
A Theranostic Small-Molecule Prodrug Conjugate for Neuroendocrine Prostate Cancer

 , , and
, , and 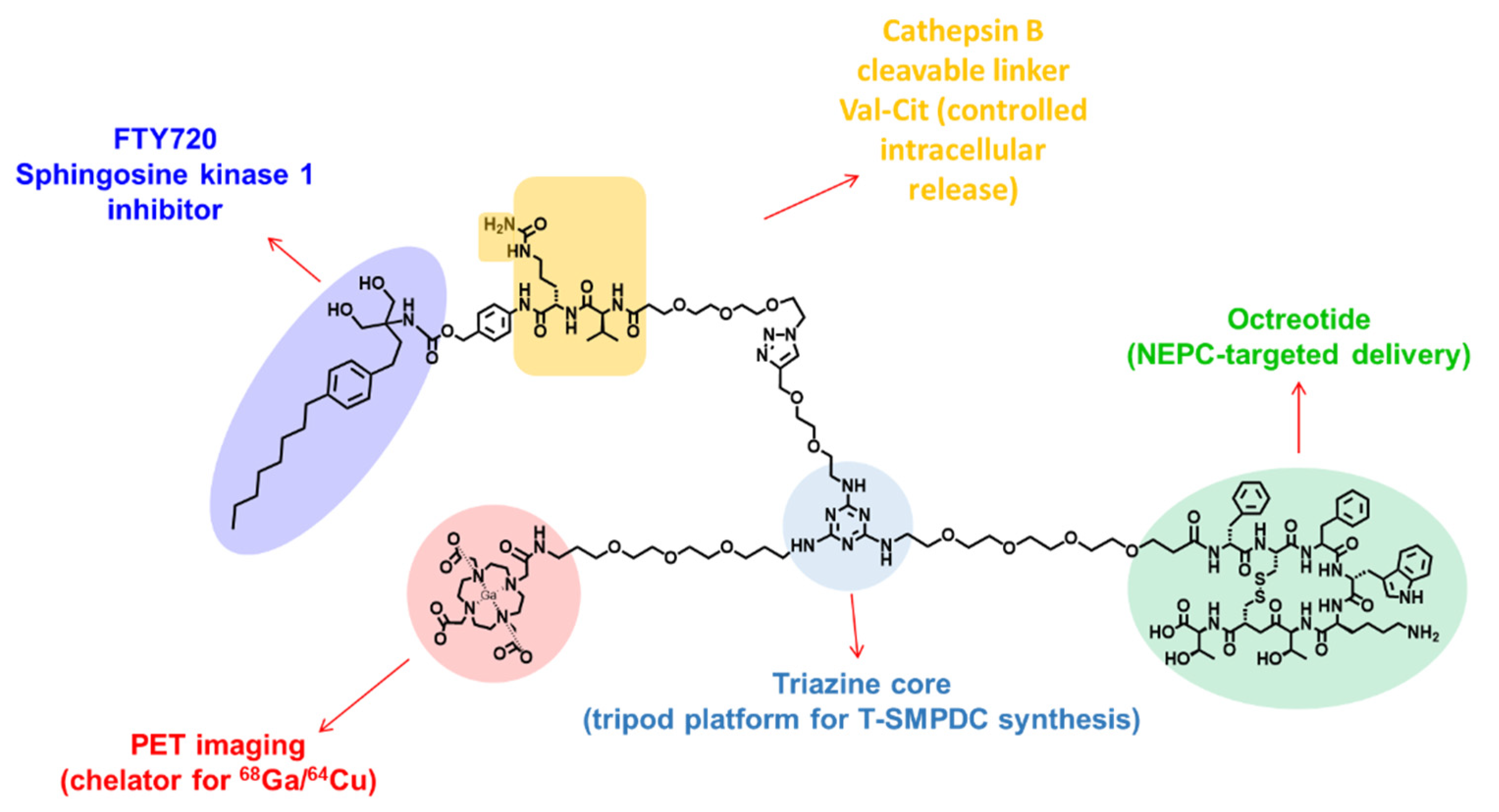{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Materials and Procedures
2.2. Chemistry
2.2.1. Synthesis of Val-Cit-pABOC-FTY720, Compound 10
2.2.2. Synthesis of TZ-PEG2-Trz-PEG3-Val-Cit-pABOC-FTY720, Compound 11
2.2.3. Synthesis of DOTA-PEG3-TZ-PEG2-Trz-PEG3-Val-Cit-pABOC-FTY720, Compound 12
2.2.4. Synthesis of DOTA-PEG3-TZ(PEG4-Octr)-PEG2-Trz-PEG3-Val-Cit-pABOC-FTY720, Compound 13
2.3. Radiochemistry
2.4. Cell Culture and Animal Models
2.5. Cell Viability Assay
2.6. Cell Uptake Assay
2.7. Internalization Assay
2.8. In Vitro Cathepsin-B-Mediated Release Assay
2.9. Small Animal PET/CT Imaging
2.10. Western Blot Analysis
2.11. Immunohistochemistry (IHC)
3. Results
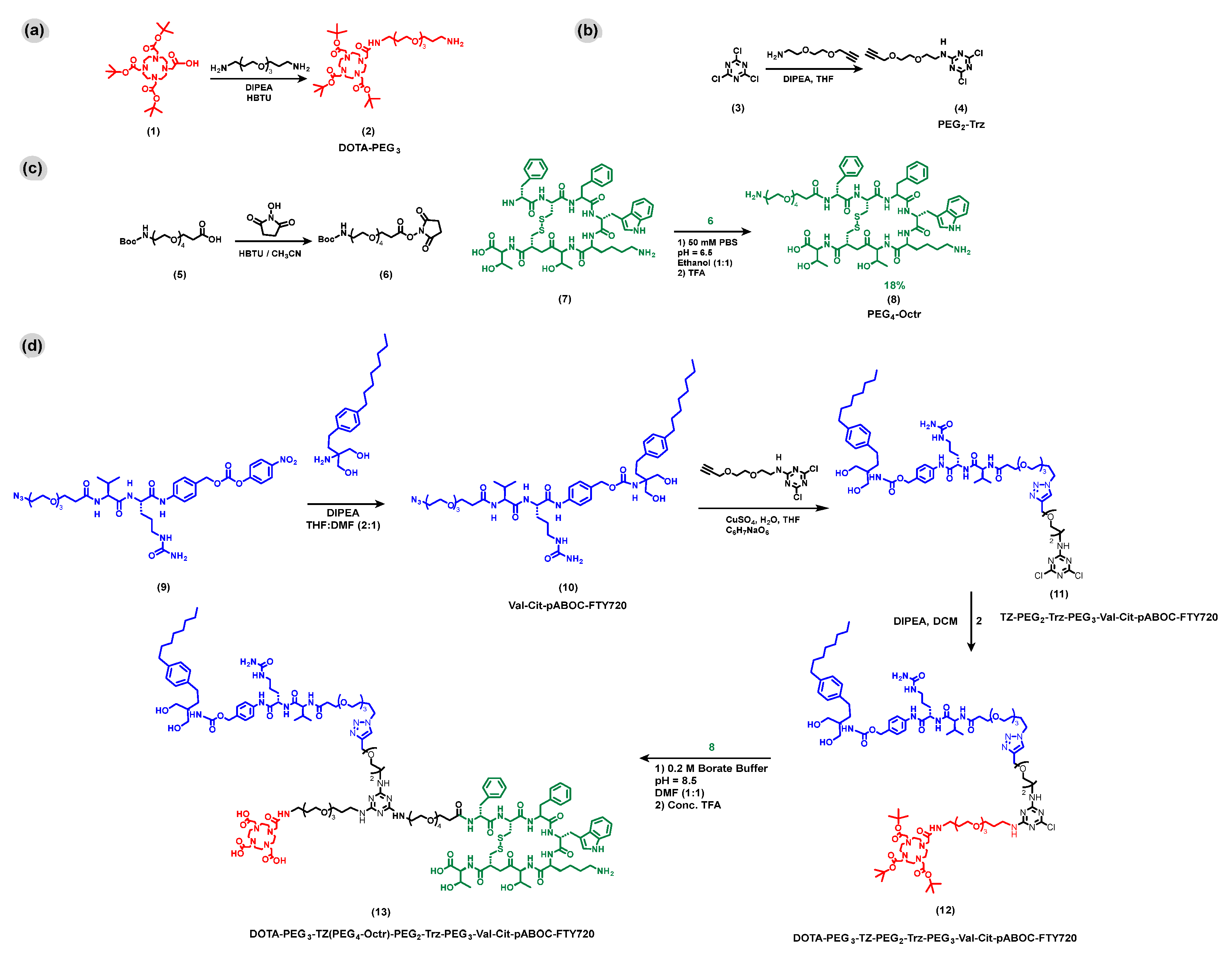
3.1. Modular Synthesis of DOTA-PEG3-TZ(PEG4-Octr)-PEG2-Trz-PEG3-Val-Cit-pABOC-FTY720
3.2. Radiochemistry
3.3. In Vitro Assays of DOTA-PEG3-TZ(PEG4-Octr)-PEG2-Trz-PEG3-Val-Cit-pABOC-FTY720
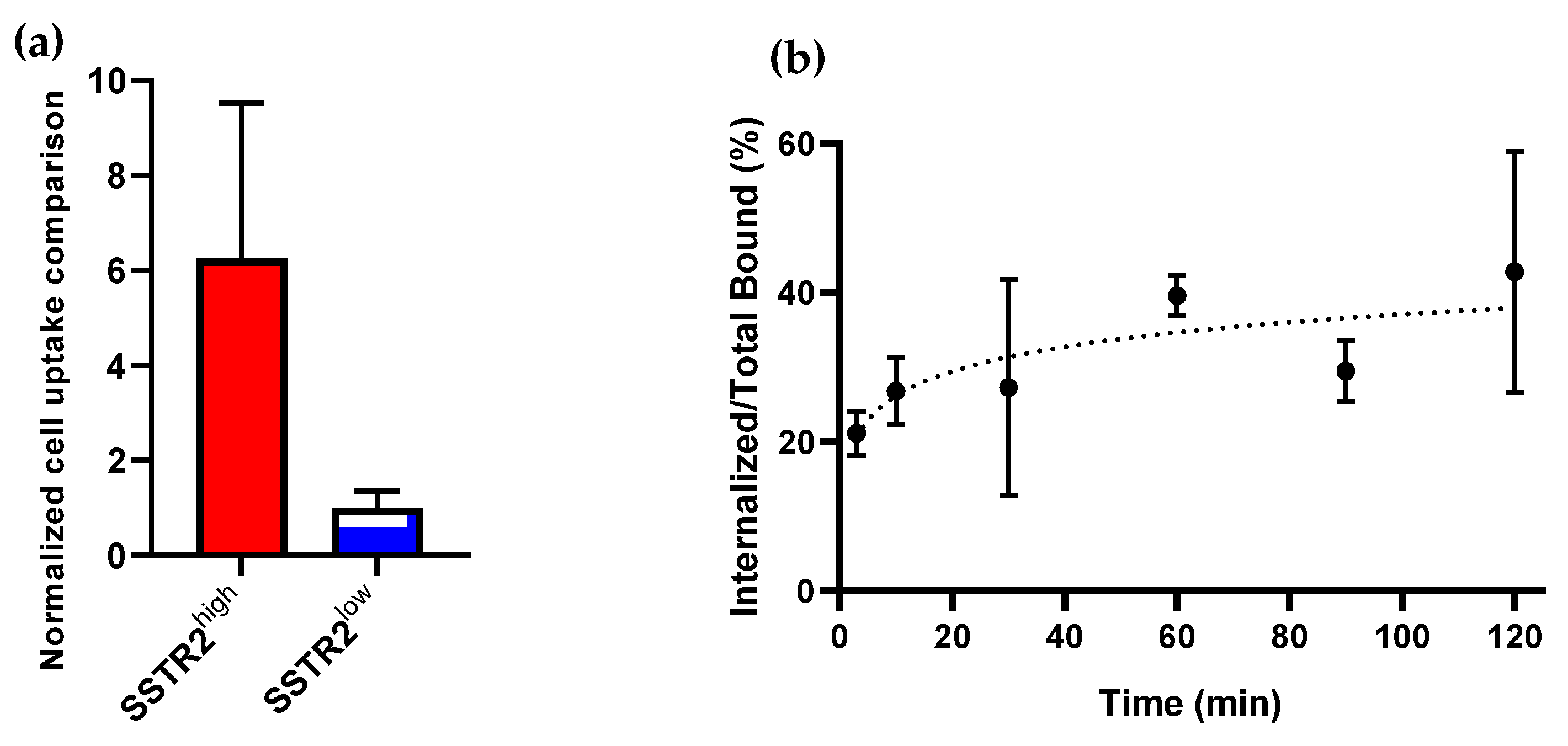
3.3.1. SSTR2-Selective Cell Uptake and SSTR2-Mediated Internalization of [64Cu]Cu- DOTA-PEG3-TZ(PEG4-Octr)-PEG2-Trz-PEG3-Val-Cit-pABOC-FTY720
3.3.2. Cathepsin-B-Mediated Release of FTY720 from DOTA-PEG3-TZ(PEG4-Octr)-PEG2-Trz-PEG3-Val-Cit-pABOC-FTY720
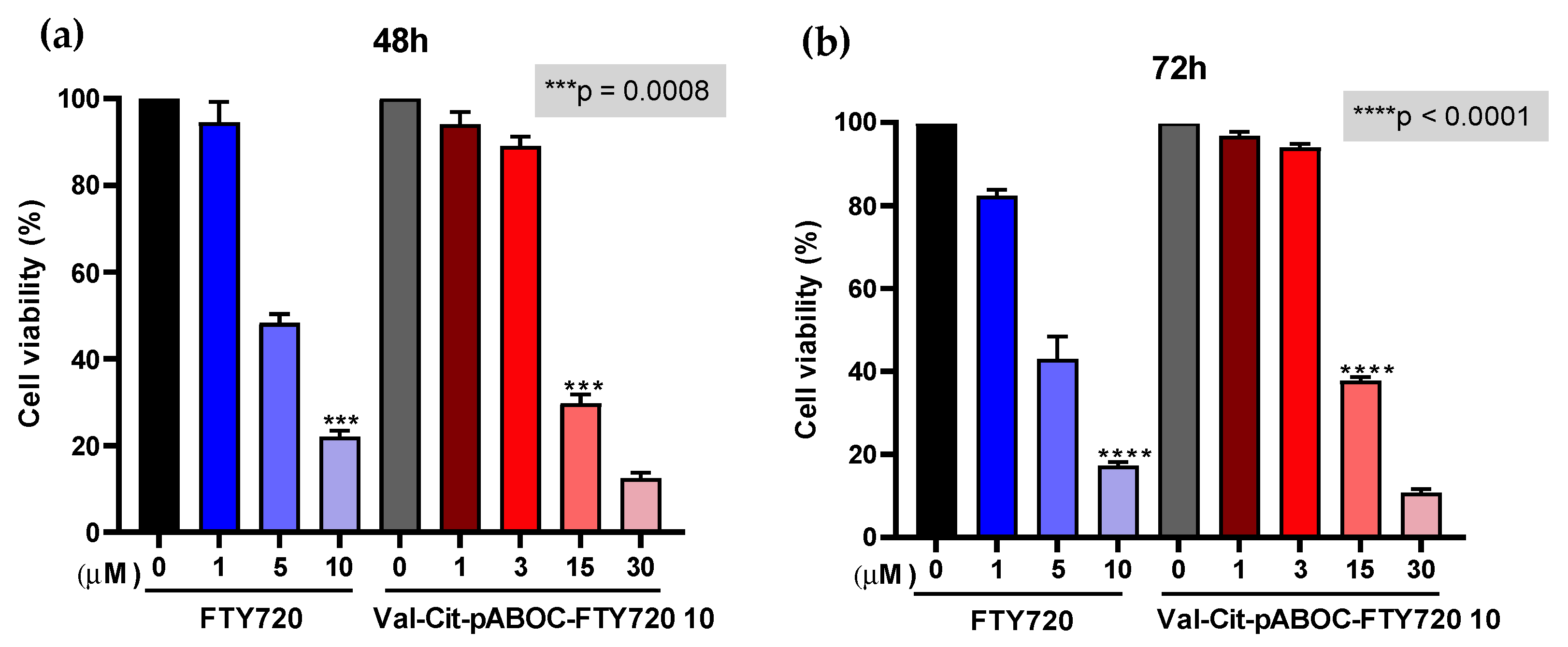
3.3.3. The Cytotoxicity of FTY720 Was Masked by Its Conjugation with the Cathepsin B Prodrug Linkage, Val-Cit
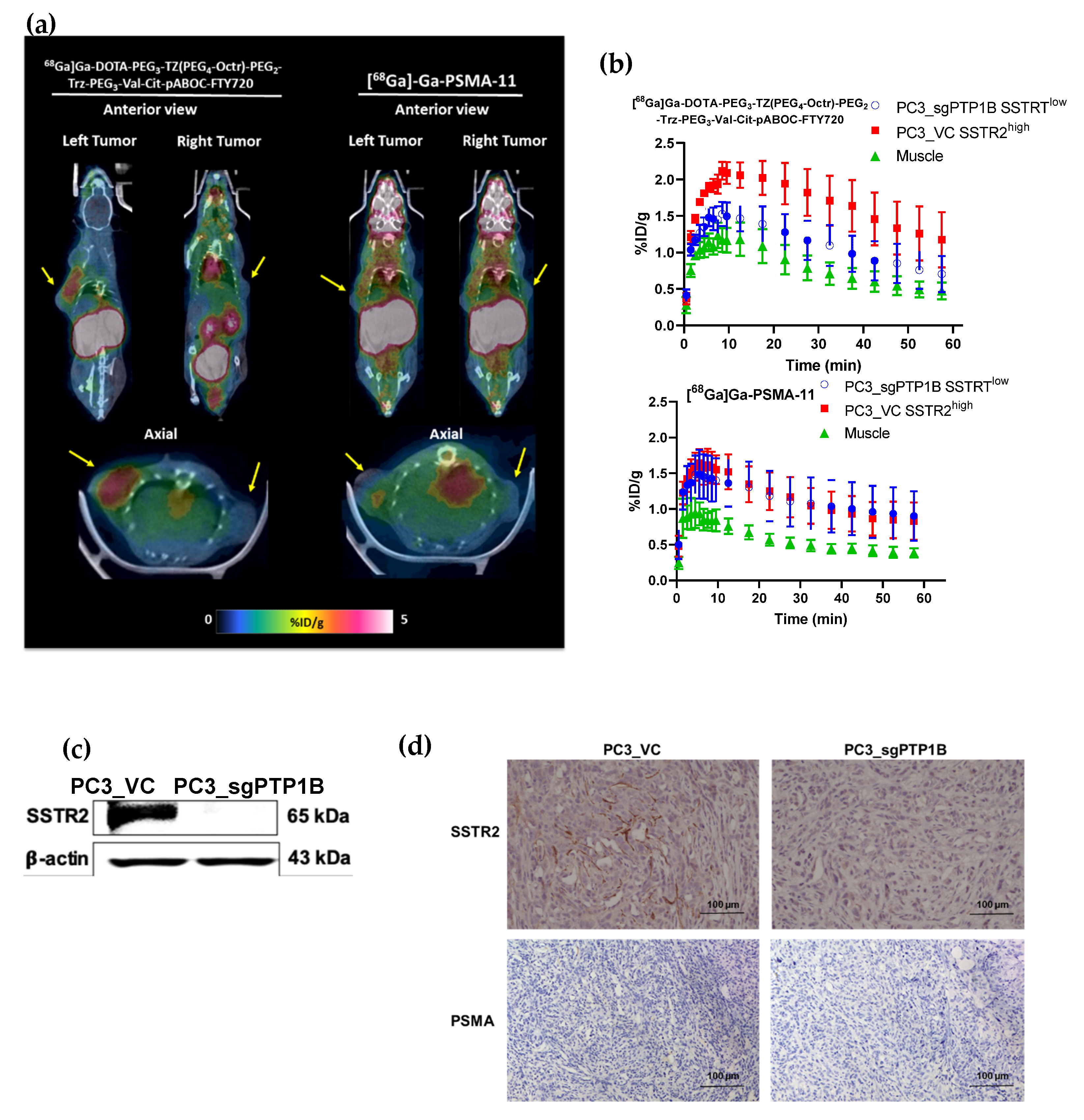
3.4. In Vivo Evaluation of [68Ga]Ga-DOTA-PEG3-TZ(PEG4-Octr)-PEG2-Trz-PEG3-Val-Cit-pABOC-FTY720
PET Imaging of Mice Showed That [68Ga]Ga-DOTA-PEG3-TZ(PEG4-Octr)-PEG2-Trz-PEG3-Val-Cit-pABOC-FTY720 had SSTR2-Selective Tumor Uptake and It Was Mainly Excreted through the Kidneys
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taher, A.; Jensen, C.T.; Yedururi, S.; Surasi, D.S.; Faria, S.C.; Bathala, T.K.; Mujtaba, B.; Bhosale, P.; Wagner-bartak, N.; Morani, A.C. Imaging of Neuroendocrine Prostatic Carcinoma. Cancers 2021, 13, 5765. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Ci, X.; Choi, S.Y.C.; Crea, F.; Lin, D.; Wang, Y. Molecular Events in Neuroendocrine Prostate Cancer Development. Nat. Rev. Urol. 2021, 18, 581–596. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.S.; Tripathi, M.; Agarwal, K.K.; Kumar, R.; Vijay, M.K.; Bal, C. Metastatic Poorly Differentiated Prostatic Carcinoma with Neuroendocrine Differentiation: Negative on 68Ga-PSMA PET/CT. Clin. Nucl. Med. 2015, 40, e163–e166. [Google Scholar] [CrossRef] [PubMed]
- Tosoian, J.J.; Gorin, M.A.; Rowe, S.P.; Andreas, D.; Szabo, Z.; Pienta, K.J.; Pomper, M.G.; Lotan, T.L.; Ross, A.E. Correlation of PSMA-Targeted 18F-DCFPyL PET/CT Findings with Immunohistochemical and Genomic Data in a Patient with Metastatic Neuroendocrine Prostate Cancer. Clin. Genitourin. Cancer 2017, 15, e65–e68. [Google Scholar] [CrossRef] [PubMed]
- Parida, G.K.; Tripathy, S.; Datta Gupta, S.; Singhal, A.; Kumar, R.; Bal, C.; Shamim, S.A. Adenocarcinoma Prostate with Neuroendocrine Differentiation: Potential Utility of 18F-FDG PET/CT and 68Ga-DOTANOC PET/CT Over 68Ga-PSMA PET/CT. Clin. Nucl. Med. 2018, 43, 248–249. [Google Scholar] [CrossRef]
- Debnath, S.; Zhou, N.; McLaughlin, M.; Rice, S.; Pillai, A.K.; Hao, G.; Sun, X. PSMA-Targeting Imaging and Theranostic Agents—Current Status and Future Perspective. Int. J. Mol. Sci. 2022, 23, 1158. [Google Scholar] [CrossRef]
- Niaz, M.J.; Sun, M.; Skafida, M.; Niaz, M.O.; Ivanidze, J.; Osborne, J.R.; O’Dwyer, E. Review of Commonly Used Prostate Specific PET Tracers Used in Prostate Cancer Imaging in Current Clinical Practice. Clin. Imaging 2021, 79, 278–288. [Google Scholar] [CrossRef]
- Bhagirath, D.; Liston, M.; Akoto, T.; Lui, B.; Bensing, B.A.; Sharma, A.; Saini, S. Novel, Non-Invasive Markers for Detecting Therapy Induced Neuroendocrine Differentiation in Castration-Resistant Prostate Cancer Patients. Sci. Rep. 2021, 11, 8279. [Google Scholar] [CrossRef]
- Zhu, J.; Liang, X.; Wu, D.; Chen, S.; Yang, B.; Mao, W.; Shen, D. Clinicopathological Characteristics and Survival Outcomes in Neuroendocrine Prostate Cancer: A Population-Based Study. Medicine 2021, 100, e25237. [Google Scholar] [CrossRef]
- Armstrong, A.J. Biomarkers in Castration-Resistant Prostate Cancer. Curr. Dev. Oncol. Drug Res. 2014, 12, 115–118. [Google Scholar]
- Beltran, H.; Demichelis, F. Therapy Considerations in Neuroendocrine Prostate Cancer: What Next? Endocr. Relat. Cancer 2021, 28, T67–T78. [Google Scholar] [CrossRef] [PubMed]
- Clermont, P.-L.; Ci, X.; Pandha, H.; Wang, Y.; Crea, F. Treatment-Emergent Neuroendocrine Prostate Cancer: Molecularly Driven Clinical Guidelines. Int. J. Endocr. Oncol. 2019, 6, IJE20. [Google Scholar] [CrossRef]
- Beltran, H.; Jendrisak, A.; Landers, M.; Mosquera, J.M.; Kossai, M.; Louw, J.; Krupa, R.; Graf, R.P.; Schreiber, N.A.; Nanus, D.M.; et al. The Initial Detection and Partial Characterization of Circulating Tumor Cells in Neuroendocrine Prostate Cancer. Clin. Cancer Res. 2016, 22, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Parent, E.E.; Kase, A.M. A Treatment Paradigm Shift: Targeted Radionuclide Therapies for Metastatic Castrate Resistant Prostate Cancer. Cancers 2022, 14, 4276. [Google Scholar] [CrossRef]
- Rosellini, M.; Santoni, M.; Mollica, V.; Rizzo, A.; Cimadamore, A.; Scarpelli, M.; Storti, N.; Battelli, N.; Montironi, R.; Massari, F. Treating Prostate Cancer by Antibody-Drug Conjugates. Int. J. Mol. Sci. 2021, 22, 1551. [Google Scholar] [CrossRef]
- DeLucia, D.C.; Cardillo, T.M.; Ang, L.; Labrecque, M.P.; Zhang, A.; Hopkins, J.E.; de Sarkar, N.; Coleman, I.; Gil da Costa, R.M.; Corey, E.; et al. Regulation of CEACAM5 and Therapeutic Efficacy of an Anti-CEACAM5-SN38 Antibody-Drug Conjugate in Neuroendocrine Prostate Cancer. Clin. Cancer Res. 2021, 27, 759–774. [Google Scholar] [CrossRef]
- Korsen, J.A.; Kalidindi, T.M.; Khitrov, S.; Samuels, Z.V.; Chakraborty, G.; Gutierrez, J.A.; Poirier, J.T.; Rudin, C.M.; Chen, Y.; Morris, M.J.; et al. Molecular Imaging of Neuroendocrine Prostate Cancer by Targeting Delta-Like Ligand 3. J. Nucl. Med. 2022, 63, 1401–1407. [Google Scholar] [CrossRef]
- Mckertish, C.M.; Kayser, V. Advances and Limitations of Antibody Drug Conjugates for Cancer. Biomedicines 2021, 9, 872. [Google Scholar] [CrossRef]
- Nejadmoghaddam, M.; Minai-tehrani, A.; Ghahremanzadeh, R.; Mahmoudi, M.; Dinarvand, R.; Zarnani, A.-H. Antibody-Drug Conjugates: Possibilities and Challenges. Avicenna J. Med. Biotechnol. 2019, 11, 3–23. [Google Scholar]
- Debnath, S.; Hao, G.; Guan, B.; Thapa, P.; Hao, J.; Hammers, H.; Sun, X. Theranostic Small-Molecule Prodrug Conjugates for Targeted Delivery and Controlled Release of Toll-like Receptor 7 Agonists. Int. J. Mol. Sci. 2022, 23, 7160. [Google Scholar] [CrossRef]
- Kumar, A.; Mastren, T.; Wang, B.; Hsieh, J.T.; Hao, G.; Sun, X. Design of a Small-Molecule Drug Conjugate for Prostate Cancer Targeted Theranostics. Bioconjug. Chem. 2016, 27, 1681–1689. [Google Scholar] [CrossRef] [PubMed]
- Bakht, M.K.; Derecichei, I.; Li, Y.; Ferraiuolo, R.M.; Dunning, M.; Oh, S.W.; Hussein, A.; Youn, H.; Stringer, K.F.; Jeong, C.W.; et al. Neuroendocrine Differentiation of Prostate Cancer Leads to PSMA Suppression. Endocr. Relat. Cancer 2019, 26, 131–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, B.; Zhou, N.; Wu, C.Y.; Li, S.; Chen, Y.A.; Debnath, S.; Hofstad, M.; Ma, S.; Raj, G.V.; He, D.; et al. Validation of Sv2a-Targeted Pet Imaging for Noninvasive Assessment of Neuroendocrine Differentiation in Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 13085. [Google Scholar] [CrossRef]
- Lee, C.; Chen, Y.; Hernandez, E.; Pong, R.; Ma, S.; Hofstad, M.; Kapur, P.; Zhau, H.; Chung, L.W.; Lai, C.; et al. The Central Role of Sphingosine Kinase 1 in the Development of Neuroendocrine Prostate Cancer (NEPC): A New Targeted Therapy of NEPC. Clin. Transl. Med. 2022, 12, 695. [Google Scholar] [CrossRef] [PubMed]
- Dayon, A.; Brizuela, L.; Martin, C.; Mazerolles, C.; Pirot, N.; Doumerc, N.; Nogueira, L.; Golzio, M.; Teissié, J.; Serre, G.; et al. Sphingosine Kinase-1 Is Central to Androgen-Regulated Prostate Cancer Growth and Survival. PLoS ONE 2009, 4, e8048. [Google Scholar] [CrossRef]
- Gupta, P.; Taiyab, A.; Hussain, A.; Alajmi, M.F.; Islam, A.; Hassan, M.I. Targeting the Sphingosine Kinase/Sphingosine-1-Phosphate Signaling Axis in Drug Discovery for Cancer Therapy. Cancers 2021, 13, 1898. [Google Scholar] [CrossRef]
- Han, H.; Lee, S.O.; Xu, Y.; Kim, J.E.; Lee, H.J. SPHK/HIF-1α Signaling Pathway Has a Critical Role in Chrysin-Induced Anticancer Activity in Hypoxia-Induced PC-3 Cells. Cells 2022, 11, 2787. [Google Scholar] [CrossRef]
- Behjati, M.; Etemadifar, M.; Abdar Esfahani, M. Cardiovascular Effects of Fingolimod: A Review Article. Iran. J. Neurol. 2014, 13, 119–126. [Google Scholar]
- Cohen, J.A.; Chun, J. Mechanisms of Fingolimod’s Efficacy and Adverse Effects in Multiple Sclerosis. Ann. Neurol. 2011, 69, 759–777. [Google Scholar] [CrossRef]
- Yao, J.; Liu, Y.; Liang, X.; Shao, J.; Zhang, Y.; Yang, J.; Zheng, M. Neuroendocrine Carcinoma as an Independent Prognostic Factor for Patients with Prostate Cancer: A Population-Based Study. Front. Endocrinol. 2021, 12, 778758. [Google Scholar] [CrossRef]
- Gofrit, O.N.; Frank, S.; Meirovitz, A.; Nechushtan, H.; Orevi, M. PET/CT with 68Ga-DOTA-TATE for Diagnosis of Neuroendocrine: Differentiation in Patients with Castrate-Resistant Prostate Cancer. Clin. Nucl. Med. 2017, 42, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Qie, Y.; Cheng, Z.; Wu, Z.; Qi, F.; Li, B.; Wu, S.; Chu, T.; Lu, Z.; Li, S.; Nie, G. Identification of Protein Kinase C Beta as a Therapeutic Target for Neuroendocrine Prostate Cancer and Development of a Nanoparticle-Based Therapeutic Strategy. Nano Today 2023, 48, 101705. [Google Scholar] [CrossRef]
- Banerjee, S.R.; Foss, C.A.; Castanares, M.; Mease, R.; Byun, Y.; Fox, J.J.; Hilton, J.; Lupold, S.E.; Kozikowski, A.P.; Pomper, M.G. Synthesis and Evaluation of Technetium-99m- and Rhenium- Labeled Inhibitors of the Prostate-Specific Membrane Antigen (PSMA). J. Med. Chem 2012, 23, 4504–4517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.K.; Chen, Y.; Kang, J.; Lisok, A.; Minn, I.; Pomper, M.G.; Boctor, E.M. Prostate-Specific Membrane Antigen-Targeted Photoacoustic Imaging of Prostate Cancer in Vivo. J. Biophotonics 2018, 11, e201800021. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Guan, B.; Nham, K.; Hao, G.; Sun, X.; Simanek, E.E. Tumor Uptake of Triazine Dendrimers Decorated with Four, Sixteen, and Sixty-Four PSMA-Targeted Ligands: Passive versus Active Tumor Targeting. Biomolecules 2019, 9, 421. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Lo, S.T.; Hill, S.; Pavan, G.M.; Sun, X.; Simanek, E.E. Antitumor Activity and Molecular Dynamics Simulations of Paclitaxel-Laden Triazine Dendrimers. Mol. Pharm. 2012, 9, 404–412. [Google Scholar] [CrossRef]
- Sharma, A.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Exploring the Orthogonal Chemoselectivity of 2,4,6-Trichloro-1,3,5-Triazine (TCT) as a Trifunctional Linker with Different Nucleophiles: Rules of the Game. Front. Chem. 2018, 6, 516. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a Strategy for Improving Nanoparticle-Based Drug and Gene Delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Turecek, P.L.; Bossard, M.J.; Schoetens, F.; Ivens, I.A. PEGylation of Biopharmaceuticals: A Review of Chemistry and Nonclinical Safety Information of Approved Drugs. J. Pharm. Sci. 2016, 105, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Ferdani, R.; Anderson, C.J. Preparation and Biological Evaluation of 64Cu Labeled Tyr 3-Octreotate Using a Phosphonic Acid-Based Cross-Bridged Macrocyclic Chelator. Bioconjug. Chem. 2012, 23, 1470–1477. [Google Scholar] [CrossRef]
- Staben, L.R.; Koenig, S.G.; Lehar, S.M.; Vandlen, R.; Zhang, D.; Chuh, J.; Yu, S.F.; Ng, C.; Guo, J.; Liu, Y.; et al. Targeted Drug Delivery through the Traceless Release of Tertiary and Heteroaryl Amines from Antibody-Drug Conjugates. Nat. Chem. 2016, 8, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Florento, L.; Matias, R.; Tuaño, E.; Santiago, K.; Cruz, F.D.; Tuazon, A. Comparison of Cytotoxic Activity of Anticancer Drugs against Various Human Tumor Cell Lines Using in Vitro Cell-Based Approach. Int. J. Biomed. Sci. 2012, 8, 76–80. [Google Scholar]
- Institute, N.C. Targeted Therapy to Treat Cancer. Available online: https://www.cancer.gov/about-cancer/treatment/types/targeted-therapies (accessed on 30 November 2022).
- Marsilje, T.H.; Pei, W.; Chen, B.; Lu, W.; Uno, T.; Jin, Y.; Jiang, T.; Kim, S.; Li, N.; Warmuth, M.; et al. Synthesis, Structure−Activity Relationships, and in Vivo Efficacy of the Novel Potent and Selective Anaplastic Lymphoma Kinase (ALK) Inhibitor 5-Chloro-N2-(2-Isopropoxy-5-Methyl-4-(Piperidin-4- Yl)Phenyl)-N4-(2-(Isopropylsulfonyl)Phenyl)Pyrimidine-2,4- diamine (LDK378) Currently in Phase 1 and Phase 2 Clinical Trials. J. Med. Chem. 2013, 56, 5675–5690. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.T.; Stern, S.; Clogston, J.D.; Zheng, J.; Adiseshaiah, P.P.; Dobrovolskaia, M.; Lim, J.; Patri, A.K.; Sun, X.; Simanek, E.E. Biological Assessment of Triazine Dendrimer: Toxicological Profiles, Solution Behavior, Biodistribution, Drug Release and Efficacy in a PEGylated, Paclitaxel Construct. Mol. Pharm. 2010, 7, 993–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puranik, A.D.; Dromain, C.; Fleshner, N.; Sathekge, M.; Pavel, M.; Eberhardt, N.; Zengerling, F.; Marienfeld, R.; Grunert, M.; Prasad, V. Target Heterogeneity in Oncology: The Best Predictor for Differential Response to Radioligand Therapy in Neuroendocrine Tumors and Prostate Cancer. Cancers 2021, 13, 3607. [Google Scholar] [CrossRef]
- Abdelsalam, S.S.; Korashy, H.M.; Zeidan, A.; Agouni, A. The Role of Protein Tyrosine Phosphatase (PTP)-1B in Cardiovascular Disease and Its Interplay with Insulin Resistance. Biomolecules 2019, 9, 286. [Google Scholar] [CrossRef]
- He, R.J.; Yu, Z.H.; Zhang, R.Y.; Zhang, Z.Y. Protein Tyrosine Phosphatases as Potential Therapeutic Targets. Acta Pharmacol. Sin. 2014, 35, 1227–1246. [Google Scholar] [CrossRef]
- Usmani, S.; Ahmed, N.; Marafi, F.; Rasheed, R.; Amanguno, H.G.; Al Kandari, F. Molecular Imaging in Neuroendocrine Differentiation of Prostate Cancer. Clin. Nucl. Med. 2017, 42, 410–413. [Google Scholar] [CrossRef]
- Patel, G.K.; Chugh, N.; Tripathi, M. Neuroendocrine Differentiation of Prostate Cancer—An Intriguing Example of Tumor Evolution at Play. Cancers 2019, 11, 1405. [Google Scholar] [CrossRef]
- Pchejetski, D.; Bohler, T.; Brizuela, L.; Sauer, L.; Doumerc, N.; Golzio, M.; Salunkhe, V.; Teissié, J.; Malavaud, B.; Waxman, J.; et al. FTY720 (Fingolimod) Sensitizes Prostate Cancer Cells to Radiotherapy by Inhibition of Sphingosine Kinase-1. Cancer Res. 2010, 70, 8651–8661. [Google Scholar] [CrossRef]
- Mansi, R.; Abid, K.; Nicolas, G.P.; Del Pozzo, L.; Grouzmann, E.; Fani, M. A New 68Ga-Labeled Somatostatin Analog Containing Two Iodo-Amino Acids for Dual Somatostatin Receptor Subtype 2 and 5 Targeting. EJNMMI Res. 2020, 10, 90. [Google Scholar] [CrossRef] [PubMed]
- Riga, S.; Cicoria, G.; Pancaldi, D.; Zagni, F.; Vichi, S.; Dassenno, M.; Mora, L.; Lodi, F.; Morigi, M.P.; Marengo, M. Production of Ga-68 with a General Electric PETtrace Cyclotron by Liquid Target. Phys. Medica 2018, 55, 116–126. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez, P.; Debnath, S.; Chen, Y.-A.; Hernandez, E.; Jha, P.; Dakanali, M.; Hsieh, J.-T.; Sun, X. A Theranostic Small-Molecule Prodrug Conjugate for Neuroendocrine Prostate Cancer. Pharmaceutics 2023, 15, 481. https://doi.org/10.3390/pharmaceutics15020481
Gonzalez P, Debnath S, Chen Y-A, Hernandez E, Jha P, Dakanali M, Hsieh J-T, Sun X. A Theranostic Small-Molecule Prodrug Conjugate for Neuroendocrine Prostate Cancer. Pharmaceutics. 2023; 15(2):481. https://doi.org/10.3390/pharmaceutics15020481
Chicago/Turabian StyleGonzalez, Paulina, Sashi Debnath, Yu-An Chen, Elizabeth Hernandez, Preeti Jha, Marianna Dakanali, Jer-Tsong Hsieh, and Xiankai Sun. 2023. "A Theranostic Small-Molecule Prodrug Conjugate for Neuroendocrine Prostate Cancer" Pharmaceutics 15, no. 2: 481. https://doi.org/10.3390/pharmaceutics15020481
APA StyleGonzalez, P., Debnath, S., Chen, Y. -A., Hernandez, E., Jha, P., Dakanali, M., Hsieh, J. -T., & Sun, X. (2023). A Theranostic Small-Molecule Prodrug Conjugate for Neuroendocrine Prostate Cancer. Pharmaceutics, 15(2), 481. https://doi.org/10.3390/pharmaceutics15020481