

Characteristics of Environmental Klebsiella pneumoniae and Klebsiella oxytoca Bacteriophages and Their Therapeutic Applications
 , , ,
, , ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Bacteriophage Origin
2.3. Phage Isolation from Environmental Samples
2.4. Phage Propagation and Titer Determination
2.5. The Influence of Physical and Chemical Factors on the Lytic Activity of Phages
2.6. Host Range Determination
2.7. One-Step Growth Experiments
2.8. Electron Microscope Study
2.8.1. Phage Preparation
2.8.2. Phage Particles Measurements
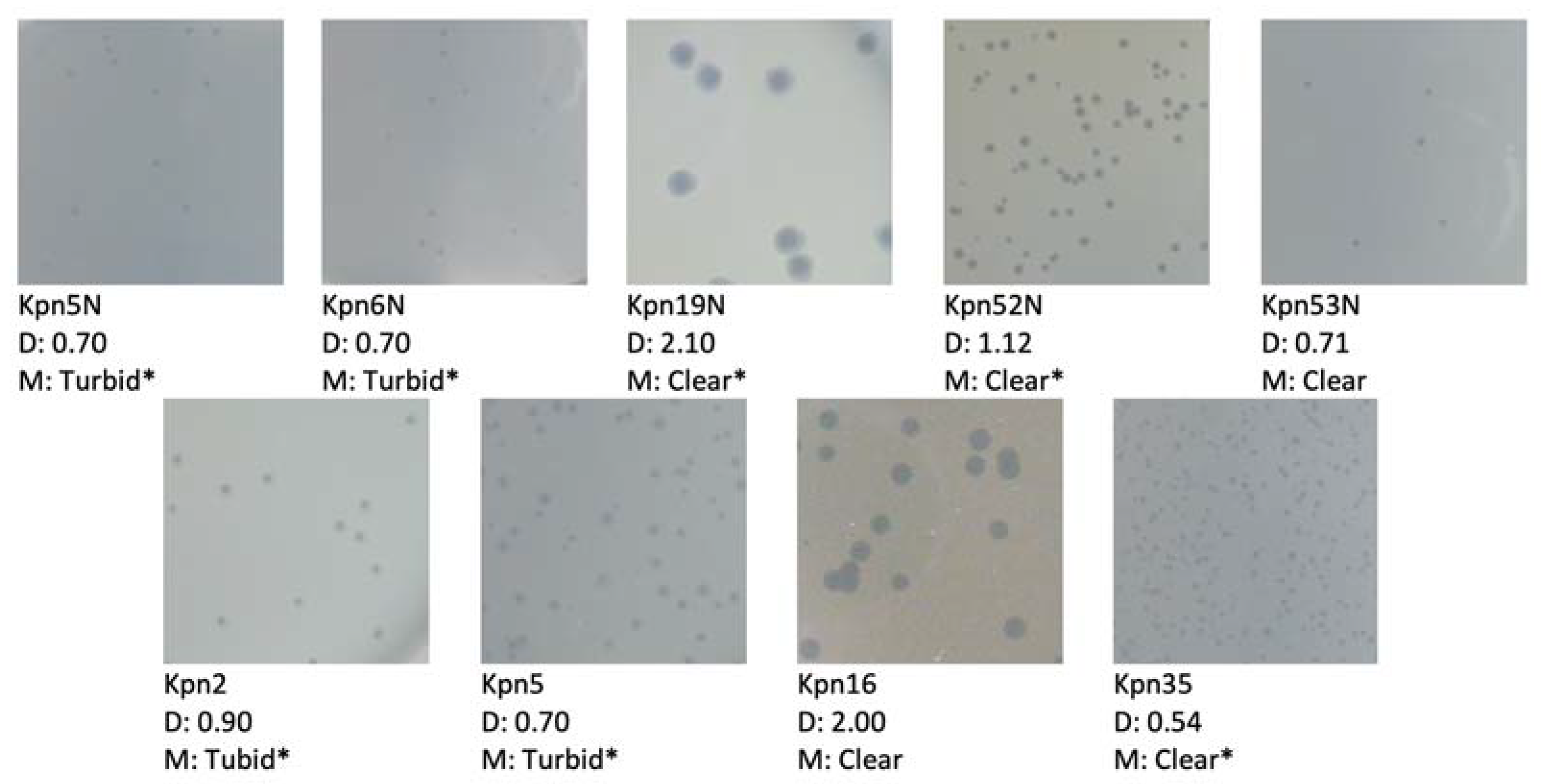
2.9. Plaque Morphology Analysis
2.10. Phage DNA Isolation, Phage Genome Sequencing and Bioinformatic Analysis
2.11. GenBank Accession Numbers
3. Results and Discussion
3.1. Bacteriophage Presence in Environmental and Sewage Samples
3.2. Bacteriophage Host Range Determination
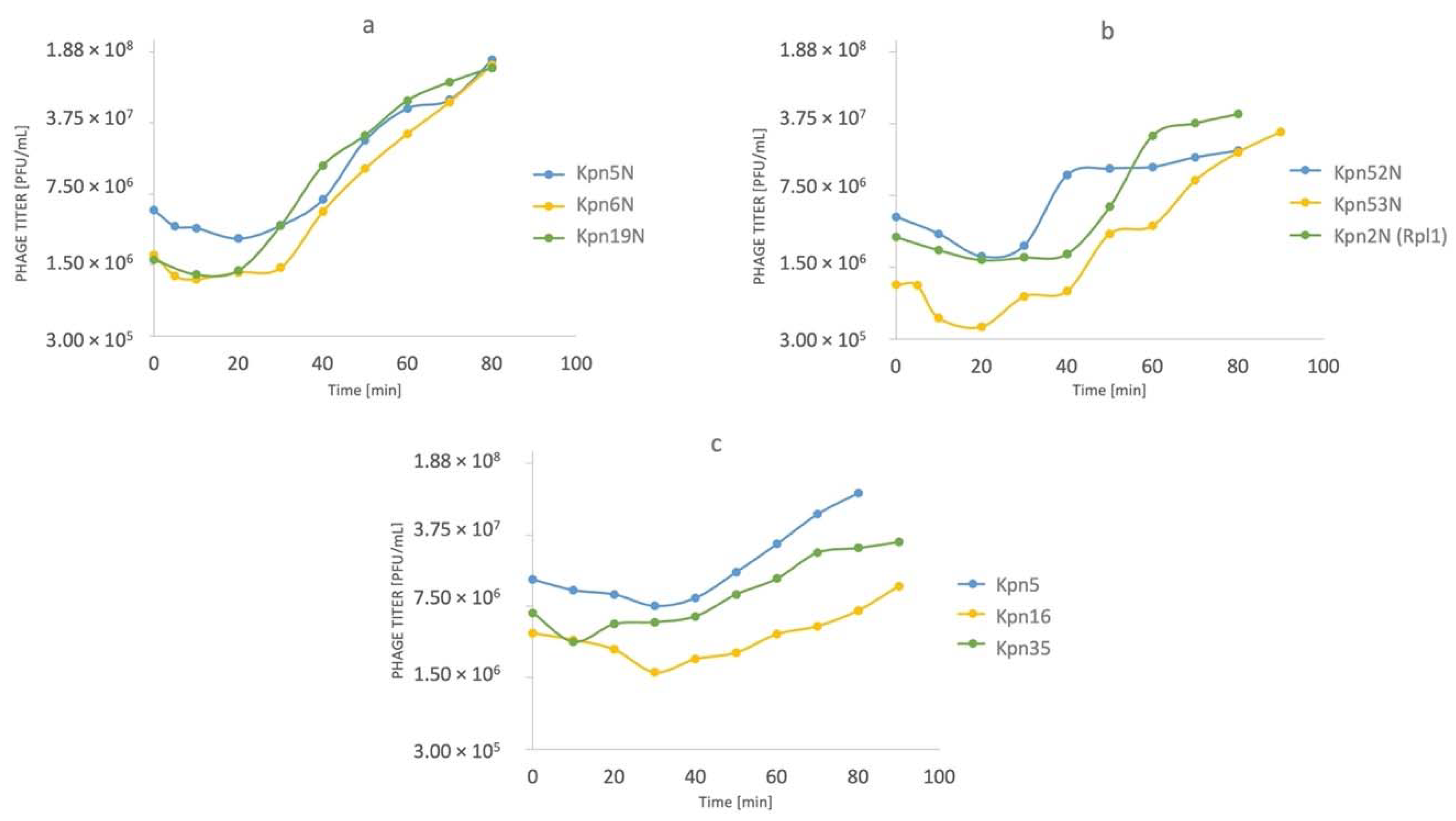
3.3. One-Step Growth Experiments
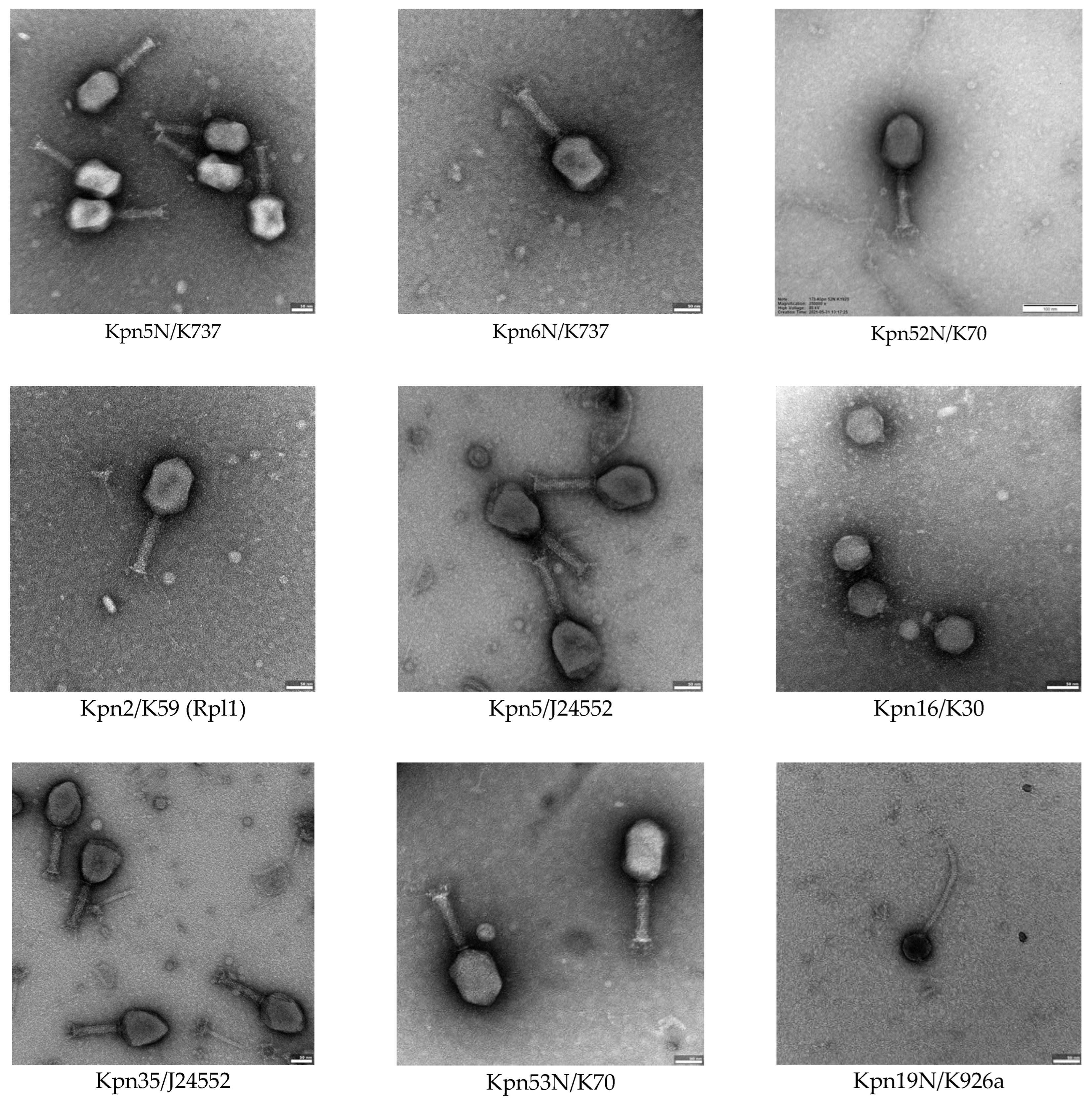
3.4. Bacteriophage Morphology Using Electron Microscopy
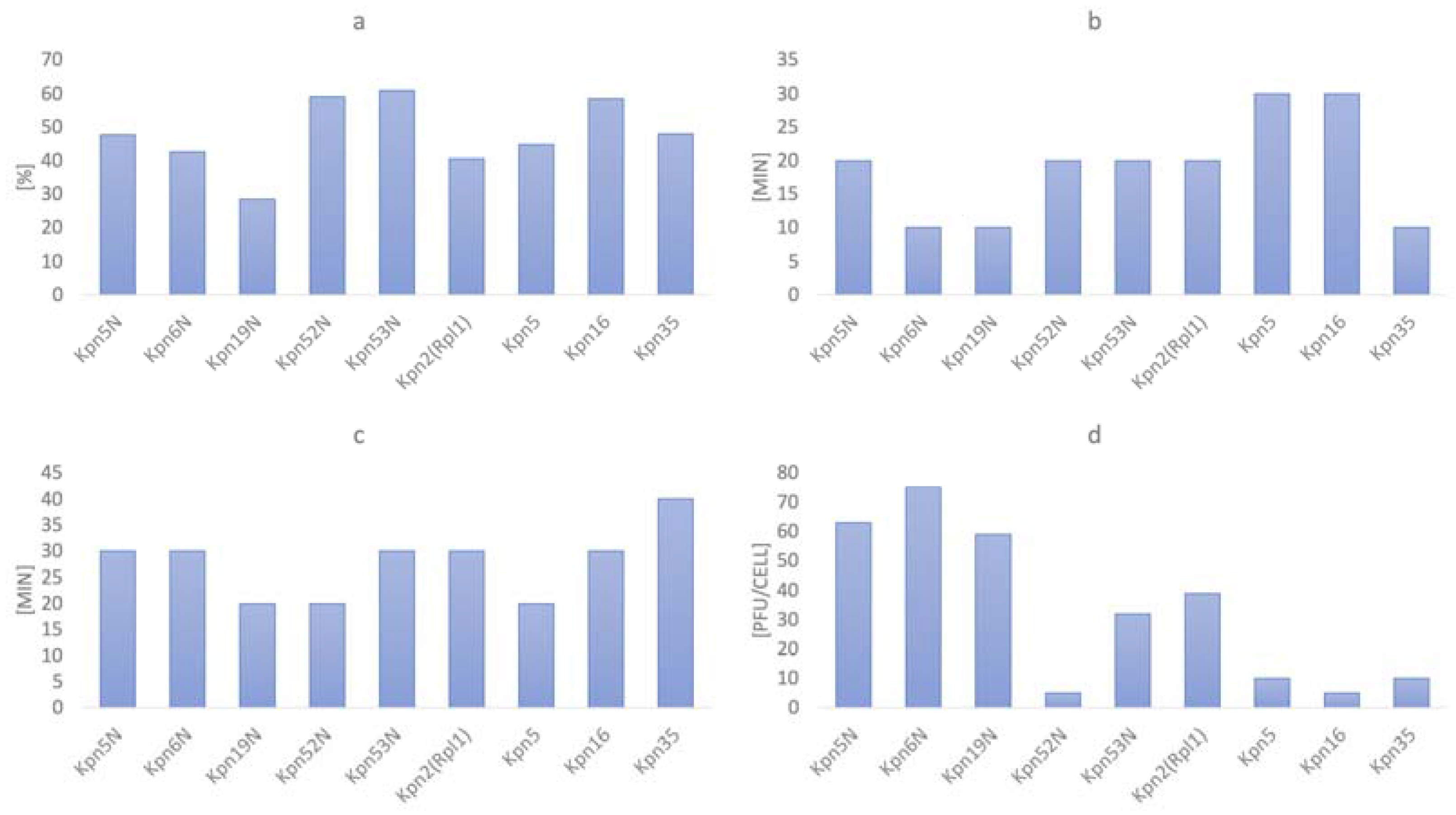
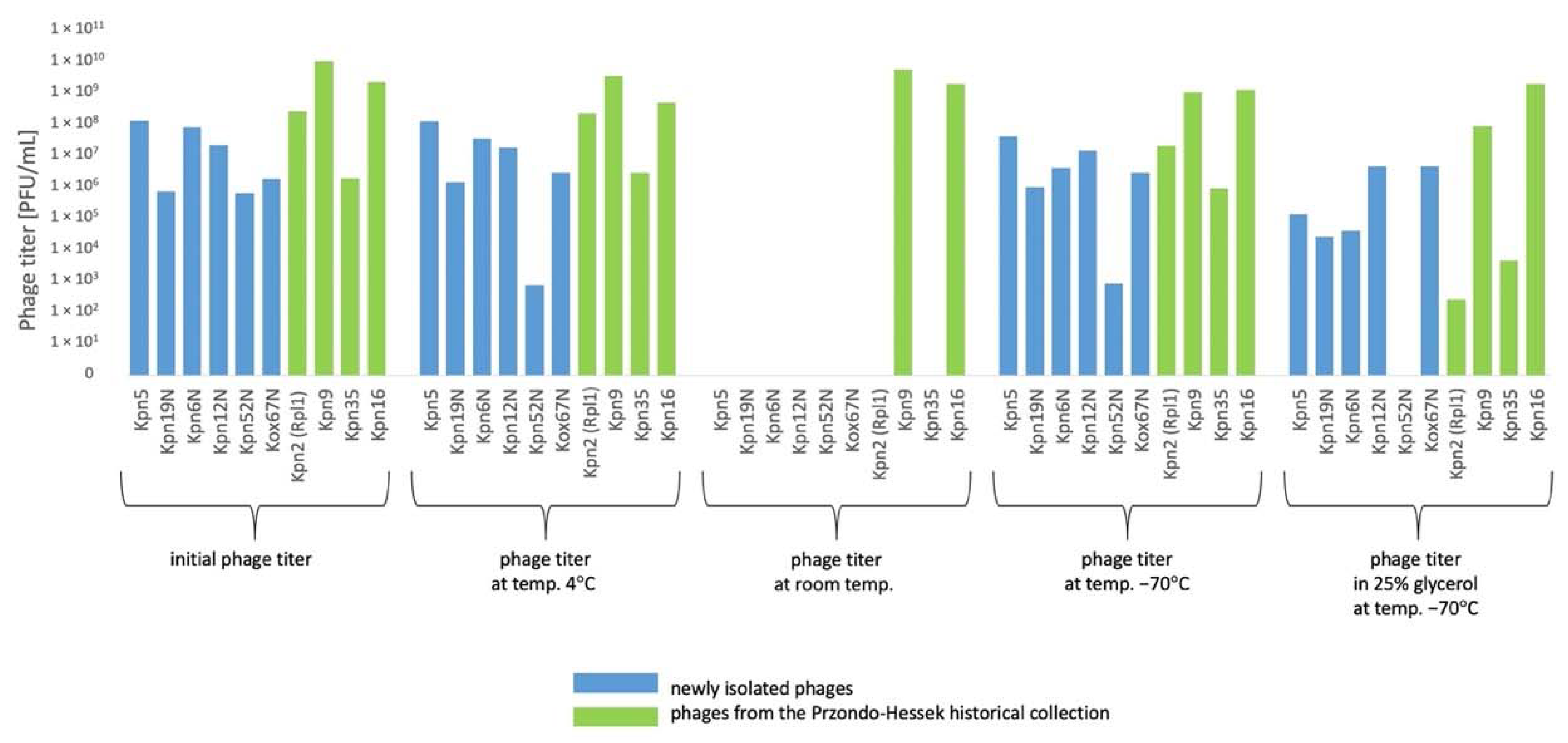
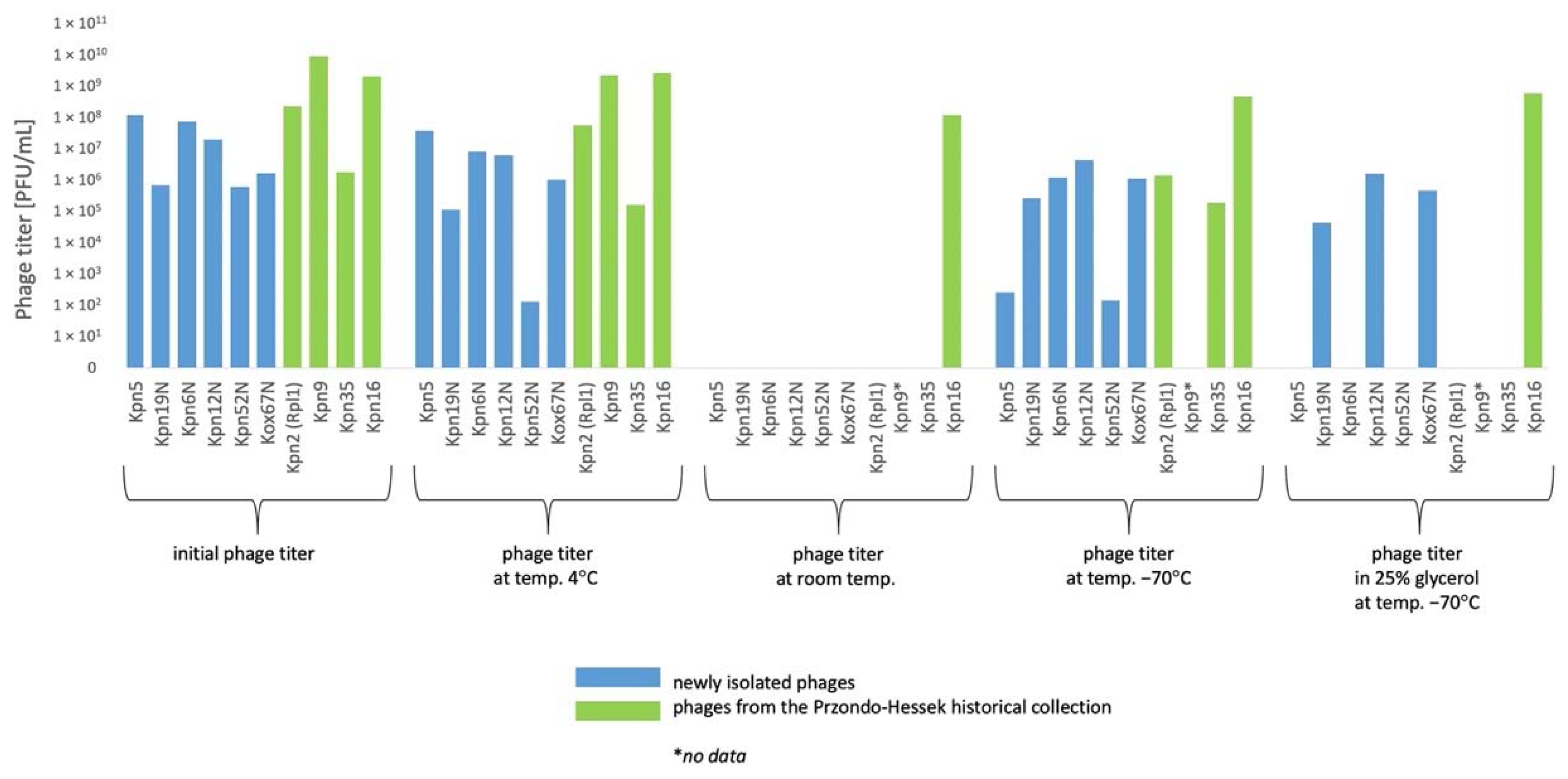
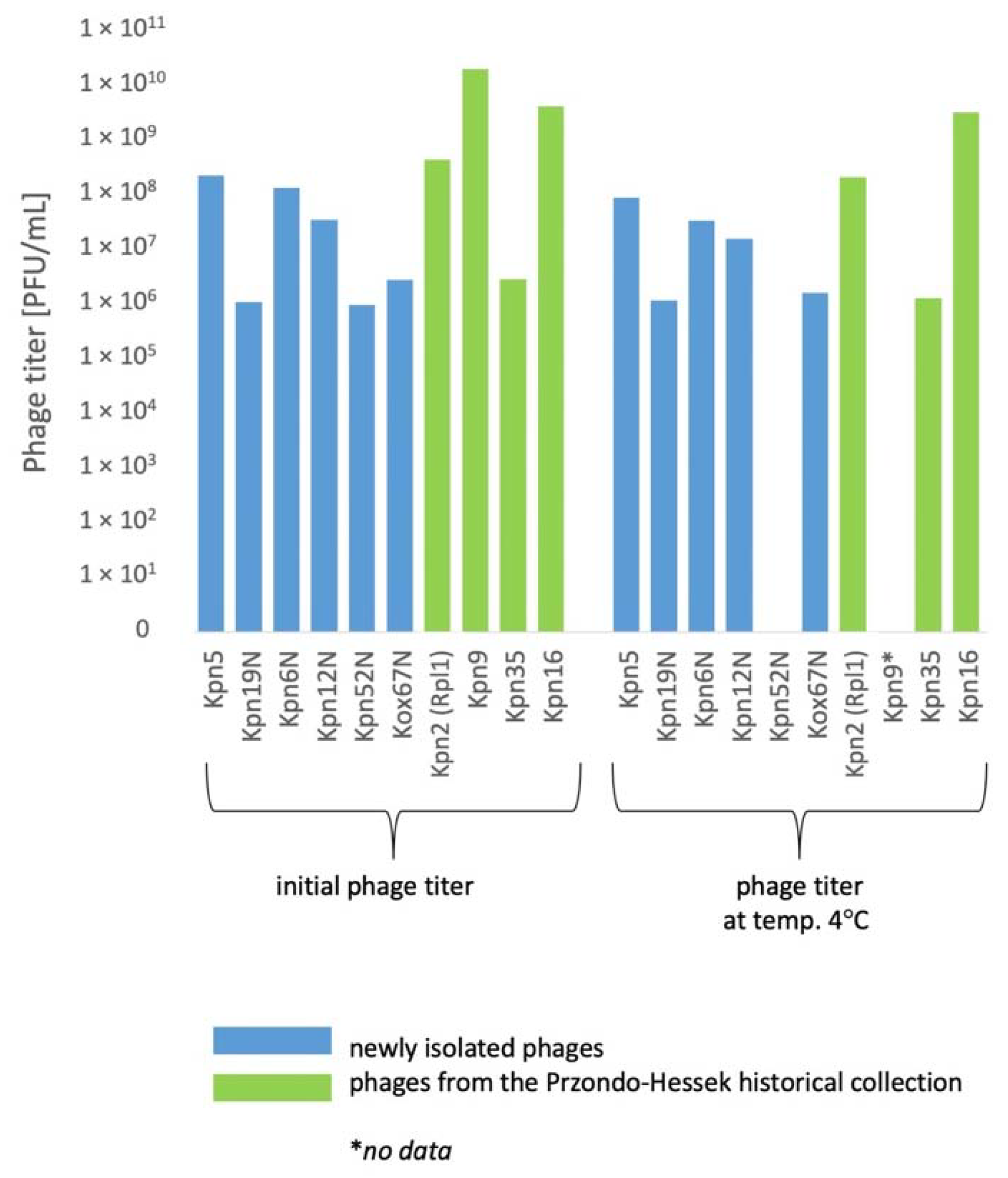
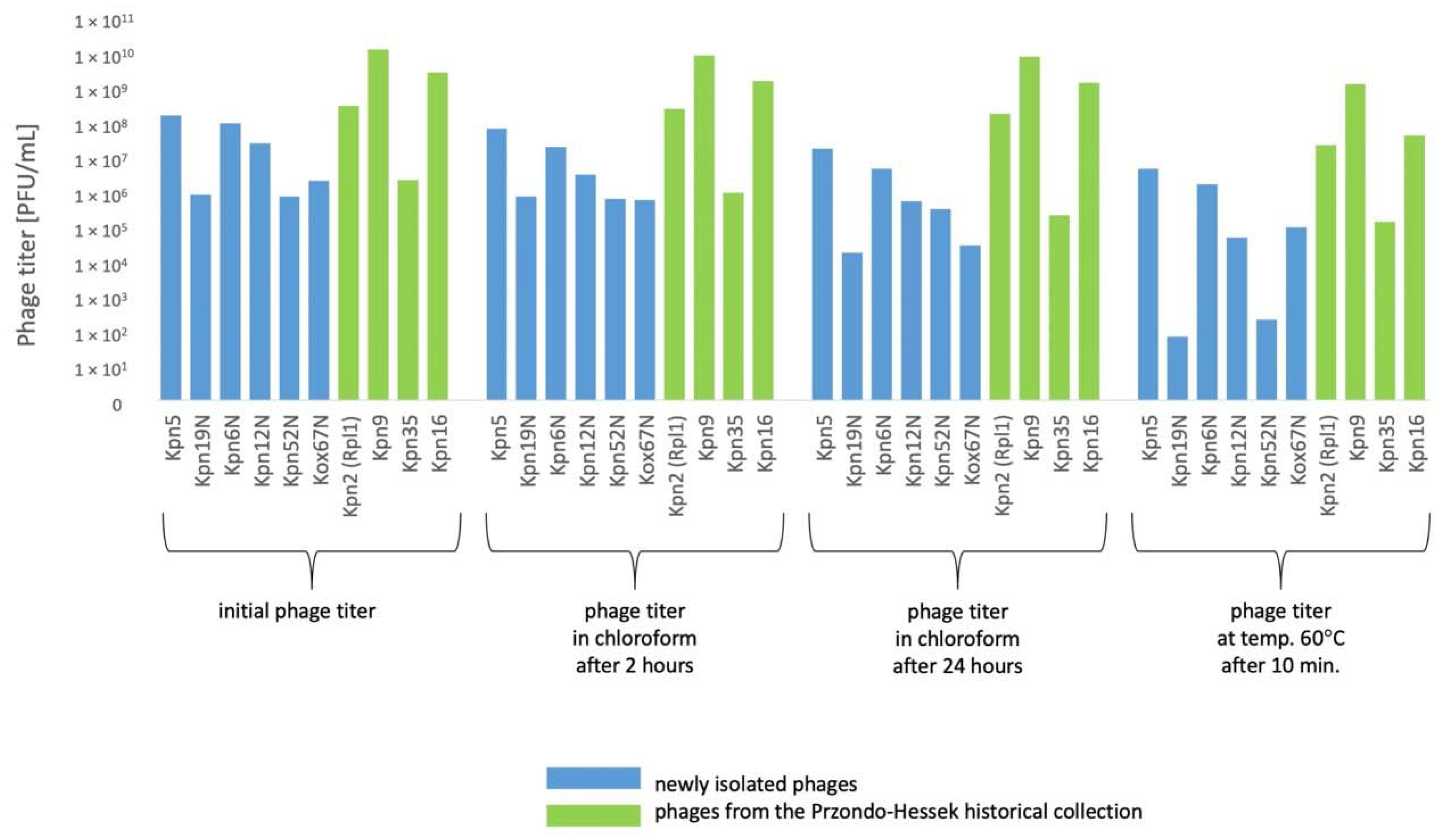
3.5. The Influence of Physico-Chemical Factors on Phage Lytic Activity
3.5.1. Klebsiella phages from the Przondo-Hessek Collection
3.5.2. Newly Isolated Klebsiella Phages
3.6. General Characteristic of Kpn35, Rpl1, Kpn5N and Kpn6N Genomic Sequences

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage Preparation | Kpn35/ | Kpn35/ | Rpl1 | Kpn5N | Kpn6N |
|---|---|---|---|---|---|
| Phage name | Klebsiella phage Kpn35c1 | Klebsiella phage Kpn35c2 | Raoltella phage Rpl1 | Klebsiella phage Kpn5N | Klebsiella phage Kpn6N |
| Short name | Kpn35c1 | Kpn35c2 | Rpl1 | Kpn5 | Kpn6 |
| Morphology | myovirus | myovirus | myovirus | myovirus | myovirus |
| Genus (Species) * | Slopekvirus | Jiaodavirus | Slopekvirus | Jiaodavirus | Jiaodavirus |
| Family | Straboviridae | Straboviridae | Straboviridae | Straboviridae | Straboviridae |
| Subfamily | - | Tevenvirinae | - | Tevenvirinae | Tevenvirinae |
| Host(s) | Klebsiella pneumoniae | Klebsiella pneumoniae | Raoltella planticola/Klebsiella pneumoniae | Klebsiella pneumoniae | Klebsiella pneumoniae |
| Genome size (bp) | 171,960 | >167,021 | 174,270 | 164,863 | 163,912 |
| Virion DNA/packaging strategy | terminally redundant, circularly permuted/headful packaging, | terminally redundant, circularly permuted/headful packaging, | terminally redundant, circularly permuted/headful packaging, | terminally redundant, circularly permuted/headful packaging, predicted preferred pac cuts between pos. 457–480 of genomic sequence | terminally redundant, circularly permuted/headful packaging, predicted preferred pac cuts between pos. 457–480 of genomic sequence |
| Best Blast hit (coverage in %/identity in % | Klebsiella phage K15 (98/99.6) | NT | Klebsiella phage K15 (97/98.64) | Klebsiella phage Kpn6 (97/96.2) | Klebsiella phage KP185 (97/95.5) |
| G + C content (%) | 41.9 | NT | 41.9 | 39.5 | 39.6 |
| Number of predicted protein-coding genes | 270 | NT | 271 | 271 | 271 |
| tRNA genes | 2 | NT | 2 | 16 | 16 |
| Number of genes encoding homologs of T4 proteins | 102 | NT | 102 | 159 | 159 |
| Homing endonucleases | 2: gp11, gp134 | NT | 1: gp134 | 3: 130, 133, 261 | 0 |
| Predicted receptor binding proteins | 1: gp56 | NT | 1: gp56 | 1: gp80 | 1: gp80 |
| Possible depolymerase domain containing proteins | 3: gp54, gp56, gp262 | NT | 3: gp54, gp56, gp262 | 4: gp78, gp79, gp80, gp262 | 4: gp78, gp79, gp80, gp262 |
3.7. Evaluation of the Coding Potential of New Phages with Respect to Their Safety in Therapeutic Applications
3.7.1. Lifestyle and Probability to Participate in DNA Transfer between Bacteria
3.7.2. Potential to Encode Toxins or Other Virulence-Associated Proteins
3.8. Evaluation of the Coding Potential of New Phages with Respect to the Efficacy in Infecting Various Klebsiella Strains
3.8.1. Ability to Penetrate through External Anti-Phage Cell Barriers
3.8.2. Ability to Overcome Intracellular Phage Defense Mechanisms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. Available online: https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 17 December 2022).
- Fang, C.-T.; Lai, S.-Y.; Yi, W.-C.; Hsueh, P.-R.; Liu, K.-L.; Chang, S.-C. Klebsiella pneumoniae Genotype K1: An Emerging Pathogen That Causes Septic Ocular or Central Nervous System Complications from Pyogenic Liver Abscess. Clin. Infect. Dis. 2007, 45, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Herridge, W.P.; Shibu, P.; O’Shea, J.; Brook, T.C.; Hoyles, L. Bacteriophages of Klebsiella spp., their diversity and potential therapeutic uses. J. Med. Microbiol. 2020, 69, 176–194. [Google Scholar] [CrossRef] [PubMed]
- Choby, J.E.; Howard-Anderson, J.; Weiss, D.S. Hypervirulent Klebsiella pneumoniae—Clinical and molecular perspectives. J. Intern. Med. 2020, 287, 283–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, R.M.; Bachman, M.A. Colonization, Infection, and the Accessory Genome of Klebsiella pneumoniae. Front. Cell. Infect. Microbiol. 2018, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Hernández-García, M.; Pérez-Viso, B.; León-Sampedro, R.; Francisco, C.N.-S.; López-Fresneña, N.; Díaz-Agero, C.; Morosini, M.I.; Ruiz-Garbajosa, P.; Cantón, R. Outbreak of NDM-1+CTX-M-15+DHA-1-producing Klebsiella pneumoniae high-risk clone in Spain owing to an undetectable colonised patient from Pakistan. Int. J. Antimicrob. Agents 2019, 54, 233–239. [Google Scholar] [CrossRef]
- Struve, C.; Krogfelt, K.A. Pathogenic potential of environmental Klebsiella pneumoniae isolates. Environ. Microbiol. 2004, 6, 584–590. [Google Scholar] [CrossRef]
- Struve, C.; Roe, C.C.; Stegger, M.; Stahlhut, S.G.; Hansen, D.S.; Engelthaler, D.M.; Andersen, P.S.; Driebe, E.M.; Keim, P.; Krogfelt, K.A. Mapping the Evolution of Hypervirulent Klebsiella pneumoniae. mBio 2015, 6, e00630-15. [Google Scholar] [CrossRef] [Green Version]
- Yoon, E.-J.; Kim, J.O.; Kim, D.; Lee, H.; Yang, J.W.; Lee, K.J.; Jeong, S.H. Klebsiella pneumoniae Carbapenemase Producers in South Korea between 2013 and 2015. Front. Microbiol. 2018, 9, 56. [Google Scholar] [CrossRef] [Green Version]
- Satlin, M.J.; Chen, L.; Gomez-Simmonds, A.; Marino, J.; Weston, G.; Bhowmick, T.; Seo, S.K.; Sperber, S.J.; Kim, A.C.; Eilertson, B.; et al. Impact of a Rapid Molecular Test for Klebsiella pneumoniae Carbapenemase and Ceftazidime-Avibactam Use on Outcomes After Bacteremia Caused by Carbapenem-Resistant Enterobacterales. Clin. Infect. Dis. 2022, 75, 2066–2075. [Google Scholar] [CrossRef]
- Swathi, C.; Chikala, R.; Ratnakar, K.; Sritharan, V. A structural, epidemiological & genetic overview of Klebsiella pneumoniae carbapenemases (KPCs). Indian J. Med. Res. 2016, 144, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Kamau, E.; Ranson, E.L.; Tsan, A.T.; Bergmann-Leitner, E.S.; Garner, O.B.; Yang, S. Clinical and genomic characterization of hypervirulent Klebsiella pneumoniae (hvKp) infections via passive surveillance in Southern California, 2020–2022. Front. Microbiol. 2022, 13, 1001169. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Medina, N.; Barrios-Camacho, H.; Duran-Bedolla, J.; Garza-Ramos, U. Klebsiella variicola: An emerging pathogen in humans. Emerg. Microbes Infect. 2019, 8, 973–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Medina, N.; Martínez-Romero, E.; De la Cruz, M.A.; Ares, M.A.; Valdovinos-Torres, H.; Silva-Sánchez, J.; Lozano-Aguirre, L.; Martínez-Barnetche, J.; Andrade, V.; Garza-Ramos, U. A Klebsiella variicola Plasmid Confers Hypermucoviscosity-Like Phenotype and Alters Capsule Production and Virulence. Front. Microbiol. 2020, 11, 579612. [Google Scholar] [CrossRef]
- Batinovic, S.; Wassef, F.; Knowler, S.A.; Rice, D.T.; Stanton, C.R.; Rose, J.; Tucci, J.; Nittami, T.; Vinh, A.; Drummond, G.R.; et al. Bacteriophages in Natural and Artificial Environments. Pathogens 2019, 8, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gencay, Y.E.; Gambino, M.; Prüssing, T.F.; Brøndsted, L. The genera of bacteriophages and their receptors are the major determinants of host range. Environ. Microbiol. 2019, 21, 2095–2111. [Google Scholar] [CrossRef]
- Tan, D.; Zhang, Y.; Cheng, M.; Le, S.; Gu, J.; Bao, J.; Qin, J.; Guo, X.; Zhu, T. Characterization of Klebsiella pneumoniae ST11 Isolates and Their Interactions with Lytic Phages. Viruses 2019, 11, 1080. [Google Scholar] [CrossRef] [Green Version]
- Weber-Dąbrowska, B.; Jończyk-Matysiak, E.; Żaczek, M.; Łobocka, M.; Łusiak-Szelachowska, M.; Górski, A. Bacteriophage Procurement for Therapeutic Purposes. Front. Microbiol. 2016, 7, 1177. [Google Scholar] [CrossRef] [Green Version]
- Anand, T.; Virmani, N.; Kumar, S.; Mohanty, A.K.; Pavulraj, S.; Bera, B.C.; Vaid, R.K.; Ahlawat, U.; Tripathi, B. Phage therapy for treatment of virulent Klebsiella pneumoniae infection in a mouse model. J. Glob. Antimicrob. Resist. 2020, 21, 34–41. [Google Scholar] [CrossRef]
- Assafiri, O.; Song, A.A.-L.; Tan, G.H.; Hanish, I.; Hashim, A.M.; Yusoff, K. Klebsiella virus UPM2146 lyses multiple drug-resistant Klebsiella pneumoniae in vitro and in vivo. PLoS ONE 2021, 16, e0245354. [Google Scholar] [CrossRef]
- Cano, E.J.; Caflisch, K.M.; Bollyky, P.L.; Van Belleghem, J.D.; Patel, R.; Fackler, J.; Brownstein, M.J.; Horne, B.; Biswas, B.; Henry, M.; et al. Phage Therapy for Limb-threatening Prosthetic Knee Klebsiella pneumoniae Infection: Case Report and In Vitro Characterization of Anti-biofilm Activity. Clin. Infect. Dis. 2020, 73, e144–e151. [Google Scholar] [CrossRef]
- Chadha, P.; Katare, O.P.; Chhibber, S. Liposome loaded phage cocktail: Enhanced therapeutic potential in resolving Klebsiella pneumoniae mediated burn wound infections. Burns 2017, 43, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Corbellino, M.; Kieffer, N.; Kutateladze, M.; Balarjishvili, N.; Leshkasheli, L.; Askilashvili, L.; Tsertsvadze, G.; Rimoldi, S.G.; Nizharadze, D.; Hoyle, N.; et al. Eradication of a Multidrug-Resistant, Carbapenemase-Producing Klebsiella pneumoniae Isolate Following Oral and Intra-rectal Therapy with a Custom Made, Lytic Bacteriophage Preparation. Clin. Infect. Dis. 2020, 70, 1998–2001. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-H.; Kuo, C.-F.; Wang, C.-H.; Wu, C.-M.; Tsao, N. Experimental Phage Therapy in Treating Klebsiella pneumoniae -Mediated Liver Abscesses and Bacteremia in Mice. Antimicrob. Agents Chemother. 2011, 55, 1358–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Harja, K.; Chhibber, S. Evidence to Support the Therapeutic Potential of Bacteriophage Kpn5 in Burn Wound Infection Caused by Klebsiella pneumoniae in BALB/c Mice. J. Microbiol. Biotechnol. 2010, 20, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Thiry, D.; Passet, V.; Danis-Wlodarczyk, K.; Lood, C.; Wagemans, J.; De Sordi, L.; van Noort, V.; Dufour, N.; Debarbieux, L.; Mainil, J.G.; et al. New Bacteriophages against Emerging Lineages ST23 and ST258 of Klebsiella pneumoniae and Efficacy Assessment in Galleria mellonella Larvae. Viruses 2019, 11, 411. [Google Scholar] [CrossRef] [Green Version]
- Łusiak-Szelachowska, M.; Międzybrodzki, R.; Fortuna, W.; Borysowski, J.; Górski, A. Anti-phage serum antibody responses and the outcome of phage therapy. Folia Microbiol. 2020, 66, 127–131. [Google Scholar] [CrossRef]
- Międzybrodzki, R.; Borysowski, J.; Weber-Dąbrowska, B.; Fortuna, W.; Letkiewicz, S.; Szufnarowski, K.; Pawełczyk, Z.; Rogóż, P.; Kłak, M.; Wojtasik, E.; et al. Clinical Aspects of Phage Therapy. Adv. Virus Res. 2012, 83, 73–121. [Google Scholar] [CrossRef]
- Międzybrodzki, R.; Hoyle, N.; Zhvaniya, F.; Łusiak-Szelachowska, M.; Weber-Dąbrowska, B.; Łobocka, M.; Borysowski, J.; Alavidze, Z.; Kutter, E.; Górski, A.; et al. Current Updates from the Long-Standing Phage Research Centers in Georgia, Poland, and Russia. In Bacteriophages; Harper, D.R., Abedon, S.T., Burrowes, B.H., McConville, M.L., Eds.; Springer: Cham, Switzerland, 2021; pp. 921–951. [Google Scholar] [CrossRef]
- Rostkowska, O.M.; Międzybrodzki, R.; Miszewska-Szyszkowska, D.; Górski, A.; Durlik, M. Trearment of recurrent urinary tract infections in a 60-year-old kidney transplant recipient. The use of phage therapy. Transpl. Infect. Dis. 2021, 23, e13391. [Google Scholar] [CrossRef]
- Domingo-Calap, P.; Beamud, B.; Vienne, J.; González-Candelas, F.; Sanjuán, R. Isolation of Four Lytic Phages Infecting Klebsiella pneumoniae K22 Clinical Isolates from Spain. Int. J. Mol. Sci. 2020, 21, 425. [Google Scholar] [CrossRef]
- Peng, Q.; Fang, M.; Liu, X.; Zhang, C.; Liu, Y.; Yuan, Y. Isolation and Characterization of a Novel Phage for Controlling Multidrug-Resistant Klebsiella pneumoniae. Microorganisms 2020, 8, 542. [Google Scholar] [CrossRef] [Green Version]
- Townsend, E.M.; Kelly, L.; Gannon, L.; Muscatt, G.; Dunstan, R.; Michniewski, S.; Sapkota, H.; Kiljunen, S.J.; Kolsi, A.; Skurnik, M.; et al. Isolation and Characterization of Klebsiella Phages for Phage Therapy. Phage 2021, 2, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Przondo-Hessek, A. Bacteriophages of Klebsiella bacilli. Isolation, properties and use in typing. Arch. Immunol. Ther. Exp. 1966, 14, 413–435. [Google Scholar]
- Strój, L.; Weber-Dabrowska, B.; Partyka, K.; Mulczyk, M.; Wójcik, M. Skuteczne zastosowanie bakteriofagoterapii w ropnym zapaleniu opon mózgowo-rdzeniowych u noworodka [Successful treatment with bacteriophage in purulent cerebrospinal meningitis in a newborn]. Neurol. Neurochir. Pol. 1999, 33, 693–698. [Google Scholar] [PubMed]
- de Campos, T.A.; de Almeida, F.M.; de Almeida, A.P.C.; Nakamura-Silva, R.; Oliveira-Silva, M.; de Sousa, I.F.A.; Cerdeira, L.; Lincopan, N.; Pappas, G.J.J.; Pitondo-Silva, A. Multidrug-Resistant (MDR) Klebsiella variicola Strains Isolated in a Brazilian Hospital Belong to New Clones. Front. Microbiol. 2021, 12, 604031. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. MALDI-TOF mass spectrometry: An emerging technology for microbial identification and diagnosis. Front. Microbiol. 2015, 6, 791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drancourt, M.; Bollet, C.; Carta, A.; Rousselier, P. Phylogenetic analyses of Klebsiella species delineate Klebsiella and Raoultella gen. nov., with description of Raoultella ornithinolytica comb. nov., Raoultella terrigena comb. nov. and Raoultella planticola comb. nov. Int. J. Syst. Evol. Microbiol. 2001, 51, 925–932. [Google Scholar] [CrossRef]
- Przondo-Hessek, A.; Slopek, S. Typing of Klebsiella bacilli by means of Klebsiella bacteriophages from the collection of Przondo-Hessek. Arch. Immunol. Ther. Exp. 1967, 15, 563–577. [Google Scholar]
- McLaughlin, M.R. Simple colorimetric microplate test of phage lysis in Salmonella enterica. J. Microbiol. Methods 2007, 69, 394–398. [Google Scholar] [CrossRef]
- Żaczek, M.; Łusiak-Szelachowska, M.; Jończyk-Matysiak, E.; Weber-Dąbrowska, B.; Międzybrodzki, R.; Owczarek, B.; Kopciuch, A.; Fortuna, W.; Rogóż, P.; Górski, A. Antibody Production in Response to Staphylococcal MS-1 Phage Cocktail in Patients Undergoing Phage Therapy. Front. Microbiol. 2016, 7, 1681. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.H. Enumeration of bacteriophage particles. In Bacteriophages; Adams, M.H., Ed.; Interscience Publishers: New York, NY, USA, 1959; pp. 27–30. [Google Scholar]
- Kropinski, A.M.; Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of bacteriophages by double agar overlay plaque assay. In Bacteriophages; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2009; Volume 501, pp. 69–76. [Google Scholar] [CrossRef]
- Jurczak-Kurek, A.; Gąsior, T.; Nejman-Faleńczyk, B.; Bloch, S.; Dydecka, A.; Topka, G.; Necel, A.; Jakubowska-Deredas, M.; Narajczyk, M.; Richert, M.; et al. Biodiversity of bacteriophages: Morphological and biological properties of a large group of phages isolated from urban sewage. Sci. Rep. 2016, 6, 34338. [Google Scholar] [CrossRef]
- Topka, G.; Bloch, S.; Nejman-Faleńczyk, B.; Gąsior, T.; Jurczak-Kurek, A.; Necel, A.; Dydecka, A.; Richert, M.; Węgrzyn, G.; Węgrzyn, A. Characterization of Bacteriophage vB-EcoS-95, Isolated from Urban Sewage and Revealing Extremely Rapid Lytic Development. Front. Microbiol. 2019, 9, 3326. [Google Scholar] [CrossRef] [PubMed]
- Kęsik-Szeloch, A.; Drulis-Kawa, Z.; Weber-Dąbrowska, B.; Kassner, J.; Majkowska-Skrobek, G.; Augustyniak, D.; Łusiak-Szelachowska, M.; Żaczek, M.; Górski, A.; Kropinski, A.M. Characterising the biology of novel lytic bacteriophages infecting multidrug resistant Klebsiella pneumoniae. Virol. J. 2013, 10, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedon, S.T.; Herschler, T.D.; Stopar, D. Bacteriophage Latent-Period Evolution as a Response to Resource Availability. Appl. Environ. Microbiol. 2001, 67, 4233–4241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, P.; Abedon, S.T. Practical Methods for Determining Phage Growth Parameters. In Bacteriophages; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2009; Volume 501, pp. 175–202. [Google Scholar] [CrossRef]
- Ackermann, H.-W. Basic Phage Electron Microscopy. In Bacteriophages; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2009; Volume 501, pp. 113–126. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garneau, J.R.; Depardieu, F.; Fortier, L.-C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. In Gene Prediction; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2019; Volume 1962, pp. 1–14. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021, 49, D10–D17. [Google Scholar] [CrossRef]
- Laing, C.; Buchanan, C.; Taboada, E.N.; Zhang, Y.; Kropinski, A.; Villegas, A.; Thomas, J.E.; Gannon, V.P. Pan-genome sequence analysis using Panseq: An online tool for the rapid analysis of core and accessory genomic regions. BMC Bioinform. 2010, 11, 461. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a Versatile Software Package for Scalable and Robust Microbial Pangenome Analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef] [Green Version]
- Turner, D.; Reynolds, D.; Seto, D.; Mahadevan, P. CoreGenes3.5: A webserver for the determination of core genes from sets of viral and small bacterial genomes. BMC Res. Notes 2013, 6, 140. [Google Scholar] [CrossRef] [Green Version]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Garg, A.; Gupta, D. VirulentPred: A SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinform. 2008, 9, 62. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- D’Andrea, M.M.; Marmo, P.; De Angelis, L.H.; Palmieri, M.; Ciacci, N.; Di Lallo, G.; Demattè, E.; Vannuccini, E.; Lupetti, P.; Rossolini, G.M.; et al. φBO1E, a newly discovered lytic bacteriophage targeting carbapenemase-producing Klebsiella pneumoniae of the pandemic Clonal Group 258 clade II lineage. Sci. Rep. 2017, 7, 2614. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, B.E.; Costa, A.R.; van den Berg, D.F.; van Rossum, T.; Hagedoorn, S.; Walinga, H.; Xiao, M.; Song, W.; Haas, P.-J.; Nobrega, F.L.; et al. Genomic characterization of four novel bacteriophages infecting the clinical pathogen Klebsiella pneumoniae. DNA Res. 2021, 28, dsab013. [Google Scholar] [CrossRef]
- Jończyk-Matysiak, E.; Łodej, N.; Kula, D.; Owczarek, B.; Orwat, F.; Międzybrodzki, R.; Neuberg, J.; Bagińska, N.; Weber-Dąbrowska, B.; Gorski, A. Factors determining phage stability/activity: Challenges in practical phage application. Expert Rev. Anti-Infect. Ther. 2019, 17, 583–606. [Google Scholar] [CrossRef]
- Vincent, A.T.; Paquet, V.E.; Bernatchez, A.; Tremblay, D.M.; Moineau, S.; Charette, S.J. Characterization and diversity of phages infecting Aeromonas salmonicida subsp. salmonicida. Sci. Rep. 2017, 7, 7054. [Google Scholar] [CrossRef] [Green Version]
- Merrill, B.D.; Ward, A.T.; Grose, J.H.; Hope, S. Software-based analysis of bacteriophage genomes, physical ends, and packaging strategies. BMC Genom. 2016, 17, 679. [Google Scholar] [CrossRef] [Green Version]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; Hendrickson, R.C.; et al. Recent changes to virus taxonomy ratified by the International Committee on Taxonomy of Viruses (2022). Arch. Virol. 2022, 167, 2429–2440. [Google Scholar] [CrossRef]
- Millard, A.; Puxty, R.; White, D.; Gannon, L.; Harrison, C.; Cook, R.; Adriaenssens, E.M.; Turner, D. Create two new families (Kyanoviridae and Straboviridae) (Caudoviricetes). Available online: https://ictv.global/ictv/proposals/2021.082B.R.Tevens_new_families.zip (accessed on 17 December 2022).
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Rüger, W. Bacteriophage T4 Genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.-C.; Liao, T.-L.; Lin, Y.-C.; Lai, Y.-C.; Lu, M.-C.; Chen, Y.-T. Complete Genome Sequence of Klebsiella pneumoniae 1084, a Hypermucoviscosity-Negative K1 Clinical Strain. J. Bacteriol. 2012, 194, 6316. [Google Scholar] [CrossRef] [Green Version]
- Kislichkina, A.A.; Lev, A.I.; Komisarova, E.V.; Fursova, N.K.; Myakinina, V.P.; Mukhina, T.N.; Bogun, A.A.; Volozhantsev, N.V. Genome sequencing and comparative analysis of three hypermucoviscous Klebsiella pneumoniae strains isolated in Russia. Pathog. Dis. 2017, 75, ftx024. [Google Scholar] [CrossRef] [Green Version]
- Turner, D.; Kropinski, A.; Adriaenssens, E. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Mijalis, E.M.; Lessor, L.E.; Cahill, J.L.; Rasche, E.S.; Everett, G.F.K. Complete Genome Sequence of Klebsiella pneumoniae Carbapenemase-Producing K. pneumoniae Myophage Miro. Genome Announc. 2015, 3, e01137-15. [Google Scholar] [CrossRef] [Green Version]
- Provasek, V.E.; Lessor, L.E.; Cahill, J.L.; Rasche, E.S.; Everett, G.F.K. Complete Genome Sequence of Carbapenemase-Producing Klebsiella pneumoniae Myophage Matisse. Genome Announc. 2015, 3, e01136-15. [Google Scholar] [CrossRef] [Green Version]
- Pereira, C.; Moreirinha, C.; Lewicka, M.; Almeida, P.; Clemente, C.; Romalde, J.L.; Nunes, M.L.; Almeida, A. Characterization and in vitro evaluation of new bacteriophages for the biocontrol of Escherichia coli. Virus Res. 2017, 227, 171–182. [Google Scholar] [CrossRef]
- Wu, C.H.; Lin, H.; Black, L.W. Bacteriophage T4 gene 17 amplification mutants: Evidence for initiation by the T4 terminase subunit gp16. J. Mol. Biol. 1995, 247, 523–528. [Google Scholar]
- Lin, H.; Black, L.W. DNA Requirements In Vivo for Phage T4 Packaging. Virology 1998, 242, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, N.C.; Torrents, E.; Gibb, E.A.; Sahlin, M.; Sjöberg, B.-M.; Edgell, D.R. Insertion of a homing endonuclease creates a genes-in-pieces ribonucleotide reductase that retains function. Proc. Natl. Acad. Sci. USA 2007, 104, 6176–6181. [Google Scholar] [CrossRef] [Green Version]
- North, O.I.; Davidson, A.R. Phage Proteins Required for Tail Fiber Assembly Also Bind Specifically to the Surface of Host Bacterial Strains. J. Bacteriol. 2021, 203, e00406-20. [Google Scholar] [CrossRef]
- Miroshnikov, K.; Marusich, E.I.; Cerritelli, M.E.; Cheng, N.; Hyde, C.C.; Steven, A.C.; Mesyanzhinov, V.V. Engineering trimeric fibrous proteins based on bacteriophage T4 adhesins. Protein Eng. Des. Sel. 1998, 11, 329–332. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A.; Eddy, S.R.; Mesyanzhinov, V.V. A Member of the Immunoglobulin Superfamily in Bacteriophage T4. Virus Genes 1997, 14, 163–165. [Google Scholar] [CrossRef]
- Sathaliyawala, T.; Islam, M.Z.; Li, Q.; Fokine, A.; Rossmann, M.G.; Rao, V.B. Functional analysis of the highly antigenic outer capsid protein, Hoc, a virus decoration protein from T4-like bacteriophages. Mol. Microbiol. 2010, 77, 444–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fokine, A.; Islam, M.Z.; Zhang, Z.; Bowman, V.D.; Rao, V.B.; Rossmann, M.G. Structure of the Three N-Terminal Immunoglobulin Domains of the Highly Immunogenic Outer Capsid Protein from a T4-Like Bacteriophage. J. Virol. 2011, 85, 8141–8148. [Google Scholar] [CrossRef]
- Dąbrowska, K.; Miernikiewicz, P.; Piotrowicz, A.; Hodyra, K.; Owczarek, B.; Lecion, D.; Kaźmierczak, Z.; Letarov, A.; Górski, A. Immunogenicity Studies of Proteins Forming the T4 Phage Head Surface. J. Virol. 2014, 88, 12551–12557. [Google Scholar] [CrossRef] [Green Version]
- Łobocka, M.; Hejnowicz, M.S.; Gągała, U.; Weber-Dąbrowska, B.; Węgrzyn, G.; Dadlez, M. The first step to bacteriophage. In Phage Therapy; Borysowski, J., Międzybrodzki, R., Górski, A., Eds.; Current Research and Applications; Caister Academic Press: Norfolk, UK, 2014; pp. 23–69. [Google Scholar]
- Masters, M. Transduction: Host DNA transfer by bacteriophages. In The Desk Encyclopedia of Microbiology; Schaechter, M., Ed.; Elsevier Academic Press: London, UK, 2004; pp. 1000–1012. [Google Scholar]
- Carlson, K.; Ȗvervatin, A. Bacteriophage T4 endonucleases II and IV, oppositely affected by dCMP hydroxymethylase activity, have different roles in the degradation and in the RNA polymerase-dependent replication of T4 cytosine-containing DNA. Genetics 1986, 114, 669–685. [Google Scholar] [CrossRef]
- Kutter, E.M.; Wiberg, J.S. Biological Effects of Substituting Cytosine for 5-Hydroxymethylcytosine in the Deoxyribonucleic Acid of Bacteriophage T4. J. Virol. 1969, 4, 439–453. [Google Scholar] [CrossRef] [Green Version]
- Warner, H.R.; Snustad, D.P.; Jorgensen, S.E.; Koerner, J.F. Isolation of Bacteriophage T4 Mutants Defective in the Ability to Degrade Host Deoxyribonucleic Acid. J. Virol. 1970, 5, 700–708. [Google Scholar] [CrossRef] [Green Version]
- Hercules, K.; Munro, J.L.; Mendelsohn, S.; Wiberg, J.S. Mutants in a Nonessential Gene of Bacteriophage T4 Which Are Defective in the Degradation of Escherichia coli Deoxyribonucleic Acid. J. Virol. 1971, 7, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Vetter, D.; Sadowski, P.D. Point Mutants in the D2a Region of Bacteriophage T4 Fail to Induce T4 Endonuclease IV. J. Virol. 1974, 14, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Siryaporn, A.; Kuchma, S.L.; O’Toole, G.A.; Gitai, Z. Surface attachment induces Pseudomonas aeruginosa virulence. Proc. Natl. Acad. Sci. USA 2014, 111, 16860–16865. [Google Scholar] [CrossRef] [Green Version]
- Głowacka-Rutkowska, A.; Ulatowska, M.; Empel, J.; Kowalczyk, M.; Boreczek, J.; Łobocka, M. A Kayvirus Distant Homolog of Staphylococcal Virulence Determinants and VISA Biomarker Is a Phage Lytic Enzyme. Viruses 2020, 12, 292. [Google Scholar] [CrossRef] [Green Version]
- Hampton, H.G.; Watson, B.N.J.; Fineran, P.C. The arms race between bacteria and their phage foes. Nature 2020, 577, 327–336. [Google Scholar] [CrossRef]
- Latka, A.; Leiman, P.; Drulis-Kawa, Z.; Briers, Y. Modeling the Architecture of Depolymerase-Containing Receptor Binding Proteins in Klebsiella Phages. Front. Microbiol. 2019, 10, 2649. [Google Scholar] [CrossRef]
- Knecht, L.E.; Veljkovic, M.; Fieseler, L. Diversity and Function of Phage Encoded Depolymerases. Front. Microbiol. 2020, 10, 2949. [Google Scholar] [CrossRef]
- Adams, M.H.; Park, B.H. An enzyme produced by a phage-host cell system. II. The properties of the polysaccharide depolymerase. Virology 1956, 2, 719–736. [Google Scholar] [CrossRef]
- Stirm, S.; Bessler, W.; Fehmel, F.; Freund-Mölbrt, E.; Thurow, H. Isolation of spike-formed particles from bacteriophage lysates. Virology 1971, 45, 303–308. [Google Scholar] [CrossRef]
- Schulz, E.C.; Dickmanns, A.; Urlaub, H.; Schmitt, A.; Mühlenhoff, M.; Stummeyer, K.; Schwarzer, D.; Gerardy-Schahn, R.; Ficner, R. Crystal structure of an intramolecular chaperone mediating triple–β-helix folding. Nat. Struct. Mol. Biol. 2010, 17, 210–215. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Doval, C.; Castón, J.R.; Luque, D.; Granell, M.; Otero, J.M.; Llamas-Saiz, A.L.; Renouard, M.; Boulanger, P.; Van Raaij, M.J. Structure of the Receptor-Binding Carboxy-Terminal Domain of the Bacteriophage T5 L-Shaped Tail Fibre with and without Its Intra-Molecular Chaperone. Viruses 2015, 7, 6424–6440. [Google Scholar] [CrossRef] [Green Version]
- Nikulin, N.A.; Zimin, A.A. Influence of Non-canonical DNA Bases on the Genomic Diversity of Tevenvirinae. Front. Microbiol. 2021, 12, 632686. [Google Scholar] [CrossRef]
- Hirano, N.; Ohshima, H.; Takahashi, H. Biochemical analysis of the substrate specificity and sequence preference of endonuclease IV from bacteriophage T4, a dC-specific endonuclease implicated in restriction of dC-substituted T4 DNA synthesis. Nucleic Acids Res. 2006, 34, 4743–4751. [Google Scholar] [CrossRef]
- Petrov, V.M.; Ratnayaka, S.; Nolan, J.M.; Miller, E.S.; Karam, J.D. Genomes of the T4-related bacteriophages as windows on microbial genome evolution. Virol. J. 2010, 7, 292. [Google Scholar] [CrossRef] [Green Version]
- Drivdahl, R.H.; Kutter, E.M. Inhibition of transcription of cytosine-containing DNA in vitro by the alc gene product of bacteriophage T4. J. Bacteriol. 1990, 172, 2716–2727. [Google Scholar] [CrossRef] [Green Version]
- Kashlev, M.; Nudler, E.; Goldfarb, A.; White, T.; Kutter, E. Bacteriophage T4 Alc protein: A transcription termination factor sensing local modification of DNA. Cell 1993, 75, 147–154. [Google Scholar] [CrossRef]
- Snyder, L.; Kaufmann, G. T4 phage exclusion mechanisms. In Molecular Biology of Bacteriophage T4; Karam, J., Drake, J.W., Kreuzer, K.N., Mosig, G., Hall, D.H., Eiserling, F.A., Black, L.W., Spicer, E.K., Kutter, E., Carlson, K., et al., Eds.; American Society for Microbiology: Washington, DC, USA, 1994; pp. 391–396. [Google Scholar]
- Wong, S.; Alattas, H.; Slavcev, R.A. A snapshot of the λ T4rII exclusion (Rex) phenotype in Escherichia coli. Curr. Genet. 2021, 67, 739–745. [Google Scholar] [CrossRef]
- Smith-Zaitlik, T.; Shibu, P.; McCartney, A.L.; Foster, G.; Hoyles, L.; Negus, D. Extended genomic analyses of the broad-host-range phages vB_KmiM-2Di and vB_KmiM-4Dii reveal slopekviruses have highly conserved genomes. Microbiology 2022, 168, 001247. [Google Scholar] [CrossRef]
- De Angelis, L.H.; Poerio, N.; Di Pilato, V.; De Santis, F.; Antonelli, A.; Thaller, M.; Fraziano, M.; Rossolini, G.; D’Andrea, M. Phage Resistance Is Associated with Decreased Virulence in KPC-Producing Klebsiella pneumoniae of the Clonal Group 258 Clade II Lineage. Microorganisms 2021, 9, 762. [Google Scholar] [CrossRef]
- Jean, S.-S.; Harnod, D.; Hsueh, P.-R. Global Threat of Carbapenem-Resistant Gram-Negative Bacteria. Front. Cell. Infect. Microbiol. 2022, 12, 823684. [Google Scholar] [CrossRef]
- Ross, A.; Ward, S.; Hyman, P. More Is Better: Selecting for Broad Host Range Bacteriophages. Front. Microbiol. 2016, 7, 1352. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Wu, N.; Bao, J.; Shi, X.; Ou, H.; Ye, S.; Zhao, W.; Wei, Z.; Cai, J.; Li, L.; et al. Heterogeneous Klebsiella pneumoniae Co-infections Complicate Personalized Bacteriophage Therapy. Front. Cell. Infect. Microbiol. 2021, 10, 608402. [Google Scholar] [CrossRef]
- Górski, A.; Międzybrodzki, R.; Łobocka, M.; Głowacka-Rutkowska, A.; Bednarek, A.; Borysowski, J.; Jończyk-Matysiak, E.; Łusiak-Szelachowska, M.; Weber-Dąbrowska, B.; Bagińska, N.; et al. Phage Therapy: What Have We Learned? Viruses 2018, 10, 288. [Google Scholar] [CrossRef] [Green Version]
- Górski, A.; Międzybrodzki, R.; Jończyk-Matysiak, E.; Borysowski, J.; Letkiewicz, S.; Weber-Dąbrowska, B. The fall and rise of phage therapy in modern medicine. Expert Opin. Biol. Ther. 2019, 19, 1115–1117. [Google Scholar] [CrossRef] [Green Version]
- Górski, A.; Międzybrodzki, R.; Jończyk-Matysiak, E.; Żaczek, M.; Borysowski, J. Phage-specific diverse effects of bacterial viruses on the immune system. Future Microbiol. 2019, 14, 1171–1174. [Google Scholar] [CrossRef] [Green Version]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics. 2017, 33, 2379–2380. [Google Scholar] [CrossRef]










| Bacterial Strain | Number of Strains | Strain Origin |
|---|---|---|
| Klebsiella pneumoniae PCM 1 (ATCC 13886 ) | 1 | PCM 1 (ATCC 13886) |
| Klebsiella pneumoniae | 14 | Environmental sample |
| Klebsiella pneumoniae | 262 | Clinical strain |
| Klebsiella oxytoca | 144 | Clinical strain |
| Klebsiella oxytoca | 15 | Environmental sample |
| Klebsiella variicola | 2 | Clinical strain |
| Klebsiella ozenae | 9 | Clinical strain |
| Raoultella planticola | 2 | Clinical strain |
| Klebsiella aerogenes | 2 | Clinical strain |
| Pantoea agglomerans | 2 | Clinical strain |
| Pluralibacter gergoviae | 5 | Clinical strain |
| Enterobacter cowanii | 3 | Clinical strain |
| Escherichia coli B | 1 | HIIET collection |
| Place of Sample Collection | Sample Type | Year of Isolation | Isolated Phages | |
|---|---|---|---|---|
| Klebsiella pneumoniae | Klebsiella oxytoca | |||
| Wastewater treatment plant in Opole | Incubated city sewage | 2008 | 6N, 7N, 8N, 10N | 9N, 13N |
| 2009 | 19N, 33N, 38N, 47N | 18N, 42N | ||
| Crude concentrated city sewage | 2009 | 37N | 15N | |
| 2010 | 21N, 48N, 61N | 16N, 49N | ||
| University Teaching Hospital in Opole | Incubated hospital sewage | 2010 | 52N, 60N | 63N |
| 2011 | 65N | - | ||
| 2013 | - | 67N | ||
| 2015 | - | B40 | ||
| 4th Military Hospital in Wrocław | Incubated hospital sewage | 2010 | 53N | - |
| Wastewater treatment plant in Kraków-Płaszów | Incubated city sewage | 2008 | 5N | 2N, 3N |
| 2009 | - | 41N, 46N | ||
| Crude concentrated city sewage | 2009 | 11N, 20N, 34N, 36N, 39N | 14N, 17N | |
| 2010 | 55N | - | ||
| Wastewater treatment plant in Pleszew (Greater Poland province) | Crude city sewage | 2010 | 12N, 35N, 50N, 54N, 62N | 56N, 64N |
| Wastewater treatment plant in Grodzisk Mazowiecki (Mazovian Province) | Crude water sample | 2012 | - | 68N |
| 2015 | - | WI42, WI43, WI105, WI106 | ||
| Incubated water sample | 2015 | - | WII43 | |
| Wrocław, irrigated pool | Crude sewage from irrigated pool No. 1 | 2008 | 4N | - |
| Chojnów (Lower Silesia province), water intake | Crude water sample | 2009 | 32N | - |
| Czeczotki (Mazovian Province), water intake | Crude water sample | 2015 | - | WI53 |
| Vistula river (Lesser Poland Province) | Crude water sample | 2009 | 15N, 27N, 28N, 29N, 30N, 41N * | 42N, 43N, 44N, 45N * |
| HIIET Phage Collection | Przondo-Hessek Phage Collection | ||||
|---|---|---|---|---|---|
| Phage Symbol | ICTV Symbol | Number and Percentage of Sensitive Strains | Phage Symbol | ICTV Symbol | Number and Percentage of Sensitive Strains |
| Kpn53N | vB_KpnM-53N | 73 (35.2%) | Kpn2 * | vB_RapM-1 | 58 (28.0%) |
| Kpn5N | vB_KpnM-5N | 59 (28.5%) | Kpn16 | vB_KpnM-16 | 52 (25.1%) |
| Kpn52N | vB_KpnM-52N | 54 (26.0%) | Kpn35 | vB_KpnM-35 | 43 (20.8%) |
| Kpn6N | vB_KpnM-6N | 39 (18.9%) | Kpn5 | vB_KpnM-5 | 31 (14.9%) |
| Kpn19N | vB_KpnS-19N | 33 (16.0%) | Kpn9 | vB_KpnM-9 | 17 (8.2%) |
| Phage Symbol/ Host Symbol | ICTV Phage Symbol | Morphotype | Total Lenght | Dimension of Head [nm] | Dimension of Tail [nm] | |||
|---|---|---|---|---|---|---|---|---|
| Capsid Length | Capsid Width/Diagonal | Total Length | Tail Width | Base Plate Width | ||||
| Kpn5N/K737 | vB_KpnM-5N | A2 | 230.4 | 115.1 | 81.6/106.4 | 115.3 | 20.0 | 31.3 |
| Kpn52N/K70 | vB_KpnM-52N | A2 | 221.1 | 107.6 | 82.2/98.6 | 118.5 | 22.7 | 37.2 |
| Kpn53N/K70 | vB_KpnM-53N | A2 | 229.8 | 118.3 | 85.5/100.1 | 111.5 | 20.1 | 32.8 |
| Kpn6N/K737 | vB_KpnM-6N | A2 | 237.6 | 119.4 | 90.2/109.5 | 118.2 | 21.3 | 32.9 |
| Kpn19N/K926a | vB_KpnS-19N | B1 | 234.8 | 56.1 | 53.6/178.7 | 178.7 | 9.1 | - |
| Kpn2/K59 (Rpl1) | vB_RaM-1 | A2 | 231.7 | 113.9 | 83.6/117.8 | 117.8 | 20.6 | 32.3 |
| Kpn16/K30 | vB_KpnP-16 * | C1 | - | - | - | - | - | - |
| Kpn35/J24552 | vB_KpnM-35 | A2 | 234.8 | 111.1 | 88.5/123.7 | 127.7 | 21.7 | 35.3 |
| Kpn5/J24552 | vB_KpnM-5 | A2 | 224.7 | 108.4 | 88.8/100.1 | 116.3 | 20.7 | 31.7 |
| No. | Phage/Host | Initial Phage Titer PFU/mL | Phage Titer at pH 4 PFU/mL | Phage Titer at pH 5 PFU/mL | ||
|---|---|---|---|---|---|---|
| After 1 h | After 5 h | After 1 h | After 5 h | |||
| 1 | Kpn5N/K737 ^ | 1.5 × 108 | 9.5 × 107 | 3.6 × 107 | 9.5 × 107 | 8.2 × 107 |
| 2 | Kpn19N/K917 ^ | 8.0 × 105 | 1.1 × 105 | 0 | 8.2 × 105 | 8.7 × 103 |
| 3 | Kpn6N/K737 ^ | 3.0 × 106 | 2.6 × 106 | 1.6 × 106 | 3.0 × 106 | 3.6 × 106 |
| 4 | Kpn12N/K1195 ^ | 4.9 × 103 | 8.7 × 102 | 0 | 2.2 × 103 | 6.2 × 102 |
| 5 | Kpn52N/K1920 ^ | 7.2 × 105 | 2.6 × 105 | 2.8 × 103 | 6.7 × 105 | 5.2 × 105 |
| 6 | Kox67N/K3459 ^ | 5.0 × 104 | 1.0 × 104 | 0 | 3.4 × 104 | 7.0 × 103 |
| 7 | Kpn2/K59 (Rpl1) * | 1.7 × 108 | 1.1 × 108 | 6.5 × 107 | 1.8 × 108 | 1.6 × 108 |
| 8 | Kpn9/J24552 * | 1.2 × 1010 | 2.9 × 109 | 1.4 × 109 | 2.9 × 109 | 1.6 × 109 |
| 9 | Kpn35/J24552 * | 2.1 × 106 | 3.0 × 105 | 1.7 × 105 | 3.7 × 105 | 2.0 × 105 |
| 10 | Kpn16/K30 * | 2.0 × 109 | 1.6 × 109 | 1.4 × 107 | 1.7 × 109 | 4.7 × 108 |
| No. | Phage/host | Initial phage titer PFU/mL | Phage titer at pH 6 PFU/mL | Phage titer at pH 8 PFU/mL | ||
| After 1 h | After 5 h | After 1 h | After 5 h | |||
| 1 | Kpn5N/K737 ^ | 1.5 × 108 | 1.6 × 108 | 1.1 × 108 | 1.0 × 108 | 1.0 × 108 |
| 2 | Kpn19N/K917 ^ | 8.0 × 105 | 8.2 × 105 | 1.7 × 104 | 3.1 × 105 | 1.7 × 104 |
| 3 | Kpn6N/K737 ^ | 3.0 × 106 | 4.0 × 106 | 3.4 × 106 | 3.0 × 106 | 2.3 × 106 |
| 4 | Kpn12N/K1195 ^ | 4.9 × 103 | 5.0 × 103 | 2.8 × 103 | 3.5 × 103 | 3.0 × 103 |
| 5 | Kpn52N/K1920^ | 7.2 × 105 | 6.0 × 105 | 7.0 × 105 | 7.4 × 105 | 6.8 × 105 |
| 6 | Kox67N/K3459 ^ | 5.0 × 104 | 3.7 × 104 | 2.1 × 104 | 3.0 × 104 | 1.9 × 104 |
| 7 | Kpn2/K59 (Rpl1) * | 1.7 × 108 | 1.8 × 108 | 1.7 × 108 | 1.4 × 108 | 1.5 × 108 |
| 8 | Kpn9/J24552 * | 1.2 × 1010 | 2.9 × 109 | 2.6 × 109 | 2.9 × 109 | 2.6 × 109 |
| 9 | Kpn35/J24552 * | 2.1 × 106 | 5.7 × 105 | 3.7 × 105 | 2.7 × 105 | 3.2 × 105 |
| 10 | Kpn16/K30 * | 2.0 × 109 | 2.1 × 109 | 1.8 × 109 | 2.2 × 109 | 2.0 × 109 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weber-Dąbrowska, B.; Żaczek, M.; Łobocka, M.; Łusiak-Szelachowska, M.; Owczarek, B.; Orwat, F.; Łodej, N.; Skaradzińska, A.; Łaczmański, Ł.; Martynowski, D.; et al. Characteristics of Environmental Klebsiella pneumoniae and Klebsiella oxytoca Bacteriophages and Their Therapeutic Applications. Pharmaceutics 2023, 15, 434. https://doi.org/10.3390/pharmaceutics15020434
Weber-Dąbrowska B, Żaczek M, Łobocka M, Łusiak-Szelachowska M, Owczarek B, Orwat F, Łodej N, Skaradzińska A, Łaczmański Ł, Martynowski D, et al. Characteristics of Environmental Klebsiella pneumoniae and Klebsiella oxytoca Bacteriophages and Their Therapeutic Applications. Pharmaceutics. 2023; 15(2):434. https://doi.org/10.3390/pharmaceutics15020434
Chicago/Turabian StyleWeber-Dąbrowska, Beata, Maciej Żaczek, Małgorzata Łobocka, Marzanna Łusiak-Szelachowska, Barbara Owczarek, Filip Orwat, Norbert Łodej, Aneta Skaradzińska, Łukasz Łaczmański, Dariusz Martynowski, and et al. 2023. "Characteristics of Environmental Klebsiella pneumoniae and Klebsiella oxytoca Bacteriophages and Their Therapeutic Applications" Pharmaceutics 15, no. 2: 434. https://doi.org/10.3390/pharmaceutics15020434
APA StyleWeber-Dąbrowska, B., Żaczek, M., Łobocka, M., Łusiak-Szelachowska, M., Owczarek, B., Orwat, F., Łodej, N., Skaradzińska, A., Łaczmański, Ł., Martynowski, D., Kaszowska, M., & Górski, A. (2023). Characteristics of Environmental Klebsiella pneumoniae and Klebsiella oxytoca Bacteriophages and Their Therapeutic Applications. Pharmaceutics, 15(2), 434. https://doi.org/10.3390/pharmaceutics15020434