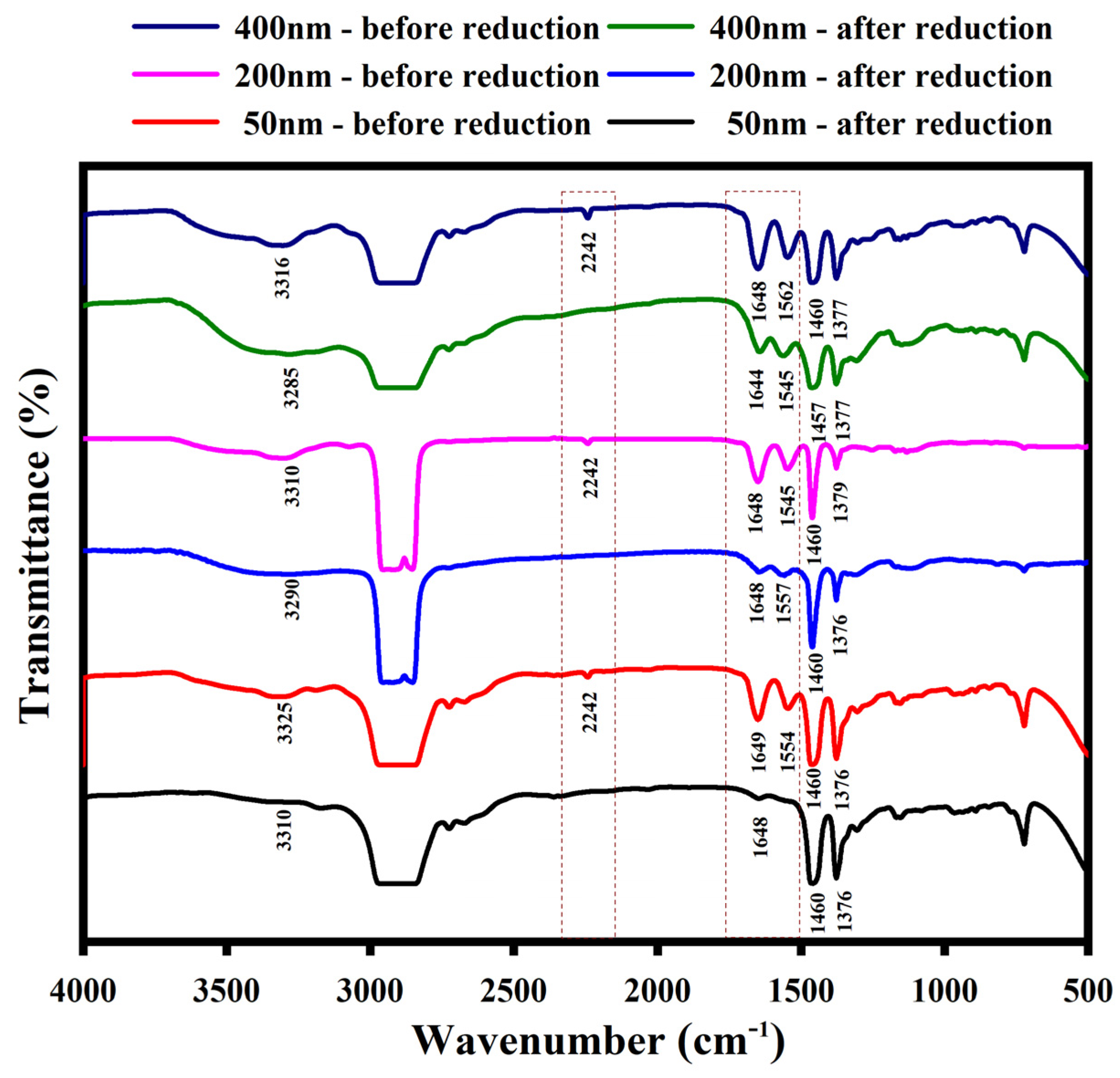
2.10. Fourier-Transform Infrared Spectroscopy (FTIR)
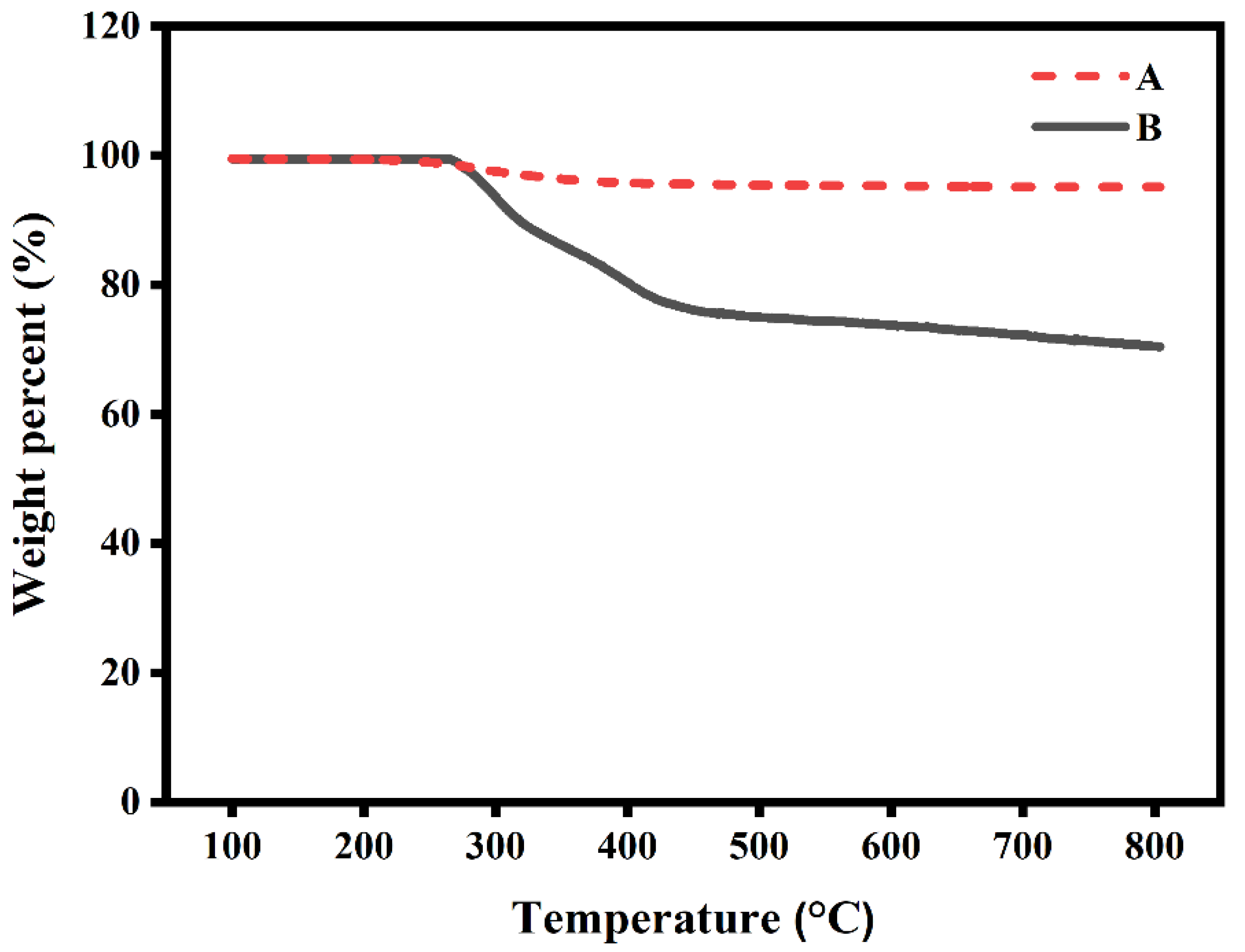
2.11. Thermogravimetric Analysis (TGA)
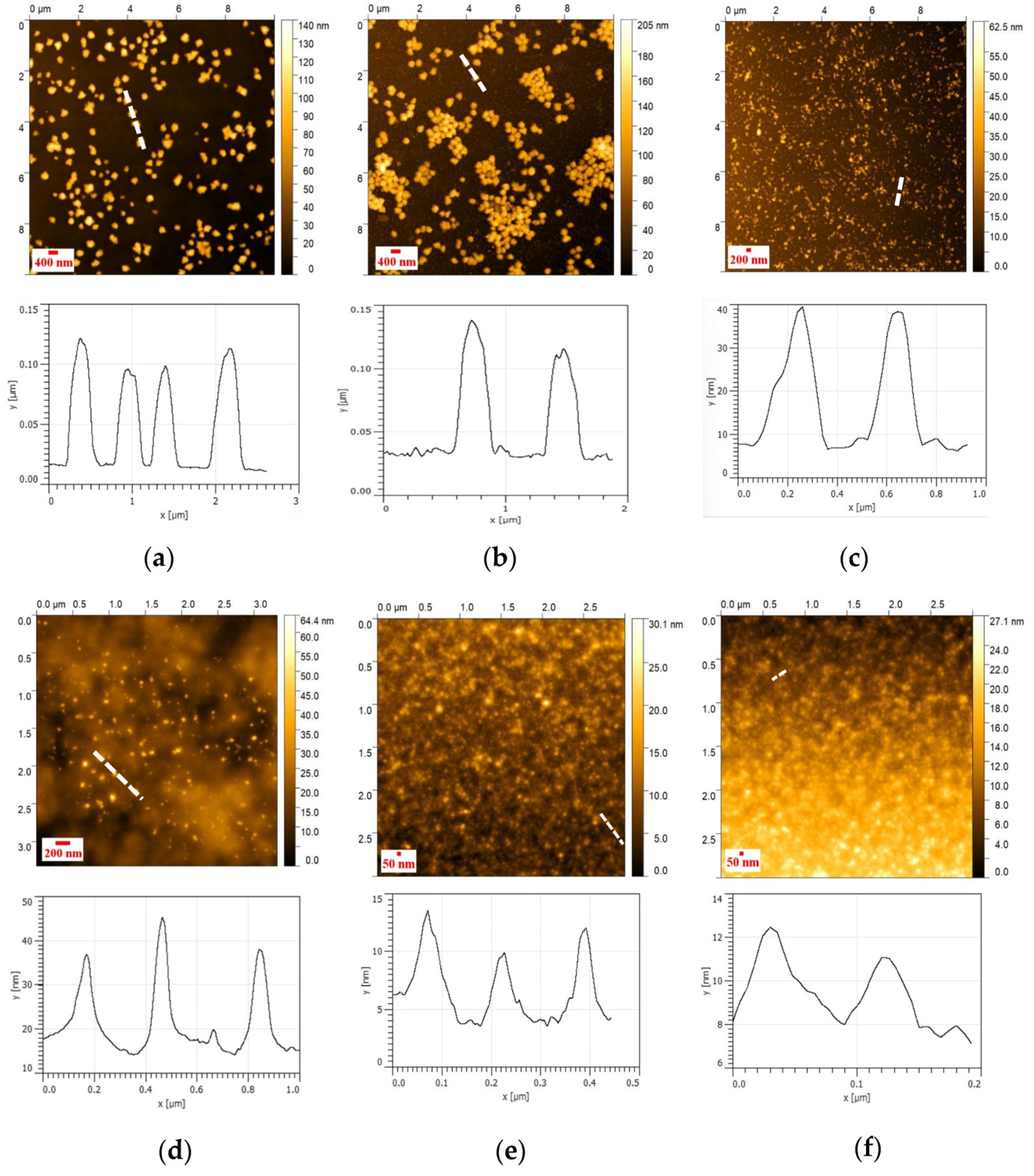
2.12. Atomic Force Microscopy (AFM)
2.12.1. Method (A)
2.12.2. Method (B)
2.13. Gel Electrophoresis
2.14. Cell Culture
2.15. Cell Proliferation Assays
3. Results and Discussion
3.1. Synthesis and Characterization of Nitrile Containing Nanohydrogels
3.2. Zeta Potentials
3.3. Fourier-Transform Infrared Spectroscopy (FTIR)
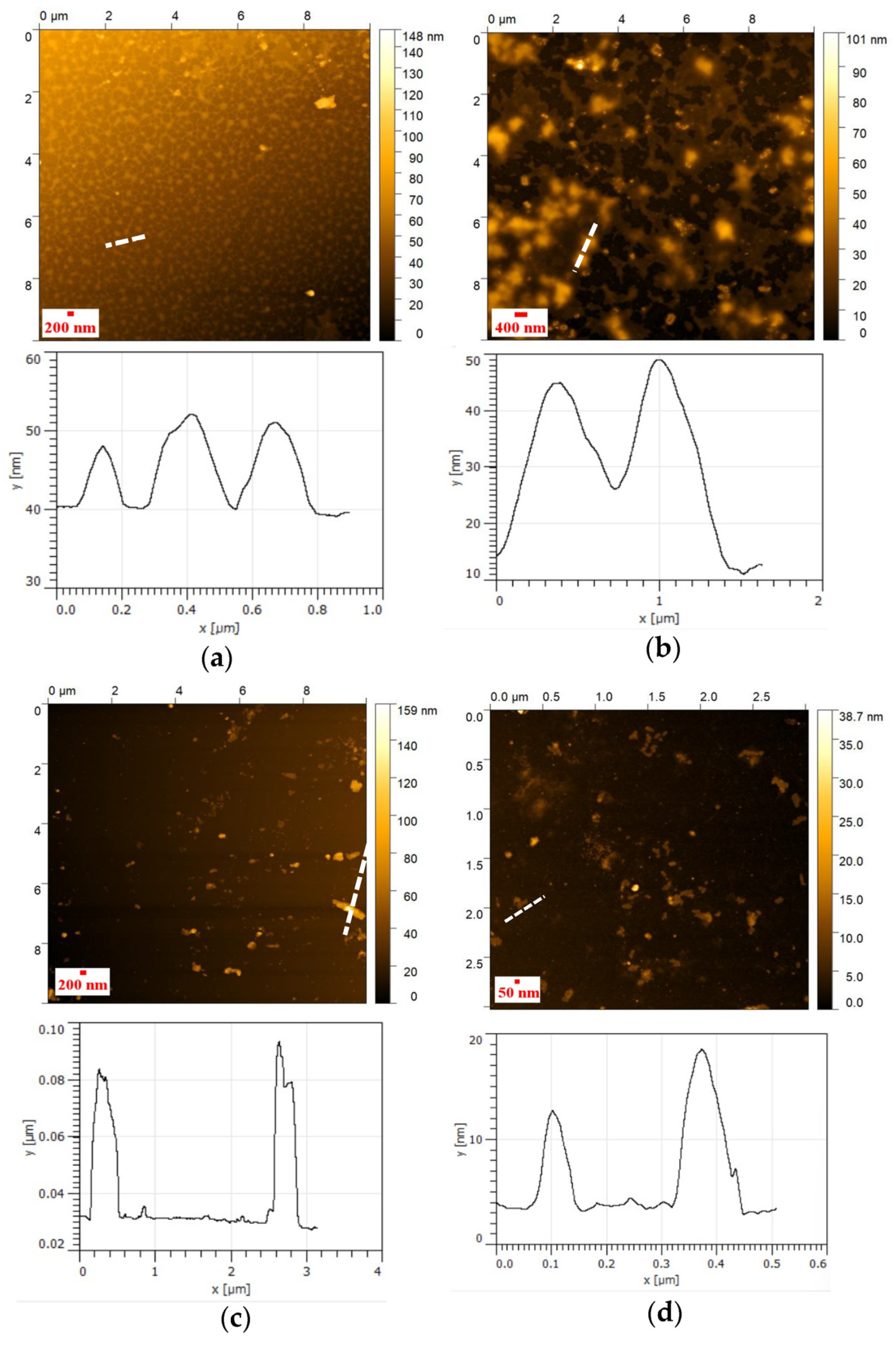
3.4. AFM Analysis of NHG’s
3.5. Complexes of NHG’s with DNA
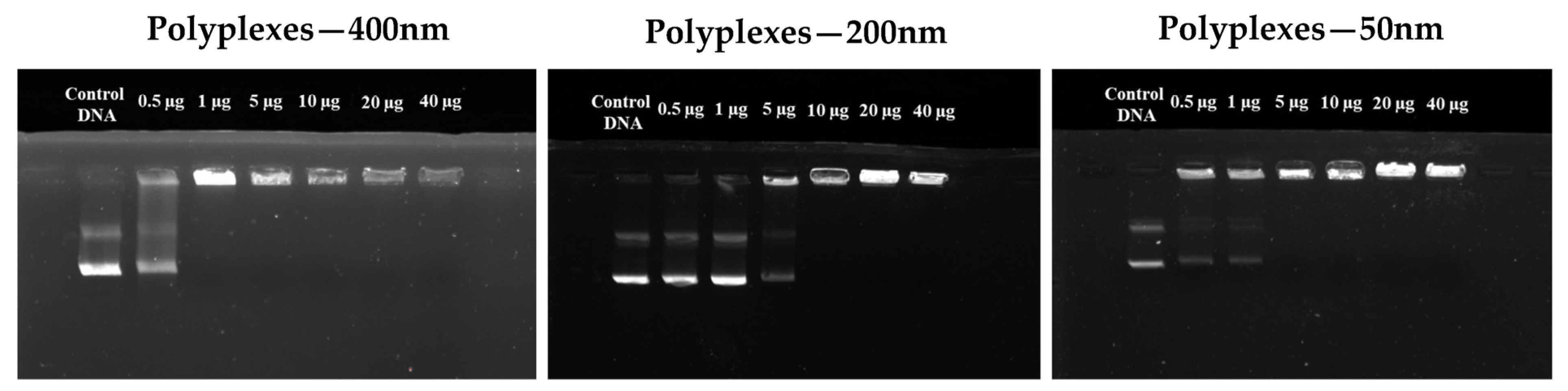
Gel Electrophoresis
3.6. Size and Zeta Potential of Polyplexes
3.6.1. AFM of Polyplexes
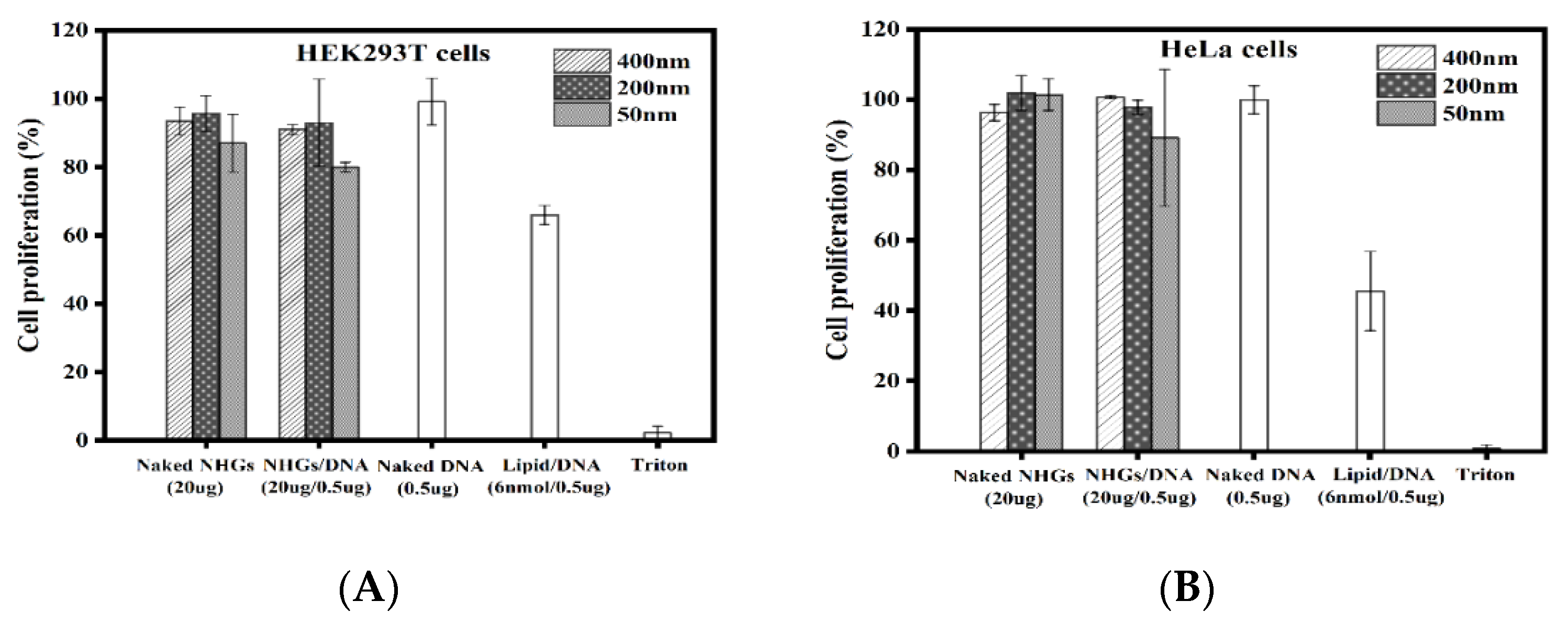
3.6.2. Cell Proliferation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | Acrylonitrile |
| BIS | N, N’-Methylenebisacrylamide |
| DLS | Dynamic Light Scattering |
| KPS | Potassium persulfate |
| NP/NPs | Nanoparticle/ Nanoparticles |
| NHG’s | Nanohydrogels |
| NIPAAM | N-Isopropylacrylamide |
| PDI | Polydispersity index |
| PEG | Poly (ethylene glycol) |
| PEO | Poly (ethylene oxide) |
| PPO | Poly (propylene oxide) |
| PVP | Polyvinylpyrrolidone |
References
- Mirza, Z.; Karim, S. Nanoparticles-based drug delivery and gene therapy for breast cancer: Recent advancements and future challenges. Semin. Cancer Biol. 2021, 69, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Luchini, A.; Vitiello, G. Understanding the nano-bio interfaces: Lipid-coatings for inorganic nanoparticles as promising strategy for biomedical applications. Front. Chem. 2019, 7, 343. [Google Scholar] [CrossRef] [PubMed]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, M.; Azadi, A.; Rafiei, P. Hydrogel nanoparticles in drug delivery. Adv. Drug Deliv. Rev. 2008, 60, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, K. PEO-related block copolymer surfactants. Colloids Surf. A Physicochem. Eng. Asp 2001, 183–185, 277–292. [Google Scholar] [CrossRef]
- Discher, B.M.; Won, Y.Y.; Ege, D.S.; Lee, J.C.-M.; Bates, F.S.; Discher, D.E.; Hammer, D.A. Polymersomes: Tough vesicles made from diblock copolymers. Science 1999, 284, 1143–1146. [Google Scholar] [CrossRef] [Green Version]
- Sprakel, J.; Leermakers, F.A.; Stuart, M.A.; Besseling, N.A. Comprehensive theory for star-like polymer micelles; combining classical nucleation and polymer brush theory. Phys. Chem. Chem. Phys. 2008, 10, 5308–5316. [Google Scholar] [CrossRef]
- Miranda, M.E.; Marcolla, C.; Rodrígues, C.A.; Wilhelm, H.M.; Sierakowski, M.R.; Bresolin, T.M.; de Freitas, R.A. Chitosan and N-carboxymethylchitosan: I. The role of N-carboxymethylation of chitosan in the thermal stability and dynamic mechanical properties of its films. Polym. Int. 2006, 55, 961–969. [Google Scholar] [CrossRef]
- Kadam, V.S.; Nicol, E.; Gaillard, C. Synthesis of flower-like poly(ethylene oxide) based macromolecular architectures by photo-cross-linking of block copolymers self-assemblies. Macromolecules 2012, 45, 410–419. [Google Scholar] [CrossRef]
- Liu, M.; Fu, J.; Li, J. Preparation of tri-block copolymer micelles loading novel organoselenium anticancer drug BBSKE and study of tissue distribution of copolymer micelles by imaging in vivo method. Int. J. Pharm. 2010, 391, 292–304. [Google Scholar] [CrossRef]
- Sahiner, N.; Alb, A.M.; Graves, R. Core-shell nanohydrogel structures as tunable delivery systems. Polymer 2007, 48, 704–711. [Google Scholar] [CrossRef]
- Khandadash, R.; Machtey, V.; Shainer, I.; Gottlieb, H.E.; Gothilf, Y.; Ebenstein, Y.; Weiss, A.; Byk, G. Novel biocompatible hydrogel nanoparticles: Generation and size-tuning of nanoparticles by the formation of micelle templates obtained from thermo-responsive monomers mixtures. J. Nanoparticle Res. 2014, 16, 2796. [Google Scholar] [CrossRef]
- Shubayev, V.I.; Pisanic, T.R.; Jin, S. Magnetic nanoparticles for theragnostics. Adv. Drug Deliv. Rev. 2009, 61, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef]
- Gref, R.; Minamitake, Y.; Peracchia, M.T.; Trubetskoy, V.; Torchilin, V.; Langer, R. Biodegradable Long-Circulating Polymeric Nanospheres. Science 1994, 263, 1600–1603. [Google Scholar] [CrossRef] [Green Version]
- Frank, M.M.; Fries, L.F. The role of complement in inflammation and phagocytosis. Immunol. Today 1991, 12, 322–326. [Google Scholar] [CrossRef]
- Van Vlerken, L.E.; Vyas, T.K.; Amiji, M.M. Poly(ethylene glycol)-modified nanocarriers for tumor-targeted and intracellular delivery. Pharm. Res. 2007, 24, 1405–1414. [Google Scholar] [CrossRef]
- Schipper, M.L.; Iyer, G.; Koh, A.L.; Cheng, Z.; Ebenstein, Y.; Aharoni, A.; Keren, S.; Bentolila, L.A.; Li, J.; Rao, J.; et al. Particle size, surface coating, and PEGylation influence the biodistribution of quantum dots in living mice. Small 2009, 5, 126–134. [Google Scholar] [CrossRef] [Green Version]
- Kazunori, K.; Glenn, S.K.; Masayuki, Y.; Teruo, O.; Yasuhisa, S. Block copolymer micelles as vehicles for drug delivery. J. Control. Release 1993, 24, 119–132. [Google Scholar] [CrossRef]
- Kataoka, K.; Shibazaki, C.; Sakurai, Y. Toxicity and Antitumor Activity against Solid Tumors of Micelle-forming Polymeric Anticancer Drug and Its Extremely Long Circulation in Blood. Cancer Res. 1991, 51, 3229–3236. [Google Scholar]
- Kwon, G.; Suwa, S.; Yokoyama, M.; Okano, T.; Sakurai, Y.; Kataoka, K. Enhanced tumor accumulation and prolonged circulation times of micelle-forming poly (ethylene oxide-aspartate) block copolymer-adriamycin conjugates. J. Control. Release 1994, 29, 17–23. [Google Scholar] [CrossRef]
- Lutz, J.F. Polymerization of oligo(ethylene glycol) (meth)acrylates: Toward new generations of smart biocompatible materials. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 3459–3470. [Google Scholar] [CrossRef]
- Tessmar, J.K.; Göpferich, A.M. Customized PEG-derived copolymers for tissue-engineering applications. Macromol. Biosci. 2007, 7, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Christensen, C.; Groth, T.; Schiødt, C.B.; Foged, N.T.; Meldal, M. Automated Sorting of Beads from a “One-Bead-Two-Compounds” Combinatorial Library of Metalloproteinase Inhibitors. QSAR Comb. Sci. 2003, 22, 737–744. [Google Scholar] [CrossRef]
- Luo, J.; Zhang, H.; Xiao, W.; Kumaresan, P.R.; Shi, C.; Pan, C.X.; Aina, O.H.; Lam, K.S. Rainbow beads: A color coding method to facilitate high-throughput screening and optimization of one-bead one-compound combinatorial libraries. J. Comb. Chem. 2008, 10, 599–604. [Google Scholar] [CrossRef]
- Gulati, N.; Rastogi, R.; Dinda, A.K.; Saxena, R.; Koul, V. Characterization and cell material interactions of PEGylated PNIPAAM nanoparticles. Colloids Surf. B Biointerfaces 2010, 79, 164–173. [Google Scholar] [CrossRef]
- Leobandung, W.; Ichikawa, H.; Fukumori, Y.; Peppas, N.A. Monodisperse nanoparticles of poly(ethylene glycol) macromers and N-isopropyl acrylamide for biomedical applications. J. Appl. Polym. Sci. 2002, 87, 1678–1684. [Google Scholar] [CrossRef]
- Yoo, J.W.; Doshi, N.; Mitragotri, S. Adaptive micro and nanoparticles: Temporal control over carrier properties to facilitate drug delivery. Adv. Drug Deliv. Rev. 2011, 63, 1247–1256. [Google Scholar] [CrossRef]
- Khandadash, R.; Machtey, V.; Weiss, A.; Byk, G. Matrix-assisted peptide synthesis on nanoparticles. J. Pept. Sci. 2014, 20, 675–679. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Amount of Macro Monomer (mg) | Size of NHG’s (nm) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Original NHG’s | Reduced NHG’s | ||||||||
| 25 °C | PDI | 45 °C | PDI | 25 °C | PDI | 45 °C | PDI | ||
| A | 200 | 401 | 0.019 | 371 | 0.132 | 407 | 0.361 | 361 | 0.355 |
| B | 400 | 199 | 0.058 | 165 | 0.265 | 200 | 0.339 | 172 | 0.34 |
| C | 500 | 54 | 0.028 | 32 | 0.33 | 71 | 0.372 | 48 | 0.362 |
| # | Size of NHG’s (nm) | ζ (mV) | |||
|---|---|---|---|---|---|
| Original NHG’s | Reduced NHG’s | ||||
| 25 °C | 45 °C | 25 °C | 45 °C | ||
| A | 400 | −18.6 | −20.5 | 22.6 | 25 |
| B | 200 | −15.4 | −17 | 19.2 | 23 |
| C | 50 | −5.8 | −8.47 | 11.6 | 14 |
| # | NHG’s/DNA (wt/wt) Ratio | 0:1 | 1:0 | 1:10 | 1:1 | 5:1 | 10:1 | 20:1 | 40:1 |
|---|---|---|---|---|---|---|---|---|---|
| Polyplexes (400 nm NHG) | Size (nm) 25 °C | AG* | 405 | AG* | AG* | 410 | 394 | 398 | 403 |
| PDI | 1.000 | 0.186 | 1.000 | 1.000 | 0.341 | 0.242 | 0.302 | 0.316 | |
| ζ (mV) 25 °C | −10 | 22 | −3.9 | 7.1 | 12.5 | 13.8 | 17.7 | 22.8 | |
| Polyplexes (200 nm NHG) | Size (nm) 25 °C | AG* | 230 | AG* | AG* | 255 | 225 | 223 | 227 |
| PDI | 1.000 | 0.242 | 1.000 | 1.000 | 0.372 | 0.259 | 0.267 | 0.292 | |
| ζ (mV) 25 °C | −8.9 | 19 | −5.7 | 4.9 | 10.7 | 12.4 | 15.7 | 18.2 | |
| Polyplexes (50 nm NHG) | Size (nm) 25 °C | AG* | 62 | AG* | AG* | 54 | 65.3 | 67.6 | 56 |
| PDI | 1.000 | 0.772 | 1.000 | 1.000 | 0.942 | 0.842 | 0.741 | 0.686 | |
| ζ (mV) 25 °C | −9.2 | 12.3 | −8.1 | 3.0 | 7.3 | 8.2 | 10.5 | 10.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eswaran, L.; Kazimirsky, G.; Byk, G. New Biocompatible Nanohydrogels of Predefined Sizes for Complexing Nucleic Acids. Pharmaceutics 2023, 15, 332. https://doi.org/10.3390/pharmaceutics15020332
Eswaran L, Kazimirsky G, Byk G. New Biocompatible Nanohydrogels of Predefined Sizes for Complexing Nucleic Acids. Pharmaceutics. 2023; 15(2):332. https://doi.org/10.3390/pharmaceutics15020332
Chicago/Turabian StyleEswaran, Lakshmanan, Gila Kazimirsky, and Gerardo Byk. 2023. "New Biocompatible Nanohydrogels of Predefined Sizes for Complexing Nucleic Acids" Pharmaceutics 15, no. 2: 332. https://doi.org/10.3390/pharmaceutics15020332
APA StyleEswaran, L., Kazimirsky, G., & Byk, G. (2023). New Biocompatible Nanohydrogels of Predefined Sizes for Complexing Nucleic Acids. Pharmaceutics, 15(2), 332. https://doi.org/10.3390/pharmaceutics15020332