
Composition-Property Relationships of pH-Responsive Poly[(2-vinylpyridine)-co-(butyl methacrylate)] Copolymers for Reverse Enteric Coatings


Abstract
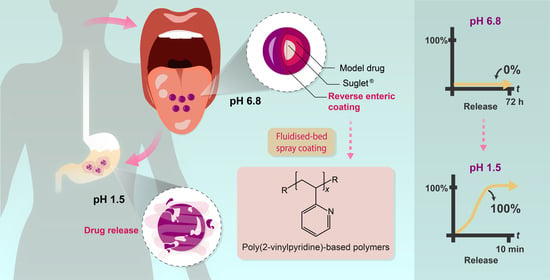
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instrumentation
2.3. Procedures
2.3.1. Synthesis of Poly[(2-vinylpyridine)x-co-(butyl methacrylate)y]
2.3.2. Synthesis of Poly[(2-vinylpyridine)x-co-(butyl methacrylate)y-co-(methyl methacrylate)z]
2.3.3. Synthesis of Poly[(2-vinyl pyridine)x-co-(butyl methacrylate)y-co-(isobornyl methacrylate)z]
2.3.4. Gas Chromatography
2.3.5. Gel Permeation Chromatography
2.3.6. Modulated Differential Scanning Calorimetry
2.3.7. Thermogravimetric Analysis
2.3.8. Copolymer Water Absorption
2.3.9. Copolymer Solubility
2.3.10. Ultraviolet-Visible Spectrophotometry
2.3.11. Fluidised-Bed Spray Coating
2.3.12. Taste-Masked Formulation Stability
2.3.13. Taste-Masked Formulation Release
3. Results and Discussion
3.1. Synthesis and Characterisation
3.2. Thermal Properties
3.3. Copolymer Solubility
3.4. Water Absorption
3.5. Fluidised-Bed Spray Coating
3.6. In Vitro Evaluation of the Copolymers as Reverse Enteric, Taste-Masking Coatings
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Felton, L.A.; Porter, S.C. An update on pharmaceutical film coating for drug delivery. Expert Opin. Drug Deliv. 2013, 10, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Cole, E.T.; Scott, R.A.; Connor, A.L.; Wilding, I.R.; Petereit, H.-U.; Schminke, C.; Beckert, T.; Cadé, D. Enteric coated HPMC capsules designed to achieve intestinal targeting. Int. J. Pharm. 2002, 231, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Patra, C.N.; Priya, R.; Swain, S.; Jena, G.K.; Panigrahi, K.C.; Ghose, D. Pharmaceutical significance of Eudragit: A review. Future J. Pharm. Sci. 2017, 3, 33–45. [Google Scholar] [CrossRef]
- Menjoge, A.R.; Kulkarni, M.G. Blends of Reverse Enteric Polymer with Enteric and pH-Independent Polymers: Mechanistic Investigations for Tailoring Drug Release. Biomacromolecules 2007, 8, 240–251. [Google Scholar] [CrossRef]
- Nollenberger, K.; Albers, J. Poly(meth)acrylate-based coatings. Int. J. Pharm. 2013, 457, 461–469. [Google Scholar] [CrossRef]
- Georgieva, Y.; Kassarova, M.; Kokova, V.; Apostolova, E.; Pilicheva, B. Taste masking of enalapril maleate by microencapsulation in Eudragit EPO® microparticles. Die Pharm. Int. J. Pharm. Sci. 2020, 75, 61–69. [Google Scholar]
- Lopalco, A.; Denora, N.; Laquintana, V.; Cutrignelli, A.; Franco, M.; Robota, M.; Hauschildt, N.; Mondelli, F.; Arduino, I.; Lopedota, A. Taste masking of propranolol hydrochloride by microbeads of EUDRAGIT® E PO obtained with prilling technique for paediatric oral administration. Int. J. Pharm. 2020, 574, 118922. [Google Scholar] [CrossRef]
- Breitkreutz, J.; Bornhöft, M.; Wöll, F.; Kleinebudde, P. Pediatric drug formulations of sodium benzoate: I. Coated granules with a hydrophilic binder. Eur. J. Pharm. Biopharm. 2003, 56, 247–253. [Google Scholar] [CrossRef]
- Wang, H.; Dumpa, N.; Bandari, S.; Durig, T.; Repka, M.A. Fabrication of Taste-Masked Donut-Shaped Tablets Via Fused Filament Fabrication 3D Printing Paired with Hot-Melt Extrusion Techniques. AAPS PharmSciTech 2020, 21, 243. [Google Scholar] [CrossRef]
- Leopold, C.S.; Eikeler, D. Eudragit® E as Coating Material for the pH-Controlled Drug Release in the Topical Treatment of Inflammatory Bowel Disease (IBD). J. Drug Target. 1998, 6, 85–94. [Google Scholar] [CrossRef]
- Aframian, D.; Davidowitz, T.; Benoliel, R. The distribution of oral mucosal pH values in healthy saliva secretors. Oral Dis. 2006, 12, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Felton, L.A. Use of polymers for taste-masking pediatric drug products. Drug Dev. Ind. Pharm. 2018, 44, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Chivate, A.; Sargar, V.; Nalawade, P.; Tawde, V. Formulation and development of oral dry suspension using taste masked Ornidazole particles prepared using Kollicoat® Smartseal 30 D. Drug Dev. Ind. Pharm. 2013, 39, 1091–1097. [Google Scholar] [CrossRef]
- Kirk-Othmer Encyclopedia of Chemical Technology: Volume 1—A to Alkaloids, 4th ed.; John Wiley and Sons: New York, NY, USA, 1996; Volume 1.
- Teunou, E.; Poncelet, D. Batch and continuous fluid bed coating—Review and state of the art. J. Food Eng. 2002, 53, 325–340. [Google Scholar] [CrossRef]
- Mehta, A.M. Processing and Equipment Considerations for Aqueous Coatings, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2008; pp. 87–124. [Google Scholar]
- Tantavichet, N.; Pritzker, M.D.; Burns, C.M. Proton uptake by poly(2-vinylpyridine) coatings. J. Appl. Polym. Sci. 2001, 81, 1493–1497. [Google Scholar] [CrossRef]
- Brandrup, J.; Immergut, E.H.; Grulke, E.A.; Abe, A.; Bloch, D.R. Polymer Handbook, 4th ed.; Wiley: New York, NY, USA, 1999; Volume 89. [Google Scholar]
- Felton, L.A. Mechanisms of polymeric film formation. Int. J. Pharm. 2013, 457, 423–427. [Google Scholar] [CrossRef]
- Yu, J.M.; Dubois, P.; Jérôme, R. Synthesis and Properties of Poly[isobornyl methacrylate (IBMA)-b-butadiene (BD)-b-IBMA] Copolymers: New Thermoplastic Elastomers of a Large Service Temperature Range. Macromolecules 1996, 29, 7316–7322. [Google Scholar] [CrossRef]
- Lin, S.Y.; Chen, K.S.; Chu, L.R. Organic esters of plasticizers affecting the water absorption, adhesive property, glass transition temperature and plasticizer permanence of Eudragit acrylic films. J. Control. Release 2000, 68, 343–350. [Google Scholar] [CrossRef]
- Wen, H.; Park, K. Oral Controlled Release Formulation Design and Drug Delivery: Theory to Practice; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Fernández–García, M.; Cuervo–Rodriguez, R.; Madruga, E.L. Glass transition temperatures of butyl acrylate–methyl methacrylate copolymers. J. Polym. Sci. Part B Polym. Phys. 1999, 37, 2512–2520. [Google Scholar] [CrossRef]
- Nguyen, G.; Nicole, D.; Matlengiewicz, M.; Roizard, D.; Henzel, N. Relation between microstructure and glass transition temperature of poly[(methyl methacrylate)-co-(ethyl acrylate)]. Polym. Int. 2001, 50, 784–791. [Google Scholar] [CrossRef]
- Ooi, S.K.; Cook, W.D.; Simon, G.P.; Such, C.H. Effects of composition on the water uptake and hydroplasticisation of the glass transition temperature of methacrylate copolymers. Eur. Polym. J. 2002, 38, 903–910. [Google Scholar] [CrossRef]
- Šoljić, I.; Jukić, A.; Janović, Z. Free radical copolymerization of N,N-dimethylaminoethyl methacrylate with styrene and methyl methacrylate: Monomer reactivity ratios and glass transition temperatures. Polym. Int. 2009, 58, 1014–1022. [Google Scholar] [CrossRef]
- Neugebauer, D.; Bury, K.; Wlazło, M. Atom transfer radical copolymerization of glycidyl methacrylate and methyl methacrylate. J. Appl. Polym. Sci. 2012, 124, 2209–2215. [Google Scholar] [CrossRef]
- Smith, L.M.; Coote, M.L. Effect of temperature and solvent on polymer tacticity in the free-radical polymerization of styrene and methyl methacrylate. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 3351–3358. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Mays, J.; Ferry, W.; Fetters, L.J. Properties and chain flexibility of poly(dl-isobornyl methacrylate). J. Polym. Sci. B Polym. Phys. 1984, 22, 1745–1751. [Google Scholar] [CrossRef]
- Ko, K.Y.; Hwang, S.H. Monomer composition effects on thermal properties of transparent poly(methyl methacrylate-co-isobornyl methacrylate-co-cyclohexyl maleimide) terpolymers. J. Ind. Eng. Chem. 2018, 59, 50–55. [Google Scholar] [CrossRef]
- Mark, J.E. Physical Properties of Polymers Handbook, 2nd ed.; Springer: New York, NY, USA, 2006. [Google Scholar]
- Elmaci, A.; Hacaloglu, J. Thermal degradation of poly(vinylpyridine)s. Polym. Degrad. Stab. 2009, 94, 738–743. [Google Scholar] [CrossRef]
- Özlem, S.; Hacaloglu, J. Thermal degradation of poly(n-butyl methacrylate), poly(n-butyl acrylate) and poly(t-butyl acrylate). J. Anal. Appl. Pyrolysis 2013, 104, 161–169. [Google Scholar] [CrossRef]
- Orhan, T.; Hacaloglu, J. Thermal degradation of poly(2-vinylpyridine) copolymers. Polym. Degrad. Stab. 2013, 98, 356–360. [Google Scholar] [CrossRef]
- Özlem-Gundogdu, S.; Gurel, E.A.; Hacaloglu, J. Pyrolysis of of poly(methy methacrylate) copolymers. J. Anal. Appl. Pyrolysis 2015, 113, 529–538. [Google Scholar] [CrossRef]
- Holland, B.J.; Hay, J.N. The kinetics and mechanisms of the thermal degradation of poly(methyl methacrylate) studied by thermal analysis-Fourier transform infrared spectroscopy. Polymer 2001, 42, 4825–4835. [Google Scholar] [CrossRef]
- Ozlem, S.; Aslan-Gürel, E.; Rossi, R.M.; Hacaloglu, J. Thermal degradation of poly(isobornyl acrylate) and its copolymer with poly(methyl methacrylate) via pyrolysis mass spectrometry. J. Anal. Appl. Pyrolysis 2013, 100, 17–25. [Google Scholar] [CrossRef]
- Matsumoto, A.; Mizuta, K.; Otsu, T. Synthesis and thermal properties of poly(cycloalkyl methacrylate)s bearing bridged- and fused-ring structures. J. Polym. Sci. Part A Polym. Chem. 1993, 31, 2531–2539. [Google Scholar] [CrossRef]
- Gryczke, A.; Schminke, S.; Maniruzzaman, M.; Beck, J.; Douroumis, D. Development and evaluation of orally disintegrating tablets (ODTs) containing Ibuprofen granules prepared by hot melt extrusion. Colloids Surf. B Biointerfaces 2011, 86, 275–284. [Google Scholar] [CrossRef]
- Bley, O.; Siepmann, J.; Bodmeier, R. Characterization of Moisture-Protective Polymer Coatings Using Differential Scanning Calorimetry and Dynamic Vapor Sorption. J. Pharm. Sci. 2009, 98, 651–664. [Google Scholar] [CrossRef]
- Bley, O.; Siepmann, J.; Bodmeier, R. Protection of moisture-sensitive drugs with aqueous polymer coatings: Importance of coating and curing conditions. Int. J. Pharm. 2009, 378, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Brewer, K.; McWhorter, T.J.; Sloper, C.; Moseby, K.; Read, J.L.; Peacock, D.; Blencowe, A. Toward Targeted Invasive Predator Control: Developing pH-Responsive Subcutaneous Implants for Native Mammals. ACS Appl. Polym. Mater. 2022, 4, 6687–6699. [Google Scholar] [CrossRef]
- Dashevskiy, A.; Mohylyuk, V.; Ahmed, A.R.; Kolter, K.; Guth, F.; Bodmeier, R. Micropellets coated with Kollicoat® Smartseal 30D for taste masking in liquid oral dosage forms. Drug Dev. Ind. Pharm. 2017, 43, 1548–1556. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymer Code | Targeted mol% of RU | Calculated mol% of RU a | Mwb (kDa) | Mnb (kDa) | Đb | Tgc (°C) |
|---|---|---|---|---|---|---|
| PVB 60/40 | 60:40 | 63:37 | 35.3 | 21.0 | 1.7 | 58.9 |
| PVB 50/50 | 50:50 | 51:49 | 37.1 | 21.1 | 1.8 | 53.1 |
| PVB 40/60 | 40:60 | 41:59 | 30.1 | 17.5 | 1.7 | 43.3 |
| PVBM 40/50/10 | 40:50:10 | 40:50:10 | 32.4 | 14.8 | 2.2 | 45.3 |
| PVBM 30/50/20 | 30:50:20 | 30:50:20 | 34.5 | 16.2 | 2.1 | 50.2 |
| PVBM 30/40/30 | 30:40:30 | 31:39:30 | 35.4 | 17.2 | 2.1 | 59.7 |
| PVBM 40/40/20 | 40:40:20 | 41:39:20 | 36.0 | 18.6 | 1.9 | 57.1 |
| PVBI 50/45/5 | 50:45:5 | 49:46:5 | 25.2 | 13.2 | 1.9 | 40.0 |
| PVBI 50/40/10 | 50:40:10 | 50:40:10 | 25.0 | 12.1 | 2.1 | 52.6 |
| PVBI 50/35/15 | 50:35:15 | 51:34:16 | 27.2 | 14.2 | 1.9 | 64.1 |
| PVBI 50/30/20 | 50:30:20 | 51:30:20 | 34.2 | 16.4 | 2.1 | 73.7 |
| PVBI 40/40/20 | 40:40:20 | 40:40:20 | 43.4 | 22.3 | 2.0 | 70.2 |
| Coating | nSuglets® | Mass Gain (% w/w) | Mass Coated (µg/Suglet®) |
|---|---|---|---|
| PEG/RhB | 1805 | 6.2 | 209 |
| PVB 60/40 | 335 | 5.2 | 186 |
| PVB 40/60 | 332 | 5.4 | 193 |
| PVBM 40/50/10 | 340 | 6.5 | 232 |
| PVBM 30/50/20 | 338 | 4.9 | 175 |
| Coating | Initial Release a (min) | Complete Release b (min) |
|---|---|---|
| PVB 60/40 | 1.3 ± 0.1 (10) | 2.1 ± 0.3 (15) |
| PVB 40/60 | 3.6 ± 1.5 (43) | 7.2 ± 2.4 (33) |
| PVBM 40/50/10 | 4.6 ± 1.4 (32) | 8.5 ± 2.0 (23) |
| PVBM 30/50/20 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brewer, K.; Blencowe, A. Composition-Property Relationships of pH-Responsive Poly[(2-vinylpyridine)-co-(butyl methacrylate)] Copolymers for Reverse Enteric Coatings. Pharmaceutics 2023, 15, 454. https://doi.org/10.3390/pharmaceutics15020454
Brewer K, Blencowe A. Composition-Property Relationships of pH-Responsive Poly[(2-vinylpyridine)-co-(butyl methacrylate)] Copolymers for Reverse Enteric Coatings. Pharmaceutics. 2023; 15(2):454. https://doi.org/10.3390/pharmaceutics15020454
Chicago/Turabian StyleBrewer, Kyle, and Anton Blencowe. 2023. "Composition-Property Relationships of pH-Responsive Poly[(2-vinylpyridine)-co-(butyl methacrylate)] Copolymers for Reverse Enteric Coatings" Pharmaceutics 15, no. 2: 454. https://doi.org/10.3390/pharmaceutics15020454
APA StyleBrewer, K., & Blencowe, A. (2023). Composition-Property Relationships of pH-Responsive Poly[(2-vinylpyridine)-co-(butyl methacrylate)] Copolymers for Reverse Enteric Coatings. Pharmaceutics, 15(2), 454. https://doi.org/10.3390/pharmaceutics15020454