
Biological Evaluation and In Vitro Characterization of ADME Profile of In-House Pyrazolo[3,4-d]pyrimidines as Dual Tyrosine Kinase Inhibitors Active against Glioblastoma Multiforme

 , ,
, ,  ,
,  ,
,  , , ,
, , ,  , ,
, , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Drugs and Materials
2.2. Enzymatic Assay
2.3. Cell Culture, Antiproliferative Effects, and Cytotoxicity on Cell-Based Assays
2.4. In Vitro ADME Assays
2.4.1. HPLC/UV–MS Method
2.4.2. Parallel Artificial Membrane Permeability Assay (PAMPA)
2.4.3. Metabolic Stability Assay
2.5. Reversible/Irreversible Effect on LN229 Cell Death
2.6. Cell-Cycle Analysis
2.7. T-Scratch Assay
2.8. Data Analysis
3. Results and Discussion
3.1. In Vitro Biological Evaluation
3.1.1. Enzymatic Assay
3.1.2. Cell Viability Assay
3.2. In Vitro ADME Evaluation
3.3. Irreversible Cytotoxic Effect on LN229 Cell Death
3.4. Cell-Cycle Analysis
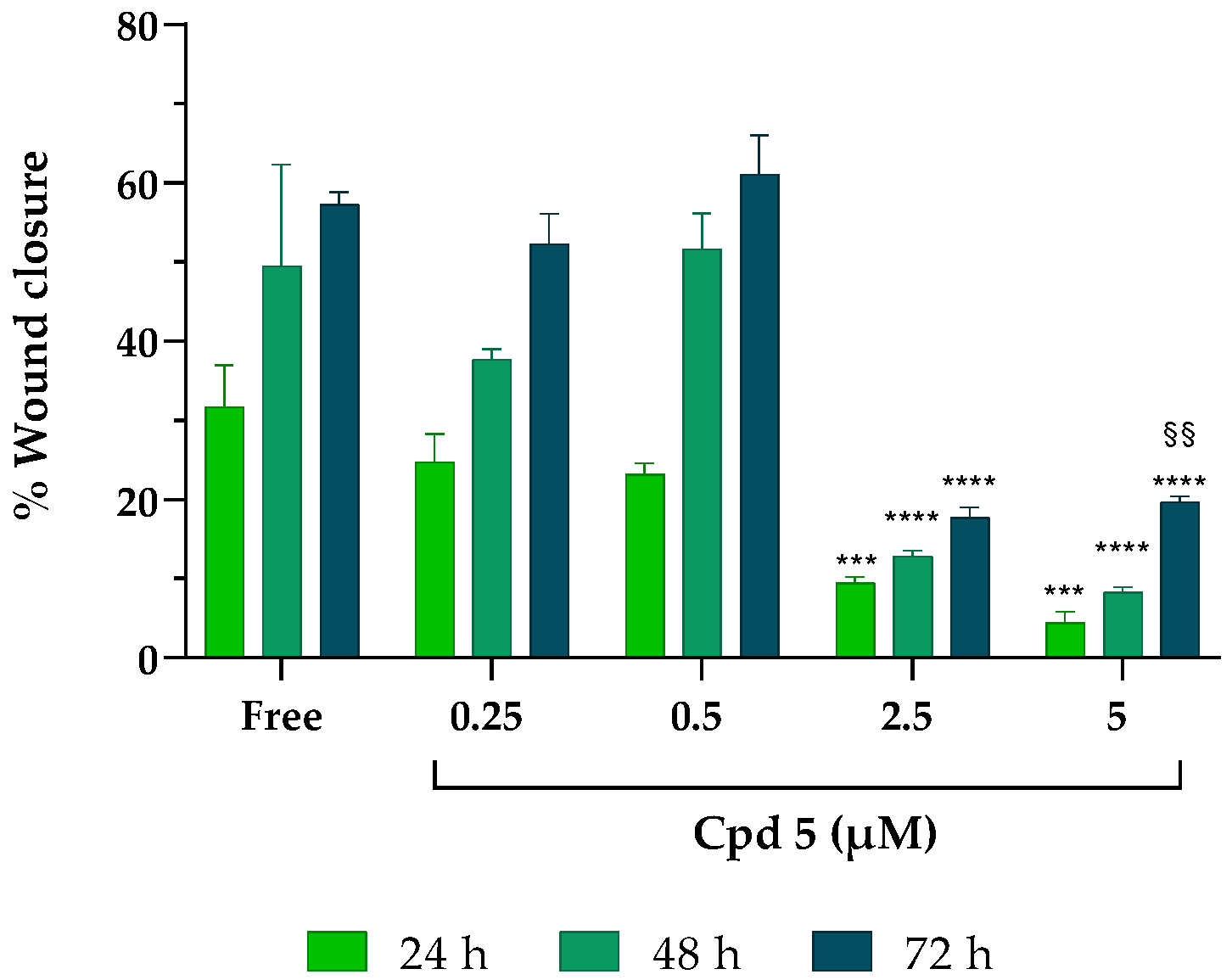
3.5. T-Scratch Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fallacara, A.L.; Zamperini, C.; Podolski-Renić, A.; Dinić, J.; Stanković, T.; Stepanović, M.; Mancini, A.; Rango, E.; Iovenitti, G.; Molinari, A.; et al. A New Strategy for Glioblastoma Treatment: In Vitroand In Vivo Preclinical Characterization of Si306,a Pyrazolo [3,4-d]Pyrimidine DualSrc/P-Glycoprotein Inhibitor. Cancers 2019, 11, 848. [Google Scholar] [CrossRef] [Green Version]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. Glioblastoma 2017, 143–153. [Google Scholar] [CrossRef]
- Ceccherini, E.; Indovina, P.; Zamperini, C.; Dreassi, E.; Casini, N.; Cutaia, O.; Forte, I.M.; Pentimalli, F.; Esposito, L.; Polito, M.S.; et al. SRC Family Kinase Inhibition through a New Pyrazolo[3,4-d]Pyrimidine Derivative as a Feasible Approach for Glioblastoma Treatment. J. Cell. Biochem. 2015, 116, 856–863. [Google Scholar] [CrossRef]
- Molinari, A.; Iovenitti, G.; Mancini, A.; Gravina, G.L.; Chebbi, M.; Caruana, M.; Vignaroli, G.; Orofino, F.; Rango, E.; Angelucci, A.; et al. AuNP Pyrazolo[3,4-d]Pyrimidine Nanosystem in Combination with Radiotherapy against Glioblastoma. ACS Med. Chem. Lett. 2020, 11, 664–670. [Google Scholar] [CrossRef]
- Cirotti, C.; Contadini, C.; Barilà, D. SRC Kinase in Glioblastoma: News from an Old Acquaintance. Cancers 2020, 12, 1558. [Google Scholar] [CrossRef]
- Lamballe, F.; Toscano, S.; Conti, F.; Arechederra, M.; Baeza, N.; Figarella-Branger, D.; Helmbacher, F.; Maina, F. Coordination of Signalling Networks and Tumorigenic Properties by ABL in Glioblastoma Cells. Oncotarget 2016, 7, 74747–74767. [Google Scholar] [CrossRef] [Green Version]
- Luttman, J.H.; Colemon, A.; Mayro, B.; Pendergast, A.M. Role of the ABL Tyrosine Kinases in the Epithelial–Mesenchymal Transition and the Metastatic Cascade. Cell Commun. Signal. 2021, 19, 59. [Google Scholar] [CrossRef]
- Spreafico, A.; Schenone, S.; Serchi, T.; Orlandini, M.; Angelucci, A.; Magrini, D.; Bernardini, G.; Collodel, G.; Di Stefano, A.; Tintori, C.; et al. Antiproliferative and Proapoptotic Activities of New Pyrazolo[3,4-d]Pyrimidine Derivative Src Kinase Inhibitors in Human Osteosarcoma Cells. FASEB J. 2008, 22, 1560–1571. [Google Scholar] [CrossRef]
- Molinari, A.; Fallacara, A.L.; Di Maria, S.; Zamperini, C.; Poggialini, F.; Musumeci, F.; Schenone, S.; Angelucci, A.; Colapietro, A.; Crespan, E.; et al. Efficient Optimization of Pyrazolo[3,4-d]Pyrimidines Derivatives as c-Src Kinase Inhibitors in Neuroblastoma Treatment. Bioorg. Med. Chem. Lett. 2018, 28, 3454–3457. [Google Scholar] [CrossRef]
- Vignaroli, G.; Iovenitti, G.; Zamperini, C.; Coniglio, F.; Calandro, P.; Molinari, A.; Fallacara, A.L.; Sartucci, A.; Calgani, A.; Colecchia, D.; et al. Prodrugs of Pyrazolo[3,4-d]Pyrimidines: From Library Synthesis to Evaluation as Potential Anticancer Agents in an Orthotopic Glioblastoma Model. J. Med. Chem. 2017, 60, 6305–6320. [Google Scholar] [CrossRef]
- Indovina, P.; Giorgi, F.; Rizzo, V.; Khadang, B.; Schenone, S.; Di Marzo, D.; Forte, I.M.; Tomei, V.; Mattioli, E.; D’urso, V.; et al. New Pyrazolo[3,4-d]Pyrimidine SRC Inhibitors Induce Apoptosis in Mesothelioma Cell Lines through P27 Nuclear Stabilization. Oncogene 2012, 31, 929–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radi, M.; Tintori, C.; Musumeci, F.; Brullo, C.; Zamperini, C.; Dreassi, E.; Fallacara, A.L.; Vignaroli, G.; Crespan, E.; Zanoli, S.; et al. Design, Synthesis, and Biological Evaluation of Pyrazolo[3,4- d ]Pyrimidines Active in Vivo on the Bcr-Abl T315I Mutant. J. Med. Chem. 2013, 56, 5382–5394. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, M.; Giorgi, F.; Marcelli, E.; Pentimalli, F.; Forte, I.M.; Schenone, S.; D’urso, V.; De Falco, G.; Botta, M.; Giordano, A.; et al. Antitumor Activity of New Pyrazolo[3,4-d]Pyrimidine SRC Kinase Inhibitors in Burkitt Lymphoma Cell Lines and Its Enhancement by WEE1 Inhibition. Cell Cycle 2012, 11, 1029–1039. [Google Scholar] [CrossRef] [Green Version]
- Schenone, S.; Bruno, O.; Bondavalli, F.; Ranise, A.; Mosti, L.; Menozzi, G.; Fossa, P.; Manetti, F.; Morbidelli, L.; Trincavelli, L.; et al. Synthesis of 1-(2-Chloro-2-Phenylethyl)-6-Methylthio-1H-Pyrazolo[3,4-d]Pyrimidines 4-Amino Substituted and Their Biological Evaluation. Eur. J. Med. Chem. 2004, 39, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Schenone, S.; Bruno, O.; Bondavalli, F.; Ranise, A.; Mosti, L.; Menozzi, G.; Fossa, P.; Donnini, S.; Santoro, A.; Ziche, M.; et al. Antiproliferative Activity of New 1-Aryl-4-Amino-1H-Pyrazolo[3,4-d]Pyrimidine Derivatives toward the Human Epidermoid Carcinoma A431 Cell Line. Eur. J. Med. Chem. 2004, 39, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Manetti, F.; Brullo, C.; Magnani, M.; Mosci, F.; Chelli, B.; Crespan, E.; Schenone, S.; Naldini, A.; Bruno, O.; Trincavelli, M.L.; et al. Structure-Based Optimization of Pyrazolo[3,4- d ]Pyrimidines as Abl Inhibitors and Antiproliferative Agents toward Human Leukemia Cell Lines. J. Med. Chem. 2008, 51, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Radi, M.; Dreassi, E.; Brullo, C.; Crespan, E.; Tintori, C.; Bernardo, V.; Valoti, M.; Zamperini, C.; Daigl, H.; Musumeci, F.; et al. Design, Synthesis, Biological Activity, and ADME Properties of Pyrazolo[3,4- d]Pyrimidines Active in Hypoxic Human Leukemia Cells: A Lead Optimization Study. J. Med. Chem. 2011, 54, 2610–2626. [Google Scholar] [CrossRef] [PubMed]
- Zamperini, C.; Dreassi, E.; Vignaroli, G.; Radi, M.; Dragoni, S.; Schenone, S.; Musumeci, F.; Valoti, M.; Antiochia, R.; Botta, M. CYP-Dependent Metabolism of Antitumor Pyrazolo[3,4-d]Pyrimidine Derivatives Is Characterized by an Oxidative Dechlorination Reaction. Drug Metab. Pharmacokinet. 2014, 29, 433–440. [Google Scholar] [CrossRef] [Green Version]
- Radi, M.; Brullo, C.; Crespan, E.; Tintori, C.; Musumeci, F.; Biava, M.; Schenone, S.; Dreassi, E.; Zamperini, C.; Maga, G.; et al. Identification of Potent C-Src Inhibitors Strongly Affecting the Proliferation of Human Neuroblastoma Cells. Bioorg. Med. Chem. Lett. 2011, 21, 5928–5933. [Google Scholar] [CrossRef]
- Chiaino, E.; Micucci, M.; Durante, M.; Budriesi, R.; Gotti, R.; Marzetti, C.; Chiarini, A.; Frosini, M. Apoptotic-Induced Effects of Acacia Catechu Willd. Extract in Human Colon Cancer Cells. Int. J. Mol. Sci. 2020, 21, 2102. [Google Scholar] [CrossRef]
- Santulli, C.; Brizi, C.; Micucci, M.; del Genio, A.; de Cristofaro, A.; Bracco, F.; Pepe, G.L.; di Perna, I.; Budriesi, R.; Chiarini, A.; et al. Castanea Sativa Mill. Bark Extract Protects U-373 MG Cells and Rat Brain Slices Against Ischemia and Reperfusion Injury. J. Cell. Biochem. 2017, 118, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Durante, M.; Frosini, M.; Fusi, F.; Neri, A.; Sticozzi, C.; Saponara, S. In Vitro Vascular Toxicity of Tariquidar, a Potential Tool for in Vivo PET Studies. Toxicol. Vitr. 2017, 44, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Chiaino, E.; Micucci, M.; Budriesi, R.; Mattioli, L.B.; Marzetti, C.; Corsini, M.; Frosini, M. Hibiscus Flower and Olive Leaf Extracts Activate Apoptosis in SH-SY5Y Cells. Antioxidants 2021, 10, 1962. [Google Scholar] [CrossRef] [PubMed]
- Tintori, C.; Brai, A.; Dasso Lang, M.C.; Deodato, D.; Greco, A.M.; Bizzarri, B.M.; Cascone, L.; Casian, A.; Zamperini, C.; Dreassi, E.; et al. Development and in Vitro Evaluation of a Microbicide Gel Formulation for a Novel Non-Nucleoside Reverse Transcriptase Inhibitor Belonging to the N -Dihydroalkyloxybenzyloxopyrimidines (N-DABOs) Family. J. Med. Chem. 2016, 59, 2747–2759. [Google Scholar] [CrossRef] [PubMed]
- Brai, A.; Riva, V.; Saladini, F.; Zamperini, C.; Trivisani, C.I.; Garbelli, A.; Pennisi, C.; Giannini, A.; Boccuto, A.; Bugli, F.; et al. DDX3X Inhibitors, an Effective Way to Overcome HIV-1 Resistance Targeting Host Proteins. Eur. J. Med. Chem. 2020, 200, 112319. [Google Scholar] [CrossRef]
- Chiaino, E.; Stella, R.; Peggion, C.; Micucci, M.; Budriesi, R.; Mattioli, L.B.; Marzetti, C.; Pessina, F.; Valoti, M.; Frosini, M. Acacia catechu Willd. Extract Protects Neuronal Cells from Oxidative Stress-Induced Damage. Antioxidants 2021, 11, 81. [Google Scholar] [CrossRef]
- Vignaroli, G.; Calandro, P.; Zamperini, C.; Coniglio, F.; Iovenitti, G.; Tavanti, M.; Colecchia, D.; Dreassi, E.; Valoti, M.; Schenone, S.; et al. Improvement of Pyrazolo[3,4-d]Pyrimidines Pharmacokinetic Properties: Nanosystem Approaches for Drug Delivery. Sci. Rep. 2016, 6, 21509. [Google Scholar] [CrossRef] [Green Version]
- Dreassi, E.; Zizzari, A.T.; Mori, M.; Filippi, I.; Belfiore, A.; Naldini, A.; Carraro, F.; Santucci, A.; Schenone, S.; Botta, M. 2-Hydroxypropyl-β-Cyclodextrin Strongly Improves Water Solubility and Anti-Proliferative Activity of Pyrazolo[3,4-d]Pyrimidines Src-Abl Dual Inhibitors. Eur. J. Med. Chem. 2010, 45, 5958–5964. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Shah, M.A. Targeting the Cell Cycle: A New Approach to Cancer Therapy. J. Clin. Oncol. 2005, 23, 9408–9421. [Google Scholar] [CrossRef]
- Stewart, Z.A.; Westfall, M.D.; Pietenpol, J.A. Cell-Cycle Dysregulation and Anticancer Therapy. Trends Pharmacol. Sci. 2003, 24, 139–145. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Sun, S.; Zhang, W.; Dong, J.; Huang, C.; Wang, X.; Jia, M.; Yang, H.; Wang, Y.; Jiang, Y.; et al. Irigenin Inhibits Glioblastoma Progression through Suppressing YAP/β-Catenin Signaling. Front. Pharmacol. 2022, 13, 1027577. [Google Scholar] [CrossRef]
- Othman, N.S.; Mohd Azman, D.K. Andrographolide Induces G2/M Cell Cycle Arrest and Apoptosis in Human Glioblastoma DBTRG-05MG Cell Line via ERK1/2 /c-Myc/P53 Signaling Pathway. Molecules 2022, 27, 6686. [Google Scholar] [CrossRef] [PubMed]
- Demircan, T.; Yavuz, M.; Kaya, E.; Akgül, S.; Altuntaş, E. Cellular and Molecular Comparison of Glioblastoma Multiform Cell Lines. Cureus 2021, 13, 43. [Google Scholar] [CrossRef]
- Hatoum, A.; Mohammed, R.; Zakieh, O. The Unique Invasiveness of Glioblastoma and Possible Drug Targets on Extracellular Matrix. Cancer Manag. Res. 2019, 11, 1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vollmann-Zwerenz, A.; Leidgens, V.; Feliciello, G.; Klein, C.A.; Hau, P. Tumor Cell Invasion in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-Targeted Cancer Therapies: Progress, Challenges and Future Directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
 | Enzymatic Data | |||||
|---|---|---|---|---|---|---|
| Compounds | R | R1 | R2 | R3 | c-Src Ki a | Abl Ki a |
| 1 b | H | Cl | NHCH2C6H5 | H | 3 * | 0.37 * |
| 2 | H | Cl | NH-cyclopropyl | H | 11.57 ± 1.08 | 8.01 ± 0.73 |
| 3 c | H | Cl | NHCH2C6H4-oF | H | 0.15 ± 0.02 | 0.73 ± 0.05 * |
| 4 c | H | Cl | NHCH2C6H4-pF | H | 0.51 ± 0.08 | 2.00 ± 0.10 * |
| 5 c | F | Cl | NHCH2C6H5 | H | 3.14 ± 0.38 | 0.44 ± 0.06 * |
| 6 c | F | Cl | NHCH2C6H4-oF | H | 0.07 ± 0.01 | 0.02 ± 0.01 * |
| 7 d | Cl | Cl | NHCH2C6H4-pF | H | 0.80 ± 0.09 | 0.08 * |
| 8 e | H | Cl | NHCH2CH2C6H5 | SCH3 | 0.70 * | 7.30 * |
| 9 f | H | Cl | NHCH2C6H5 | SCH2CH2-4-morpholino | 2.90 ± 0.30 * | 0.09 ± 0.01 * |
| 10 | Cl | Cl | NHCH2CH2C6H5 | H | 0.22 ± 0.03 | 0.31 ± 0.02 |
| 11 e | H | H | NHCH2C6H4-mF | H | 0.51 * | 4.92 * |
| 12 b | Br | Cl | NHC6H5 | H | 0.06 * | 0.61 * |
| Compounds | IC50 a | CC50 b | ||||
|---|---|---|---|---|---|---|
| U-87 | LN18 | LN229 | DBTRG | FIBRO 2-93 | HaCaT | |
| 1 c | 2.9 ± 1.1 * | 3.6 ± 0.5 | 3.4 ± 0.8 | 4.3 ± 0.8 | 86.9 ± 4.2 | 88.7 ± 5.7 |
| 2 | NA | 49.4 ± 2.8 | NA | NA | - | - |
| 3 | 40.2 ± 3.6 | 27.5 ± 2.1 | 29.2 ± 2.6 | 15.1 ± 2.1 | - | - |
| 4 | 29.8 ± 2.5 | 9.0 ± 1.2 | NA | 68.5 ± 3.8 | - | - |
| 5 | 6.8 ± 0.3 | 10.8 ± 1.4 | 6.9 ± 0.5 | 8.5 ± 1.4 | 91.7 ± 5.3 | 126.5 ± 6.4 |
| 6 | 32.4 ± 2.7 | 11.7 ± 1.5 | 33.8 ± 2.3 | 20.1 ± 1.8 | - | - |
| 7 | 22.7 ± 2.3 | 3.9 ± 0.6 | 7.1 ± 1.1 | 14.6 ± 0.9 | 28.6 ± 3.7 | 37.8 ± 4.9 |
| 8 | 37.9 ± 3.9 | 5.9 ± 1.1 | 26.1 ± 2.8 | 31.2 ± 2.5 | - | - |
| 9 | NA | 2.9 ± 0.5 | 58.2 ± 3.4 | 59.4 ± 3.7 | - | - |
| 10 | 33.9 ± 2.9 | 6.2 ± 1.0 | 27.9 ± 2.1 | 46.5 ± 4.1 | - | - |
| 11 | 56.9 ± 3.3 | 34.1 ± 2.9 | 58.4 ± 3.0 | 69.2 ± 3.9 | - | - |
| 12 c | 14.0 ± 1.9 * | NA | NA | NA | - | - |
| Compounds | QP LogS | GI Papp a,b (MR%) c | BBB Papp a,b (MR%) c | Metabolic Stability a (%) | Metabolite Formation (%) d |
|---|---|---|---|---|---|
| 1 e | −5.715 | 11.08 * | 16.5 * | 78.3 * | M1 = 14.5 * M2 < 0.1 * M3 = 2.6 * M4 = 4.5 * |
| 2 | −4.862 | 6.46 (34.7) | 4.96 (55.0) | 96.3 | M1 = 2.1 M2 = 1.6 |
| 3 | −6.358 | 12.4 (7.3) | 12.2 (4.8) | 80.4 | M1 = 14.5 M3 = 5.1 |
| 4 | −6.784 | 11.2 (7.8) | 10.6 (2.1) | 92.2 | M1 = 5.9 M2 = 1.0 M3 = 0.9 |
| 5 | −6.511 | 10.7 (9.2) | 11.2 (5.6) | 84.2 | M1 = 10.8 M2 = 1.7 M3 = 3.3 |
| 6 | −6.955 | 11.4 (10.1) | 12.4 (4.4) | 69.9 | M1 = 21.9 M2 = 3.0 M3 = 5.2 |
| 7 f | −7.533 | 16.6 * | 9.6 (4.4) | 94.0 * | M1 = 4 * M3 = 2 * |
| 8 | −6.207 | 3.2 (42.7) | 5.96 (18.3) | 96.6 | M1 = 1.9 M2 = 1.5 |
| 9 g | −5.6 * | 9.1 * | 2.6 (6.3) | 95 * | M1 = 5 * |
| 10 | −7.589 | 10.8 (17.2) | 11.3 (4.8) | 85.9 | M1 = 10.3 M3 = 3.8 |
| 11 | −5.973 | 10.1 (6.7) | 10.8 (5.2) | 93.0 | M1 = 0.4 M2 = 6.7 |
| 12 e | −6.576 | 6.64 * | 13.1 * | 96.4 * | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poggialini, F.; Vagaggini, C.; Brai, A.; Pasqualini, C.; Crespan, E.; Maga, G.; Perini, C.; Cabella, N.; Botta, L.; Musumeci, F.; et al. Biological Evaluation and In Vitro Characterization of ADME Profile of In-House Pyrazolo[3,4-d]pyrimidines as Dual Tyrosine Kinase Inhibitors Active against Glioblastoma Multiforme. Pharmaceutics 2023, 15, 453. https://doi.org/10.3390/pharmaceutics15020453
Poggialini F, Vagaggini C, Brai A, Pasqualini C, Crespan E, Maga G, Perini C, Cabella N, Botta L, Musumeci F, et al. Biological Evaluation and In Vitro Characterization of ADME Profile of In-House Pyrazolo[3,4-d]pyrimidines as Dual Tyrosine Kinase Inhibitors Active against Glioblastoma Multiforme. Pharmaceutics. 2023; 15(2):453. https://doi.org/10.3390/pharmaceutics15020453
Chicago/Turabian StylePoggialini, Federica, Chiara Vagaggini, Annalaura Brai, Claudia Pasqualini, Emmanuele Crespan, Giovanni Maga, Cecilia Perini, Noemi Cabella, Lorenzo Botta, Francesca Musumeci, and et al. 2023. "Biological Evaluation and In Vitro Characterization of ADME Profile of In-House Pyrazolo[3,4-d]pyrimidines as Dual Tyrosine Kinase Inhibitors Active against Glioblastoma Multiforme" Pharmaceutics 15, no. 2: 453. https://doi.org/10.3390/pharmaceutics15020453
APA StylePoggialini, F., Vagaggini, C., Brai, A., Pasqualini, C., Crespan, E., Maga, G., Perini, C., Cabella, N., Botta, L., Musumeci, F., Frosini, M., Schenone, S., & Dreassi, E. (2023). Biological Evaluation and In Vitro Characterization of ADME Profile of In-House Pyrazolo[3,4-d]pyrimidines as Dual Tyrosine Kinase Inhibitors Active against Glioblastoma Multiforme. Pharmaceutics, 15(2), 453. https://doi.org/10.3390/pharmaceutics15020453