
Interplay of Hydropathy and Heterogeneous Diffusion in the Molecular Transport within Lamellar Lipid Mesophases


Abstract
:1. Introduction
2. Materials and Methods
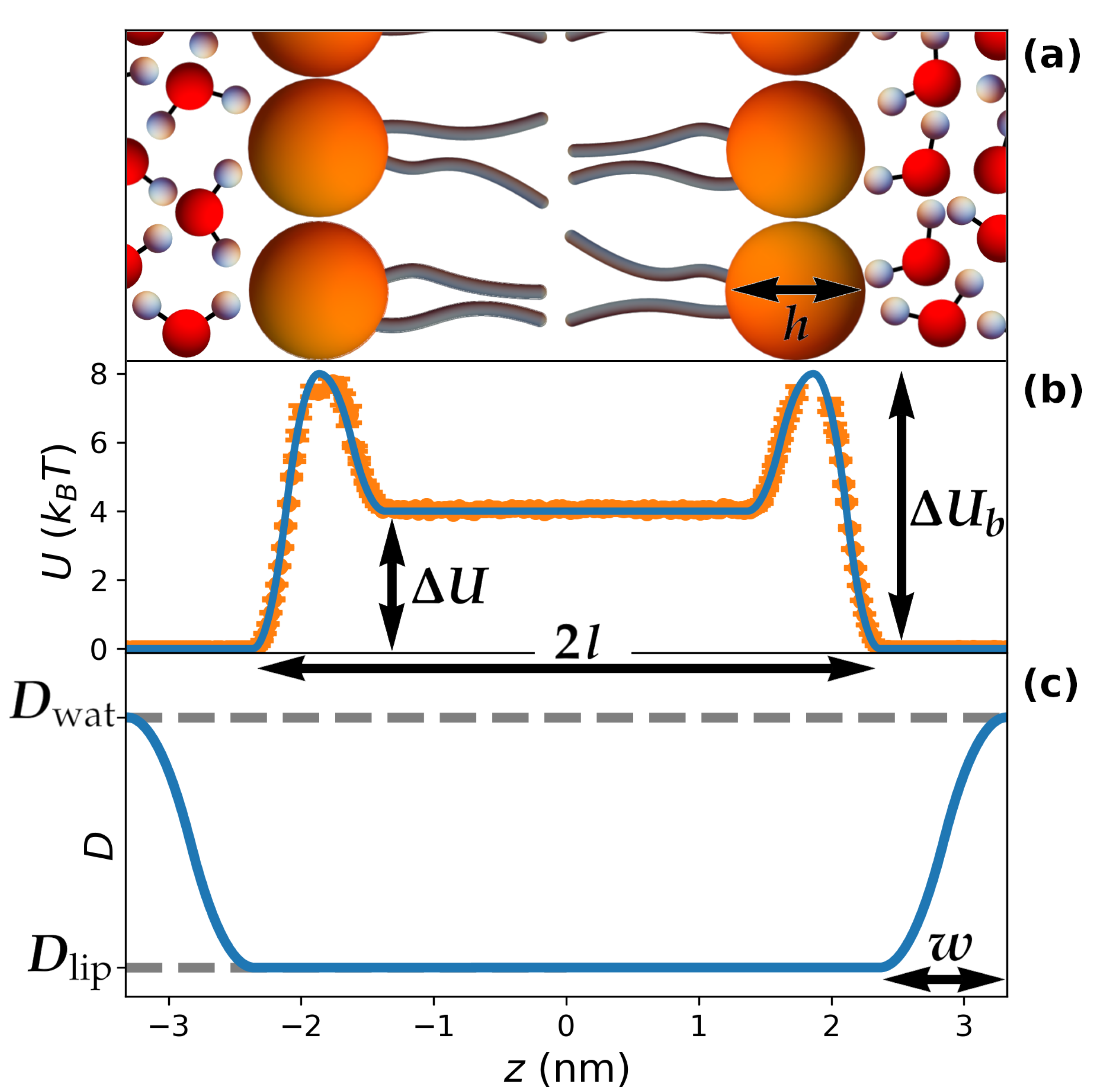
2.1. Model
2.2. Brownian Dynamics
2.3. Relationship between and
2.4. Amino Acids Simulations
3. Results
3.1. Assessing the Importance of Each Physical Ingredient
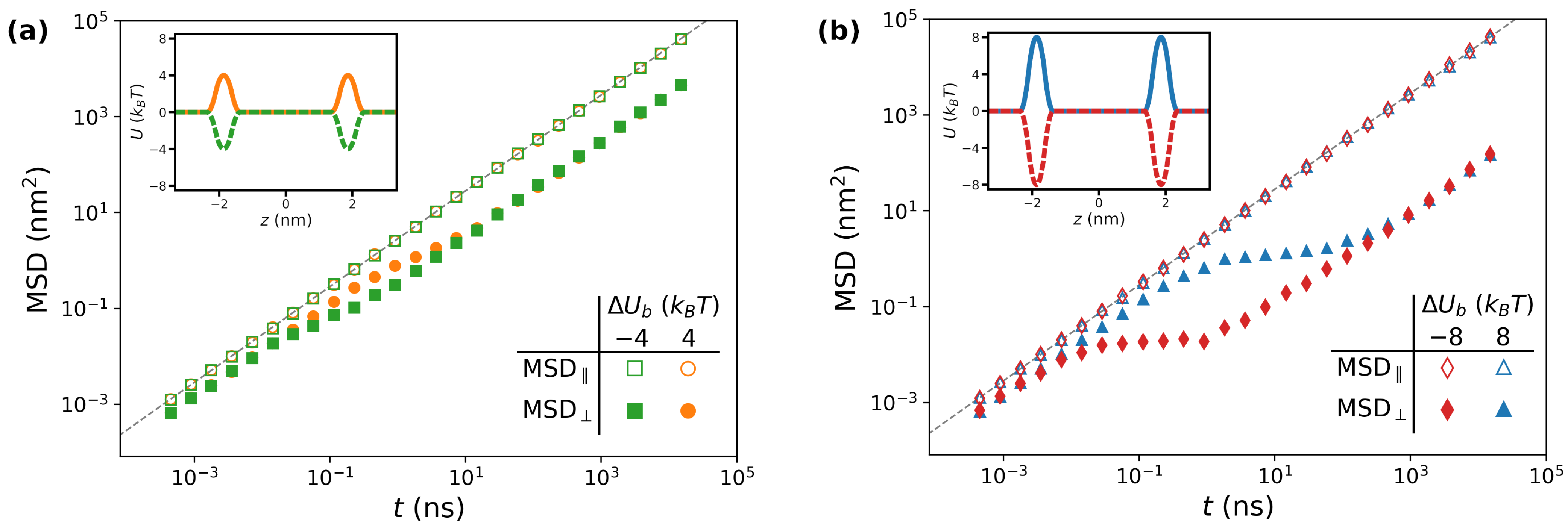
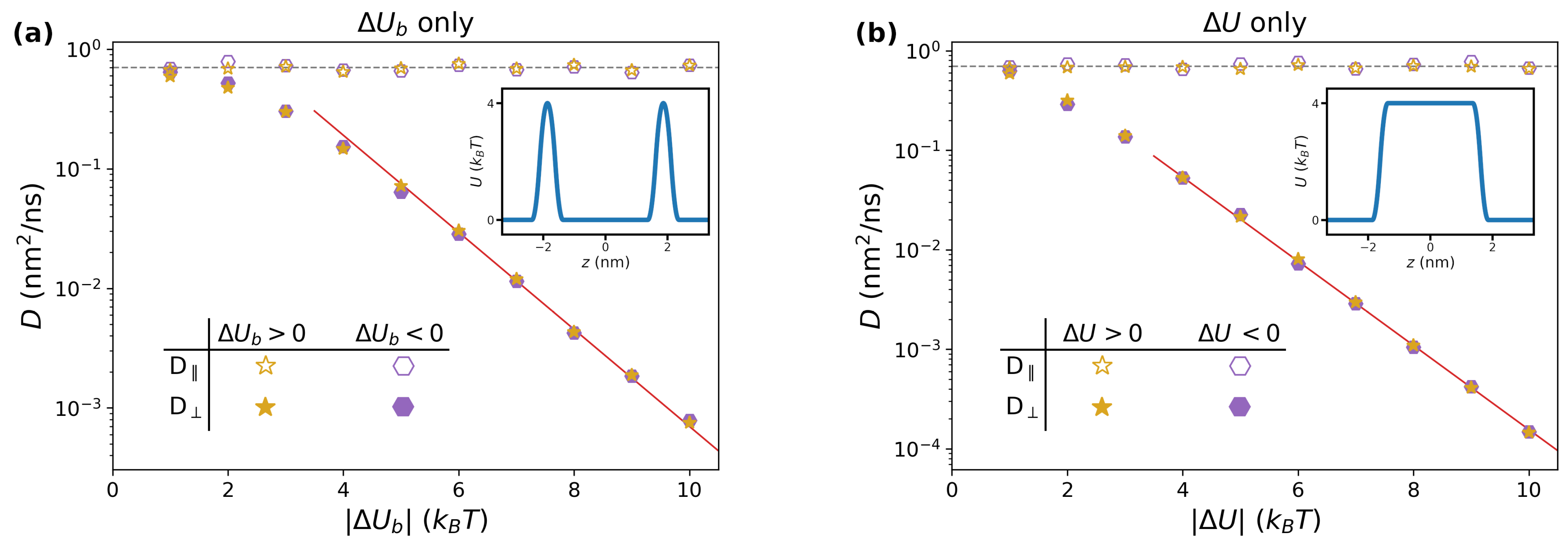
3.1.1. Impact of
3.1.2. Impact of
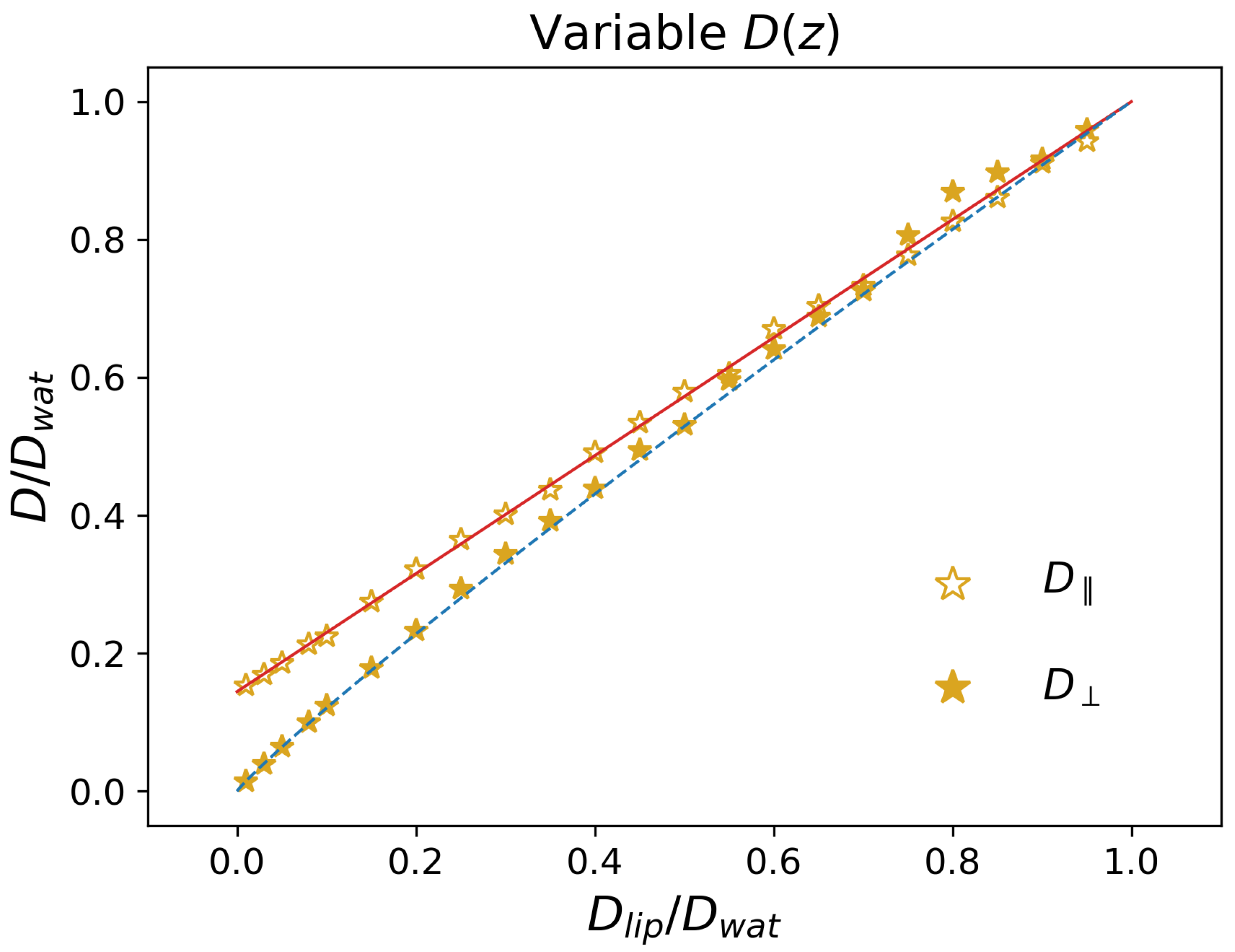
3.1.3. Impact of
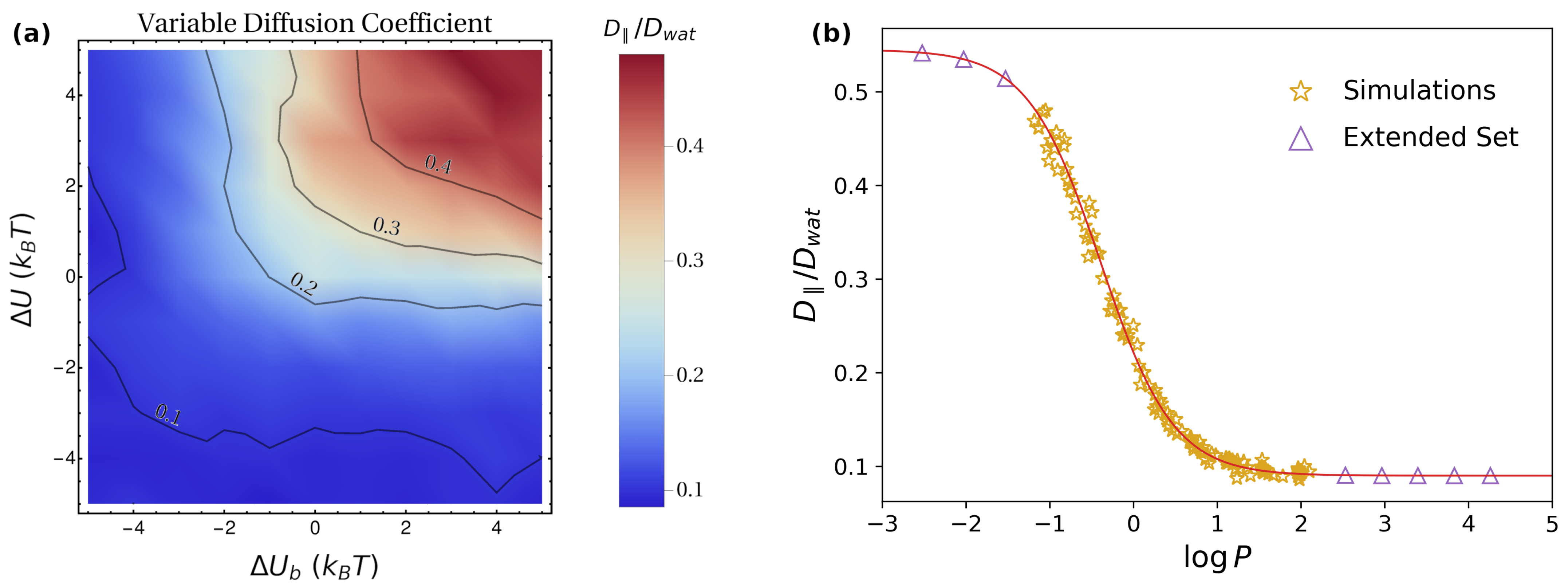
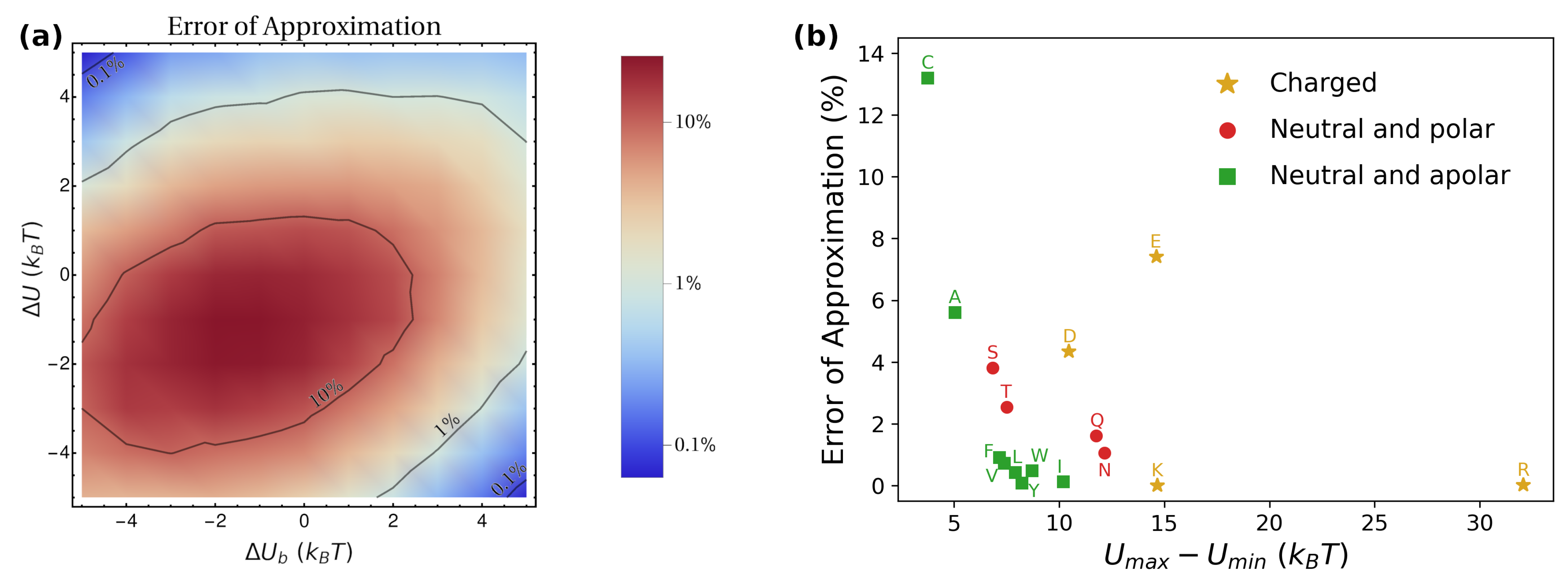
3.2. Putting the Physical Ingredients Together
3.3. Application: Large-Scale Transport of Amino Acids
4. Discussion
- The lower boundary of is equal to , obtained for highly-hydrophobic guest molecules. Together with point 1, this indicates that, for large enough barriers, parallel diffusion is dominant.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Briggs, J.; Chung, H.; Caffrey, M. The temperature-composition phase diagram and mesophase structure characterization of the monoolein/water system. J. Phys. II 1996, 6, 723–751. [Google Scholar]
- Templer, R.; Seddon, J.; Warrender, N.; Syrykh, A.; Huang, Z.; Winter, R.; Erbes, J. Inverse bicontinuous cubic phases in 2: 1 fatty acid/phosphatidylcholine mixtures. The effects of chain length, hydration, and temperature. J. Phys. Chem. B 1998, 102, 7251–7261. [Google Scholar]
- Tyler, A.I.; Barriga, H.M.; Parsons, E.S.; McCarthy, N.L.; Ces, O.; Law, R.V.; Seddon, J.M.; Brooks, N.J. Electrostatic swelling of bicontinuous cubic lipid phases. Soft Matter 2015, 11, 3279–3286. [Google Scholar] [PubMed]
- Mezzenga, R.; Meyer, C.; Servais, C.; Romoscanu, A.I.; Sagalowicz, L.; Hayward, R.C. Shear rheology of lyotropic liquid crystals: A case study. Langmuir 2005, 21, 3322–3333. [Google Scholar]
- Negrini, R.; Mezzenga, R. Diffusion, molecular separation, and drug delivery from lipid mesophases with tunable water channels. Langmuir 2012, 28, 16455–16462. [Google Scholar] [PubMed]
- Barauskas, J.; Johnsson, M.; Tiberg, F. Self-assembled lipid superstructures: Beyond vesicles and liposomes. Nano Lett. 2005, 5, 1615–1619. [Google Scholar]
- Turner, D.R.; Wang, Z.G.; Gruner, S.; Mannock, D.; Mcelhaney, R. Structural study of the inverted cubic phases of di-dodecyl alkyl-β-D-glucopyranosyl-rac-glycerol. J. Phys. II 1992, 2, 2039–2063. [Google Scholar] [CrossRef]
- Salvati Manni, L.; Assenza, S.; Duss, M.; Vallooran, J.J.; Juranyi, F.; Jurt, S.; Zerbe, O.; Landau, E.M.; Mezzenga, R. Soft biomimetic nanoconfinement promotes amorphous water over ice. Nat. Nanotechnol. 2019, 14, 609–615. [Google Scholar]
- Landau, E.M.; Rosenbusch, J.P. Lipidic cubic phases: A novel concept for the crystallization of membrane proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 14532–14535. [Google Scholar]
- Mulet, X.; Boyd, B.J.; Drummond, C.J. Advances in drug delivery and medical imaging using colloidal lyotropic liquid crystalline dispersions. J. Colloid Interface Sci. 2013, 393, 1–20. [Google Scholar]
- Mezzenga, R.; Schurtenberger, P.; Burbidge, A.; Michel, M. Understanding foods as soft materials. Nat. Mater. 2005, 4, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Fong, C.; Le, T.; Drummond, C.J. Lyotropic liquid crystal engineering–ordered nanostructured small molecule amphiphile self-assembly materials by design. Chem. Soc. Rev. 2012, 41, 1297–1322. [Google Scholar] [PubMed]
- Speziale, C.; Salvati Manni, L.; Manatschal, C.; Landau, E.M.; Mezzenga, R. A macroscopic H+ and Cl− ions pump via reconstitution of EcClC membrane proteins in lipidic cubic mesophases. Proc. Natl. Acad. Sci. USA 2016, 113, 7491–7496. [Google Scholar] [PubMed]
- Assenza, S.; Mezzenga, R. Soft condensed matter physics of foods and macronutrients. Nat. Rev. Phys. 2019, 1, 551–566. [Google Scholar] [CrossRef]
- Yao, Y.; Zhou, T.; Färber, R.; Grossner, U.; Floudas, G.; Mezzenga, R. Designing cryo-enzymatic reactions in subzero liquid water by lipidic mesophase nanoconfinement. Nat. Nanotechnol. 2021, 16, 802–810. [Google Scholar]
- Boyd, B.J.; Clulow, A.J. The influence of lipid digestion on the fate of orally administered drug delivery vehicles. Biochem. Soc. Trans. 2021, 49, 1749–1761. [Google Scholar]
- Negrini, R.; Mezzenga, R. pH-responsive lyotropic liquid crystals for controlled drug delivery. Langmuir 2011, 27, 5296–5303. [Google Scholar] [CrossRef]
- Martiel, I.; Baumann, N.; Vallooran, J.J.; Bergfreund, J.; Sagalowicz, L.; Mezzenga, R. Oil and drug control the release rate from lyotropic liquid crystals. J. Control. Release 2015, 204, 78–84. [Google Scholar]
- Zabara, A.; Mezzenga, R. Controlling molecular transport and sustained drug release in lipid-based liquid crystalline mesophases. J. Control. Release 2014, 188, 31–43. [Google Scholar]
- Negrini, R.; Fong, W.K.; Boyd, B.J.; Mezzenga, R. pH-responsive lyotropic liquid crystals and their potential therapeutic role in cancer treatment. Chem. Commun. 2015, 51, 6671–6674. [Google Scholar] [CrossRef]
- Nazaruk, E.; Miszta, P.; Filipek, S.; Gorecka, E.; Landau, E.M.; Bilewicz, R. Lyotropic cubic phases for drug delivery: Diffusion and sustained release from the mesophase evaluated by electrochemical methods. Langmuir 2015, 31, 12753–12761. [Google Scholar] [CrossRef] [PubMed]
- Clogston, J.; Craciun, G.; Hart, D.; Caffrey, M. Controlling release from the lipidic cubic phase by selective alkylation. J. Control. Release 2005, 102, 441–461. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Nguyen, T.H.; Hanley, T.; Boyd, B.J. Nanostructure of liquid crystalline matrix determines in vitro sustained release and in vivo oral absorption kinetics for hydrophilic model drugs. Int. J. Pharm. 2009, 365, 190–199. [Google Scholar] [PubMed]
- Phan, S.; Fong, W.K.; Kirby, N.; Hanley, T.; Boyd, B.J. Evaluating the link between self-assembled mesophase structure and drug release. Int. J. Pharm. 2011, 421, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Zabara, A.; Negrini, R.; Onaca-Fischer, O.; Mezzenga, R. Perforated Bicontinuous Cubic Phases with pH-Responsive Topological Channel Interconnectivity. Small 2013, 9, 3602–3609. [Google Scholar] [CrossRef]
- Meikle, T.G.; Yao, S.; Zabara, A.; Conn, C.E.; Drummond, C.J.; Separovic, F. Predicting the release profile of small molecules from within the ordered nanostructured lipidic bicontinuous cubic phase using translational diffusion coefficients determined by PFG-NMR. Nanoscale 2017, 9, 2471–2478. [Google Scholar]
- Ghanbari, R.; Assenza, S.; Saha, A.; Mezzenga, R. Diffusion of polymers through periodic networks of lipid-based nanochannels. Langmuir 2017, 33, 3491–3498. [Google Scholar]
- Assenza, S.; Mezzenga, R. Curvature and bottlenecks control molecular transport in inverse bicontinuous cubic phases. J. Chem. Phys. 2018, 148, 054902. [Google Scholar] [CrossRef]
- Ghanbari, R.; Assenza, S.; Mezzenga, R. The interplay of channel geometry and molecular features determines diffusion in lipidic cubic phases. J. Chem. Phys. 2019, 150, 094901. [Google Scholar]
- Ghanbari, R.; Assenza, S.; Zueblin, P.; Mezzenga, R. Impact of Molecular Partitioning and Partial Equilibration on the Estimation of Diffusion Coefficients from Release Experiments. Langmuir 2019, 35, 5663–5671. [Google Scholar] [CrossRef]
- Salvati Manni, L.; Duss, M.; Assenza, S.; Boyd, B.J.; Landau, E.M.; Fong, W.K. Enzymatic hydrolysis of monoacylglycerols and their cyclopropanated derivatives: Molecular structure and nanostructure determine the rate of digestion. J. Colloid Interface Sci. 2021, 588, 767–775. [Google Scholar] [PubMed]
- Chang, C.; Meikle, T.G.; Drummond, C.J.; Yang, Y.; Conn, C.E. Comparison of cubosomes and liposomes for the encapsulation and delivery of curcumin. Soft Matter 2021, 17, 3306–3313. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, S.; Zhai, J.; Drummond, C.J.; Tran, N. Novel pH-Responsive Cubosome and Hexosome Lipid Nanocarriers of SN-38 Are Prospective for Cancer Therapy. Pharmaceutics 2022, 14, 2175. [Google Scholar] [CrossRef] [PubMed]
- Dubbeldam, D.; Snurr, R. Recent developments in the molecular modeling of diffusion in nanoporous materials. Mol. Simul. 2007, 33, 305–325. [Google Scholar] [CrossRef]
- Krishna, R. Diffusion in porous crystalline materials. Chem. Soc. Rev. 2012, 41, 3099–3118. [Google Scholar] [PubMed]
- Bagchi, B. Water dynamics in the hydration layer around proteins and micelles. Chem. Rev. 2005, 105, 3197–3219. [Google Scholar] [PubMed]
- Laage, D.; Elsaesser, T.; Hynes, J.T. Water dynamics in the hydration shells of biomolecules. Chem. Rev. 2017, 117, 10694–10725. [Google Scholar]
- Bourg, I.C.; Steefel, C.I. Molecular dynamics simulations of water structure and diffusion in silica nanopores. J. Phys. Chem. C 2012, 116, 11556–11564. [Google Scholar]
- Hande, V.R.; Chakrabarty, S. How Far Is “Bulk Water” from Interfaces? Depends on the Nature of the Surface and What We Measure. J. Phys. Chem. B 2022, 126, 1125–1135. [Google Scholar]
- Vallooran, J.J.; Assenza, S.; Mezzenga, R. Spatiotemporal control of enzyme-induced crystallization under lyotropic liquid crystal nanoconfinement. Angew. Chem. 2019, 131, 7367–7371. [Google Scholar]
- Kasim, N.A.; Whitehouse, M.; Ramachandran, C.; Bermejo, M.; Lennernäs, H.; Hussain, A.S.; Junginger, H.E.; Stavchansky, S.A.; Midha, K.K.; Shah, V.P.; et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol. Pharm. 2004, 1, 85–96. [Google Scholar] [PubMed]
- Takagi, T.; Ramachandran, C.; Bermejo, M.; Yamashita, S.; Yu, L.X.; Amidon, G.L. A provisional biopharmaceutical classification of the top 200 oral drug products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 2006, 3, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Eason, T.; Ramirez, G.; Clulow, A.J.; Salim, M.; Boyd, B.J. Revisiting the Dissolution of Praziquantel in Biorelevant Media and the Impact of Digestion of Milk on Drug Dissolution. Pharmaceutics 2022, 14, 2228. [Google Scholar] [PubMed]
- Kim, J.; Lu, W.; Qiu, W.; Wang, L.; Caffrey, M.; Zhong, D. Ultrafast hydration dynamics in the lipidic cubic phase: Discrete water structures in nanochannels. J. Phys. Chem. B 2006, 110, 21994–22000. [Google Scholar] [CrossRef]
- Helfrich, W. Effect of thermal undulations on the rigidity of fluid membranes and interfaces. J. Phys. 1985, 46, 1263–1268. [Google Scholar] [CrossRef]
- Angelov, B.; Ollivon, M.; Angelova, A. X-ray diffraction study of the effect of the detergent octyl glucoside on the structure of lamellar and nonlamellar lipid/water phases of use for membrane protein reconstitution. Langmuir 1999, 15, 8225–8234. [Google Scholar]
- Nademi, Y.; Amjad Iranagh, S.; Yousefpour, A.; Mousavi, S.Z.; Modarress, H. Molecular dynamics simulations and free energy profile of Paracetamol in DPPC and DMPC lipid bilayers. J. Chem. Sci. 2014, 126, 637–647. [Google Scholar]
- Zhou, T.; Vallooran, J.J.; Assenza, S.; Szekrenyi, A.; Clapés, P.; Mezzenga, R. Efficient asymmetric synthesis of carbohydrates by aldolase nano-confined in lipidic cubic mesophases. ACS Catal. 2018, 8, 5810–5815. [Google Scholar]
- Ma, Y.; Zhu, C.; Ma, P.; Yu, K. Studies on the diffusion coefficients of amino acids in aqueous solutions. J. Chem. Eng. Data 2005, 50, 1192–1196. [Google Scholar]
- Longsworth, L. Diffusion measurements, at 25, of aqueous solutions of amino acids, peptides and sugars. J. Am. Chem. Soc. 1953, 75, 5705–5709. [Google Scholar]
- Ribeiro, A.C.; Barros, M.C.; Verissimo, L.M.; Lobo, V.M.; Valente, A.J. Binary diffusion coefficients for aqueous solutions of l-aspartic acid and its respective monosodium salt. J. Solut. Chem. 2014, 43, 83–92. [Google Scholar] [CrossRef]
- Ribeiro, A.C.; Rodrigo, M.; Barros, M.C.; Verissimo, L.M.; Romero, C.; Valente, A.J.; Esteso, M.A. Mutual diffusion coefficients of L-glutamic acid and monosodium L-glutamate in aqueous solutions at T = 298.15 K. J. Chem. Thermodyn. 2014, 74, 133–137. [Google Scholar]
- Umecky, T.; Ehara, K.; Omori, S.; Kuga, T.; Yui, K.; Funazukuri, T. Binary diffusion coefficients of aqueous phenylalanine, tyrosine isomers, and aminobutyric acids at infinitesimal concentration and temperatures from (293.2 to 333.2) K. J. Chem. Eng. Data 2013, 58, 1909–1917. [Google Scholar]
- Mendes, F.S.; Cruz, C.E.; Martins, R.N.; Ramalho, J.P.P.; Martins, L.F. On the diffusion of ketoprofen and ibuprofen in water: An experimental and theoretical approach. J. Chem. Thermodyn. 2022, 2022, 106955. [Google Scholar] [CrossRef]
- Edwards, L. The dissolution and diffusion of aspirin in aqueous media. Trans. Faraday Soc. 1951, 47, 1191–1210. [Google Scholar] [CrossRef]
- Ribeiro, A.C.; Barros, M.C.; Veríssimo, L.M.; Santos, C.I.; Cabral, A.M.; Gaspar, G.D.; Esteso, M.A. Diffusion coefficients of paracetamol in aqueous solutions. J. Chem. Thermodyn. 2012, 54, 97–99. [Google Scholar] [CrossRef]
- Antognini, L.M.; Assenza, S.; Speziale, C.; Mezzenga, R. Quantifying the transport properties of lipid mesophases by theoretical modelling of diffusion experiments. J. Chem. Phys. 2016, 145, 084903. [Google Scholar]
- Anderson, D.M.; Wennerstroem, H. Self-diffusion in bicontinuous cubic phases, L3 phases, and microemulsions. J. Phys. Chem. 1990, 94, 8683–8694. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F.; Edwards, S.F. The Theory of Polymer Dynamics; Oxford University Press: Oxford, UK, 1988; Volume 73. [Google Scholar]
- Van Kampen, N.G. Stochastic Processes in Physics and Chemistry; Elsevier: Amsterdam, The Netherlands, 1992; Volume 1. [Google Scholar]
- Marsaglia, G.; Tsang, W.W. The ziggurat method for generating random variables. J. Stat. Softw. 2000, 5, 1–7. [Google Scholar] [CrossRef]
- Martin, Y.C. Quantitative Drug Design: A Critical Introduction; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- MacCallum, J.L.; Bennett, W.D.; Tieleman, D.P. Distribution of amino acids in a lipid bilayer from computer simulations. Biophys. J. 2008, 94, 3393–3404. [Google Scholar] [CrossRef]
- Yue, Z.; Li, C.; Voth, G.A.; Swanson, J.M. Dynamic protonation dramatically affects the membrane permeability of drug-like molecules. J. Am. Chem. Soc. 2019, 141, 13421–13433. [Google Scholar] [PubMed]
- Koirala, R.P.; Bhusal, H.P.; Khanal, S.P.; Adhikari, N.P. Effect of temperature on transport properties of cysteine in water. AIP Adv. 2020, 10, 025122. [Google Scholar]
- Rodrigo, M.M.; Valente, A.J.; Barros, M.C.; Verissimo, L.M.; Romero, C.; Esteso, M.A.; Ribeiro, A.C. Mutual diffusion coefficients of L-lysine in aqueous solutions. J. Chem. Thermodyn. 2014, 74, 227–230. [Google Scholar] [CrossRef]
- Heller, W.T. Small-Angle Neutron Scattering for Studying Lipid Bilayer Membranes. Biomolecules 2022, 12, 1591. [Google Scholar] [PubMed]
- Johansson, A.C.; Lindahl, E. Position-resolved free energy of solvation for amino acids in lipid membranes from molecular dynamics simulations. Proteins Struct. Funct. Bioinform. 2008, 70, 1332–1344. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Hummer, G. Theory and simulation of ion conduction in the pentameric GLIC channel. J. Chem. Theory Comput. 2012, 8, 3759–3768. [Google Scholar] [CrossRef] [PubMed]
- Vallooran, J.J.; Bolisetty, S.; Mezzenga, R. Macroscopic alignment of lyotropic liquid crystals using magnetic nanoparticles. Adv. Mater. 2011, 23, 3932–3937. [Google Scholar] [PubMed]
- Boggara, M.B.; Krishnamoorti, R. Partitioning of nonsteroidal antiinflammatory drugs in lipid membranes: A molecular dynamics simulation study. Biophys. J. 2010, 98, 586–595. [Google Scholar] [PubMed]
- Keyvanfard, M.; Hatami, M.; Gupta, V.K.; Agarwal, S.; Sadeghifar, H.; Khalilzadeh, M.A. Liquid phase analysis of methyldopa in the presence of tyrosine using electrocatalytic effect of a catechol derivative at a surface of NiO nanoparticle modified carbon paste electrode. J. Mol. Liq. 2017, 230, 290–294. [Google Scholar] [CrossRef]
- Fagerholm, U.; Lennernäs, H. Experimental estimation of the effective unstirred water layer thickness in the human jejunum, and its importance in oral drug absorption. Eur. J. Pharm. Sci. 1995, 3, 247–253. [Google Scholar] [CrossRef]
- Lu, Y.; Li, M. Simultaneous rapid determination of the solubility and diffusion coefficients of a poorly water-soluble drug based on a novel UV imaging system. J. Pharm. Sci. 2016, 105, 131–138. [Google Scholar] [PubMed]
- Vilt, M.E.; Ho, W.W. Supported liquid membranes with strip dispersion for the recovery of Cephalexin. J. Membr. Sci. 2009, 342, 80–87. [Google Scholar] [CrossRef]
- Tajik, S.; Beitollahi, H.; Aflatoonian, M.R.; Mohtat, B.; Aflatoonian, B.; Shoaie, I.S.; Khalilzadeh, M.A.; Ziasistani, M.; Zhang, K.; Jang, H.W.; et al. Fabrication of magnetic iron oxide-supported copper oxide nanoparticles (Fe3O4/CuO): Modified screen-printed electrode for electrochemical studies and detection of desipramine. RSC Adv. 2020, 10, 15171–15178. [Google Scholar] [CrossRef]
- Beitollahi, H.; Hamzavi, M.; Torkzadeh-Mahani, M. Electrochemical determination of hydrochlorothiazide and folic acid in real samples using a modified graphene oxide sheet paste electrode. Mater. Sci. Eng. C 2015, 52, 297–305. [Google Scholar] [CrossRef]
- Pyka, A. Lipophilicity investigations of ibuprofen. J. Liquid Chromatogr. Related Technol.® 2009, 32, 723–731. [Google Scholar] [CrossRef]
- Barros, M.C.; Ribeiro, A.C.; Esteso, M.A.; Lobo, V.M.; Leaist, D.G. Diffusion of levodopa in aqueous solutions of hydrochloric acid at 25 °C. J. Chem. Thermodyn. 2014, 72, 44–47. [Google Scholar] [CrossRef]
- Ye, F.; Yaghmur, A.; Jensen, H.; Larsen, S.W.; Larsen, C.; Østergaard, J. Real-time UV imaging of drug diffusion and release from Pluronic F127 hydrogels. Eur. J. Pharm. Sci. 2011, 43, 236–243. [Google Scholar]
- Radak, B.K.; Chipot, C.; Suh, D.; Jo, S.; Jiang, W.; Phillips, J.C.; Schulten, K.; Roux, B. Constant-pH molecular dynamics simulations for large biomolecular systems. J. Chem. Theory Comput. 2017, 13, 5933–5944. [Google Scholar] [CrossRef]
- Johansson, A.C.; Lindahl, E. Titratable amino acid solvation in lipid membranes as a function of protonation state. J. Phys. Chem. B 2009, 113, 245–253. [Google Scholar]
- Enkavi, G.; Javanainen, M.; Kulig, W.; Róg, T.; Vattulainen, I. Multiscale simulations of biological membranes: The challenge to understand biological phenomena in a living substance. Chem. Rev. 2019, 119, 5607–5774. [Google Scholar]
- Prajapati, J.D.; Kleinekathöfer, U.; Winterhalter, M. How to enter a bacterium: Bacterial porins and the permeation of antibiotics. Chem. Rev. 2021, 121, 5158–5192. [Google Scholar] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | (nm2/ns) | (nm2/ns) | |
|---|---|---|---|
| Cephalexin | −0.67 | 0.70 | 0.27 |
| Hydrochlorothiazide | −0.15 | 1.69 | 0.43 |
| Levodopa | 0.00 | 0.95 | 0.21 |
| Piroxicam | 0.29 | 0.85 | 0.14 |
| Methyldopa | 0.39 | 1.14 | 0.18 |
| Paracetamol | 0.46 | 1.06 | 0.15 |
| Antipyrine | 1.01 | 1.04 | 0.11 |
| Carbamazepine | 2.93 | 1.13 | 0.10 |
| Ketoprofen | 3.31 | 0.67 | 0.06 |
| Desipramine | 3.94 | 0.46 | 0.04 |
| Ibuprofen | 3.99 | 0.77 | 0.07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bosch, A.M.; Assenza, S. Interplay of Hydropathy and Heterogeneous Diffusion in the Molecular Transport within Lamellar Lipid Mesophases. Pharmaceutics 2023, 15, 573. https://doi.org/10.3390/pharmaceutics15020573
Bosch AM, Assenza S. Interplay of Hydropathy and Heterogeneous Diffusion in the Molecular Transport within Lamellar Lipid Mesophases. Pharmaceutics. 2023; 15(2):573. https://doi.org/10.3390/pharmaceutics15020573
Chicago/Turabian StyleBosch, Antonio M., and Salvatore Assenza. 2023. "Interplay of Hydropathy and Heterogeneous Diffusion in the Molecular Transport within Lamellar Lipid Mesophases" Pharmaceutics 15, no. 2: 573. https://doi.org/10.3390/pharmaceutics15020573
APA StyleBosch, A. M., & Assenza, S. (2023). Interplay of Hydropathy and Heterogeneous Diffusion in the Molecular Transport within Lamellar Lipid Mesophases. Pharmaceutics, 15(2), 573. https://doi.org/10.3390/pharmaceutics15020573