
Influence of the Topology of Poly(L-Cysteine) on the Self-Assembly, Encapsulation and Release Profile of Doxorubicin on Dual-Responsive Hybrid Polypeptides


 ,
,  ,
, 
Abstract
:1. Introduction
2. Experiment
2.1. Materials and Methods
2.1.1. Materials
2.1.2. NMR Spectroscopy
2.1.3. FT-IR Spectroscopy
2.1.4. Size Exclusion Chromatography
2.1.5. Circular Dichroism
2.1.6. UV Spectroscopy
2.1.7. Dynamic Light Scattering
2.1.8. Static Light Scattering
2.1.9. Electrophoretic Mobility
2.1.10. Transmission Electron Microscopy
2.1.11. Cell Culture
2.2. Synthesis of the Monomers
2.2.1. Synthesis of Nim-Trityl-l-Histidine N-Carboxy Anhydride (Nim-Trityl-l-His-NCA)
2.2.2. Synthesis of S-tert-Butyl-mercapto-l-Cysteine N-Carboxy Anhydride (tBM-l-Cys-NCA)
2.2.3. Synthesis of Sarcosine N-Carboxy Anhydride (Sar-NCA)
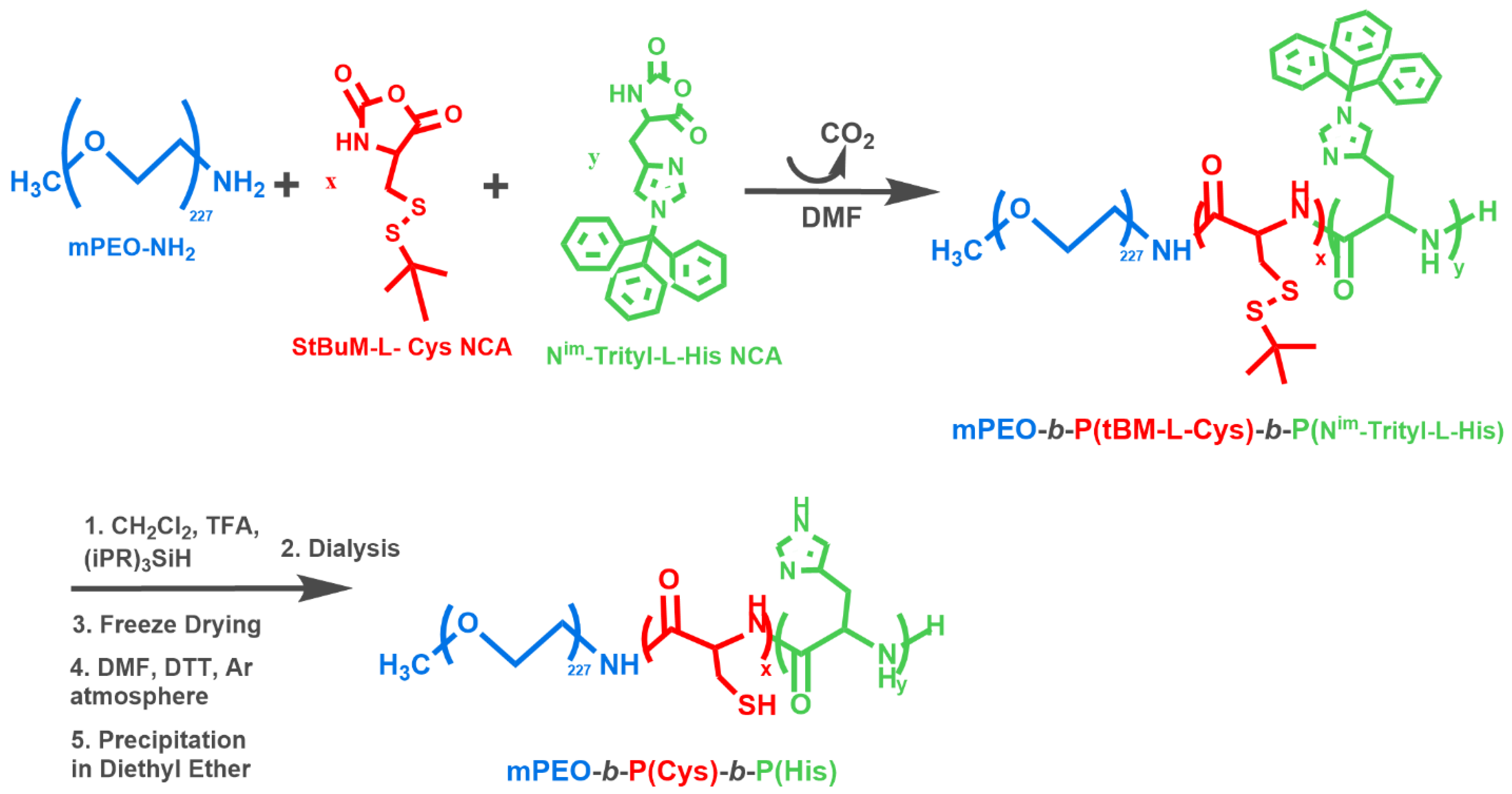
2.3. Synthesis of the Hybrid-Polypeptides
2.4. Self-Assembly of Empty NPs via Solvent Switch Method
2.5. Loading of Anticancer DOX
2.6. In Vitro Drug Release Studies
2.7. In Vitro Cytotoxic Activity: Sulforhodamine B (SRB) Assay
3. Results and Discussion
3.1. Synthesis and Characterization of the N-Carboxy Anhydrides (NCAs)
3.2. Synthesis and Characterization of the Polymers
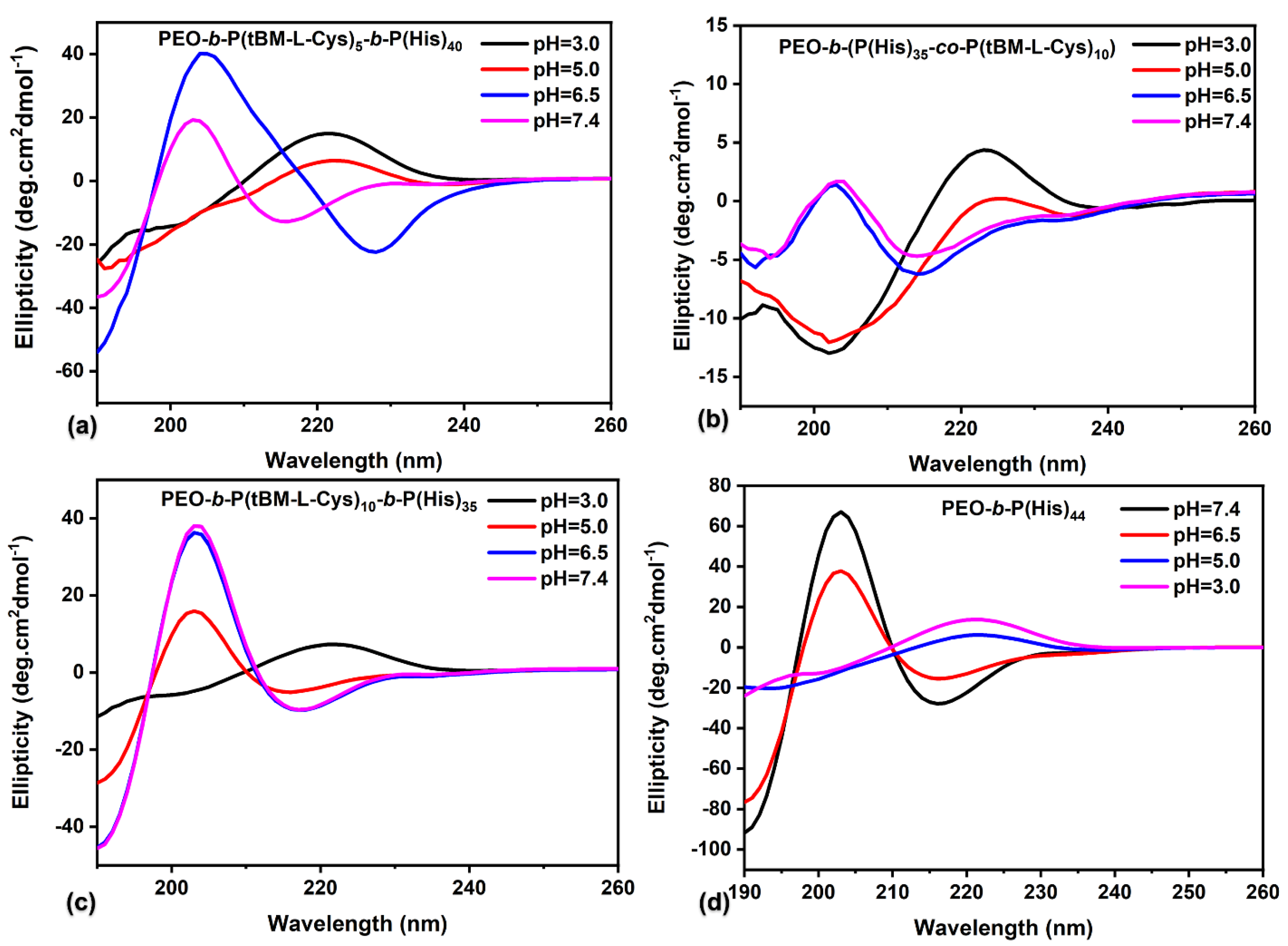
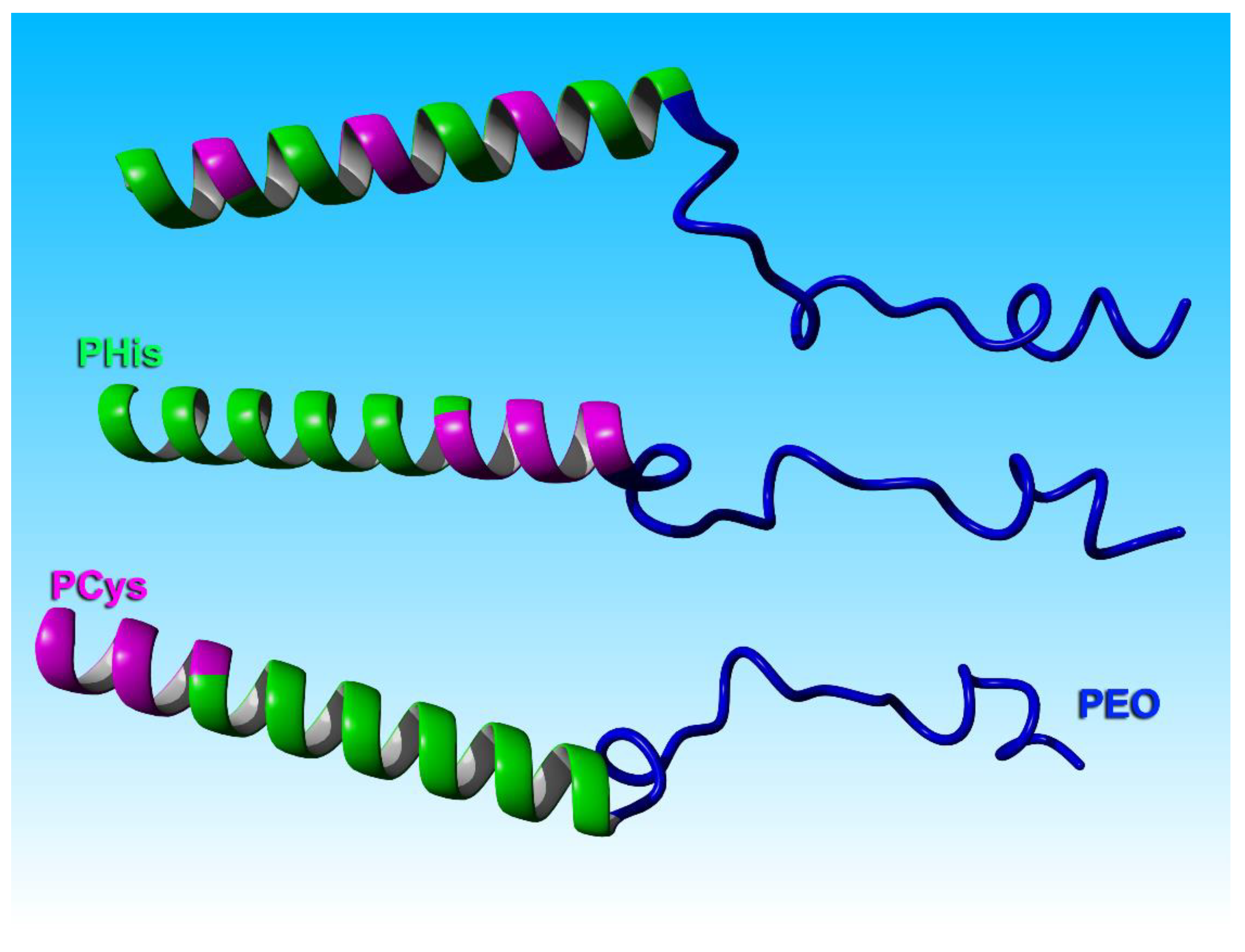
3.3. Secondary Structure through Cyclic Dichroism
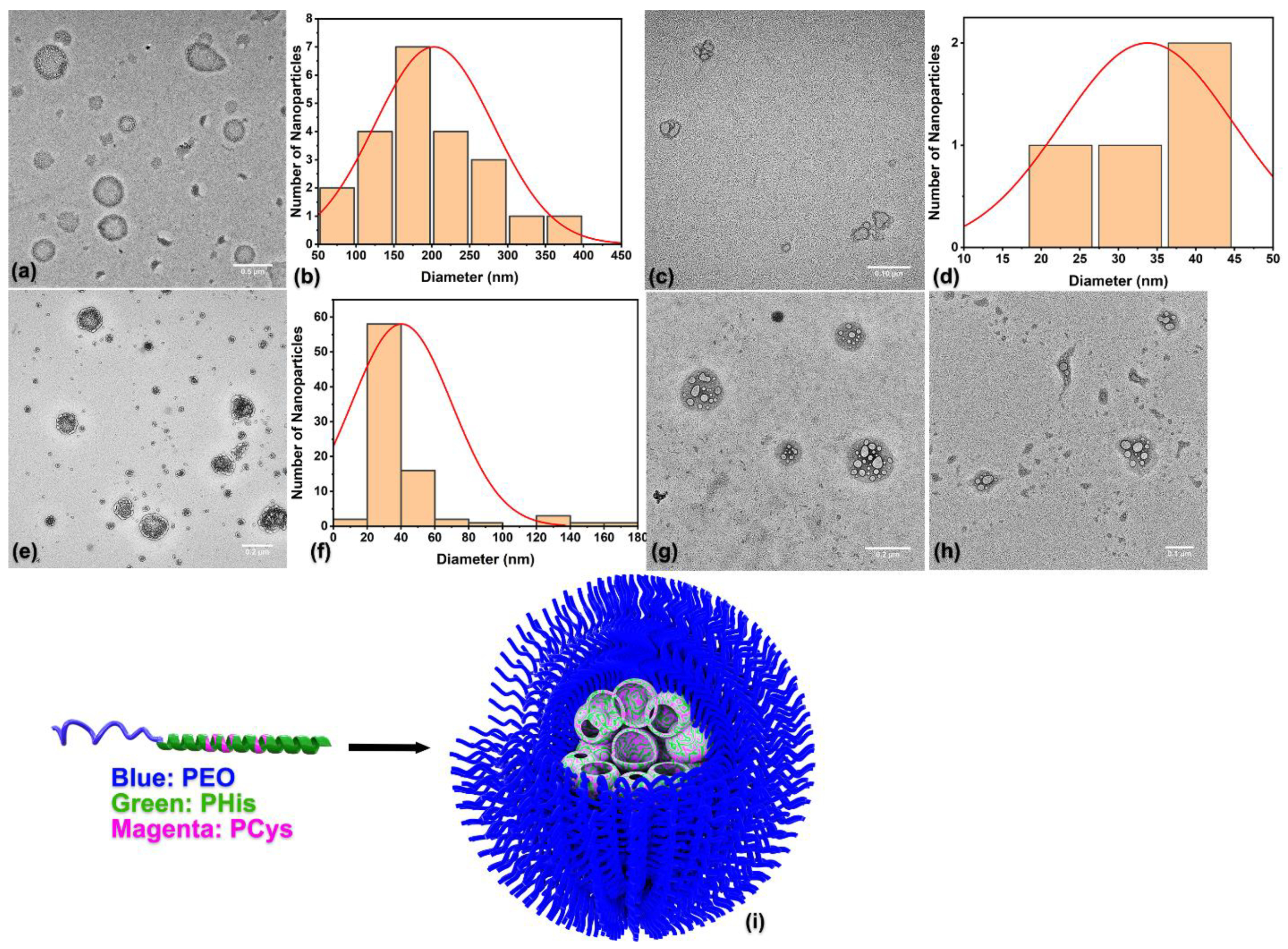
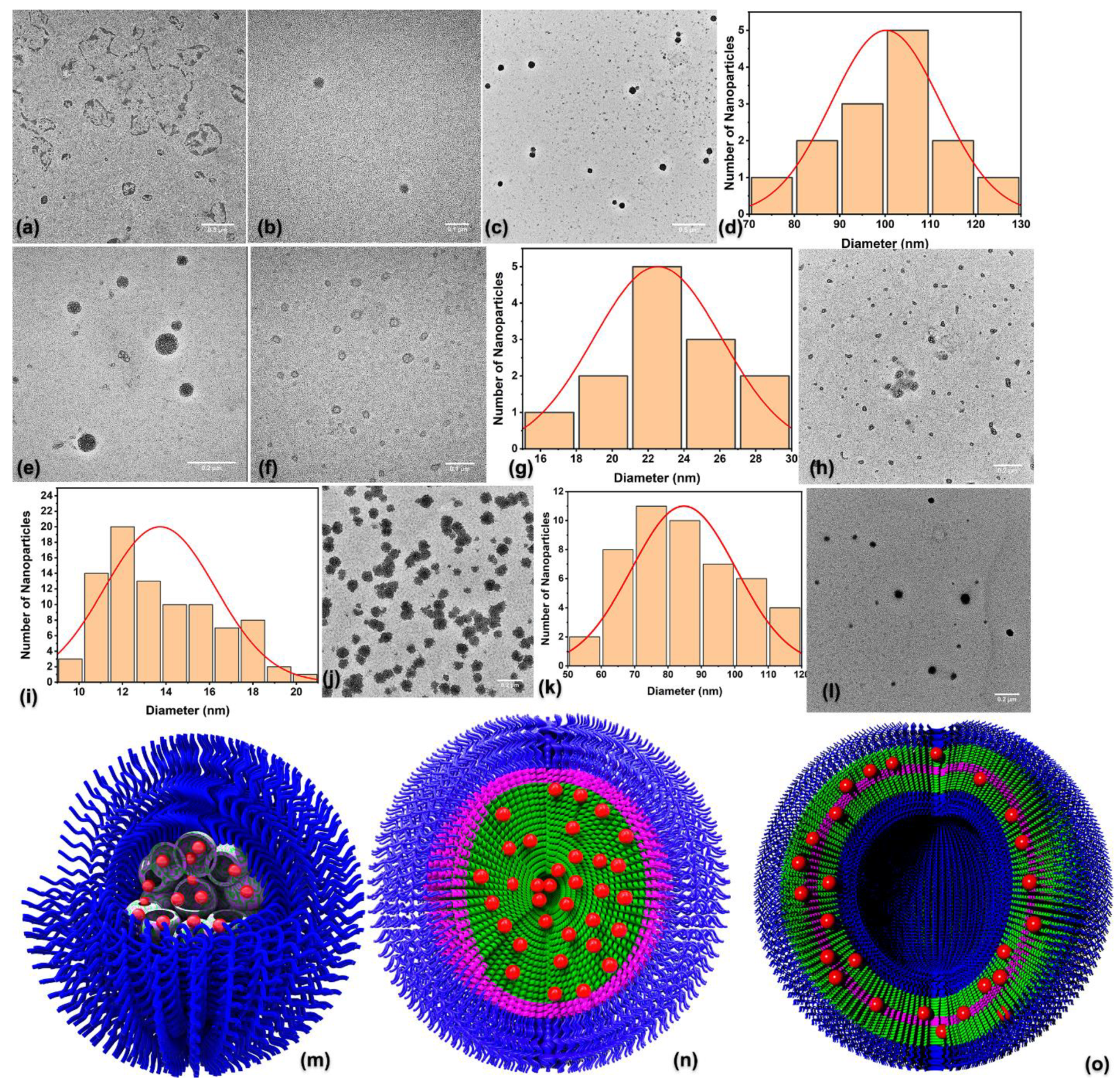
3.4. Self-Assembly of the Empty Hybrid Polymers
3.5. pH and Redox Responsiveness of the Empty NPs
3.6. Self-Assembly of the DOX-Loaded NPs
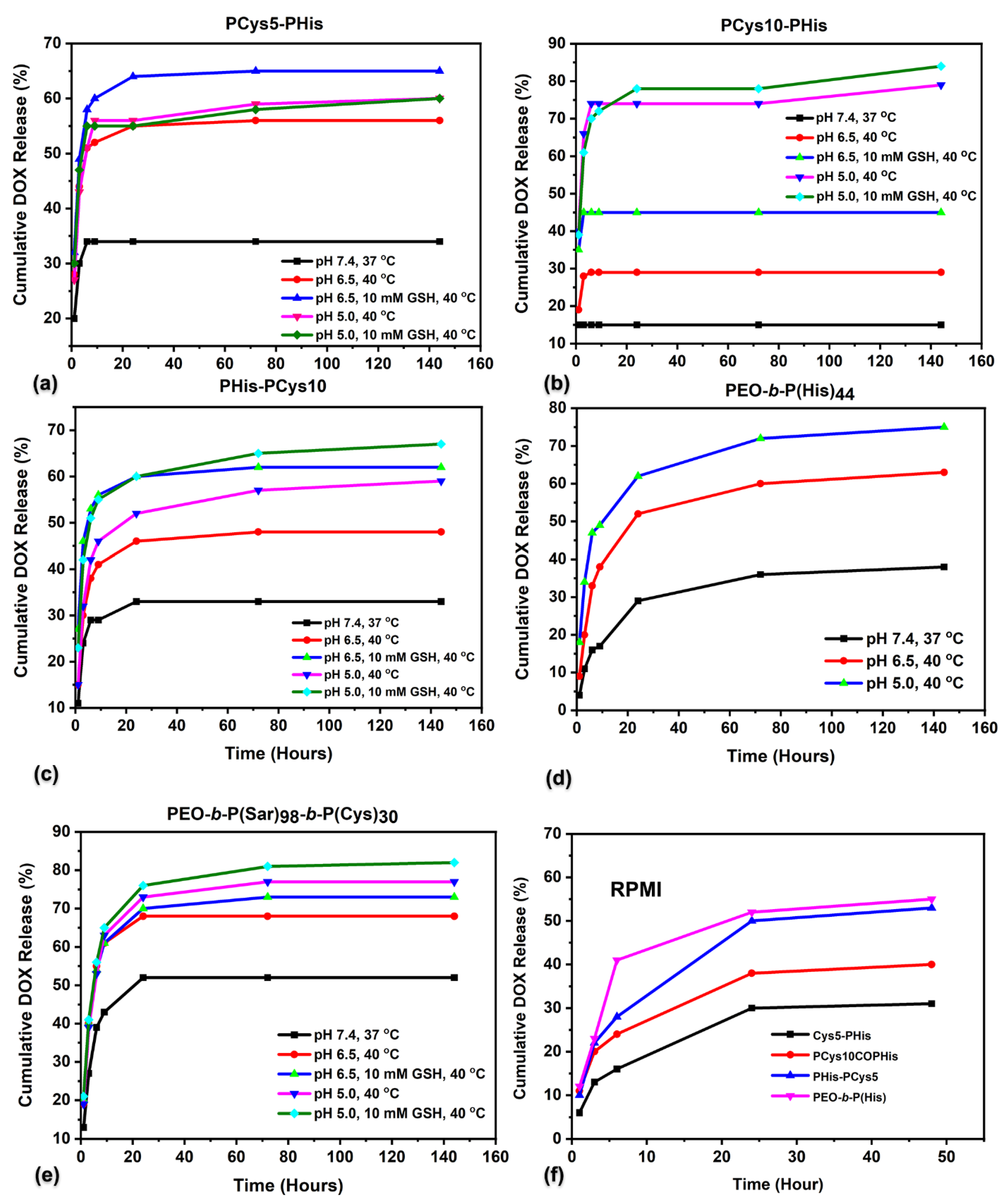
3.7. Drug Loading and In Vitro Release Studies
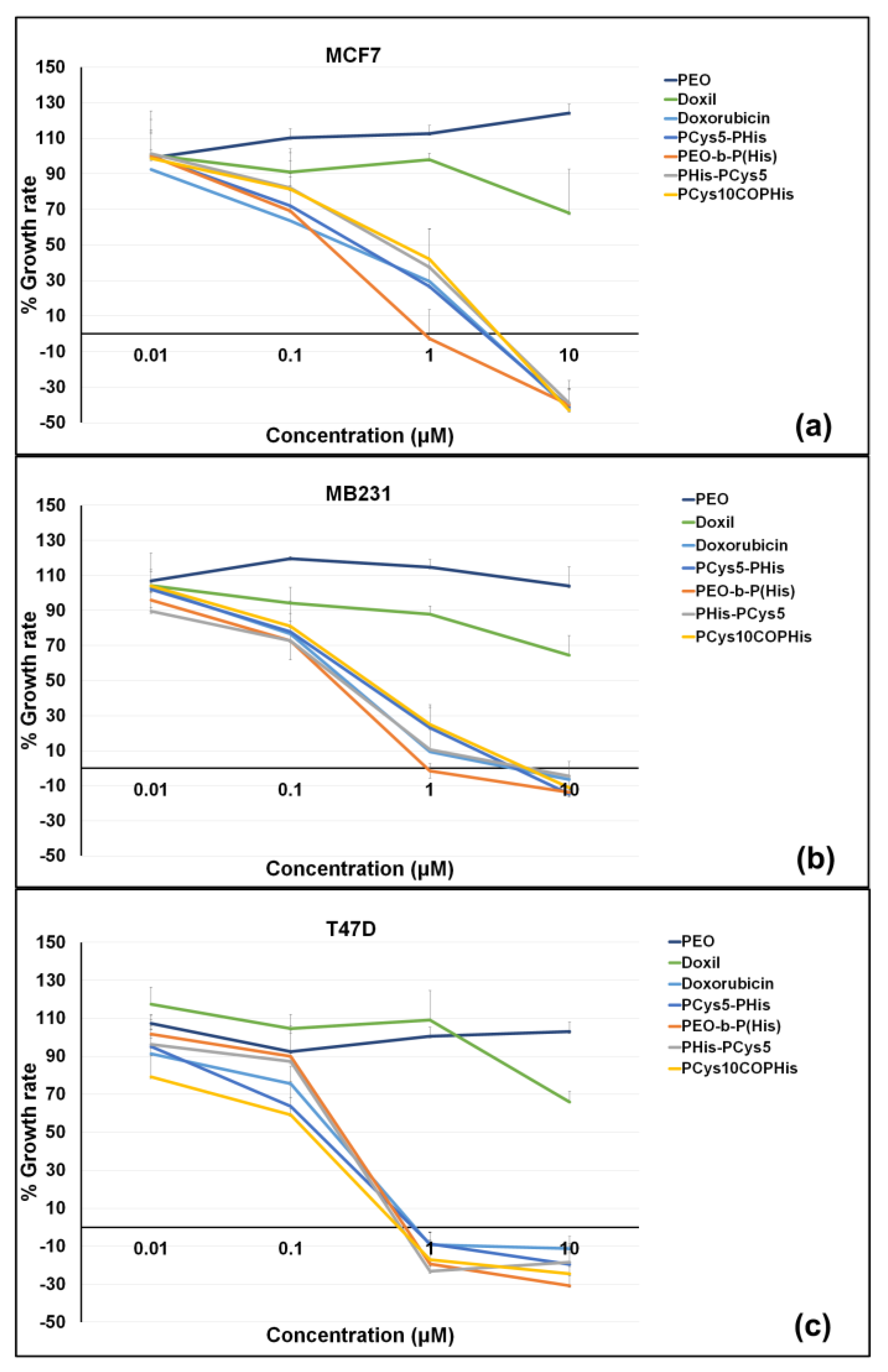
3.8. In Vitro Cytotoxic Activity
3.9. Influence of the PCys Topology on Self-Assembly, DOX Loading, In Vitro Release Profile as Well as In Vitro Cytotoxic Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Norouzi, M.; Amerian, M.; Amerian, M.; Atyabi, F. Clinical applications of nanomedicine in cancer therapy. Drug Discov. Today 2020, 25, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Jia, Y.; Wu, Y.; Shi, K.; Yang, D.; Li, P.; Qian, Z. Physical-, chemical-, and biological-responsive nanomedicine for cancer therapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnology 2020, 12, e1581. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Kessinger, C.W.; Sumer, B.D.; Gao, J. Multifunctional micellar nanomedicine for cancer therapy. Exp. Biol. Med. 2009, 234, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, Y.; Nishiyama, N.; Fukushima, S.; Koyama, H.; Yasuhiro, M.; Kataoka, K. Preparation and biological characterization of polymeric micelle drug carriers with intracellular pH-triggered drug release property: Tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy. Bioconjugate Chem. 2005, 16, 122–130. [Google Scholar] [CrossRef]
- Yoo, H.S.; Park, T.G. Folate receptor targeted biodegradable polymeric doxorubicin micelles. J. Control. Release 2004, 96, 273–283. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef]
- Liarou, E.; Varlas, S.; Skoulas, D.; Tsimblouli, C.; Sereti, E.; Dimas, K.; Iatrou, H. Smart polymersomes and hydrogels from polypeptide-based polymer systems through α-amino acid N-carboxyanhydride ring-opening polymerization. From chemistry to biomedical applications. Prog. Polym. Sci. 2018, 83, 28–78. [Google Scholar] [CrossRef]
- Skoulas, D.; Mangiapia, G.; Parisi, D.; Kasimatis, M.; Glynos, E.; Stratikos, E.; Vlassopoulos, D.; Frielinghaus, H.; Iatrou, H. Tunable Hydrogels with Improved Viscoelastic Properties from Hybrid Polypeptides. Macromolecules 2021, 54, 10786–10800. [Google Scholar] [CrossRef]
- Zhang, S.; Alvarez, J.D.; Sofroniew, V.M.; Deming, J.T. Design and synthesis of nonionic copolypeptide hydrogels with reversible thermoresponsive and tunable physical properties. Biomacromolecules 2015, 16, 1331–1340. [Google Scholar] [CrossRef] [Green Version]
- Yhee, J.Y.; Son, S.; Son, S.; Joo, M.K.; Kwon, I.C. The EPR effect in cancer therapy. In Cancer Targeted Drug Delivery; Springer: Berlin/Heidelberg, Germany, 2013; pp. 621–632. [Google Scholar]
- Li, J.; Kataoka, K. Chemo-physical Strategies to Advance the in Vivo Functionality of Targeted Nanomedicine: The Next Generation. J. Am. Chem. Soc. 2021, 143, 538–559. [Google Scholar] [CrossRef]
- Mürdter, T.E.; Friedel, G.; Backman, J.T.; McClellan, M.; Schick, M.; Gerken, M.; Bosslet, K.; Fritz, P.; Toomes, H.; Kroemer, H.K.; et al. Dose Optimization of a Doxorubicin Prodrug (HMR 1826) in Isolated Perfused Human Lungs: Low Tumor pH Promotes Prodrug Activation by β-Glucuronidase. J. Pharmacol. Exp. Ther. 2002, 301, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Thistlethwaite, A.J.; Leeper, D.B.; Moylan, D.J.; Nerlinger, R.E. pH distribution in human tumors. Int. J. Radiat. Oncol. Biol. Phys. 1985, 11, 1647–1652. [Google Scholar] [CrossRef]
- Najafi, M.; Goradel, N.H.; Farhood, B.; Salehi, E.; Solhjoo, S.; Toolee, H.; Kharazinejad, E.; Mortezaee, K. Tumor microenvironment: Interactions and therapy. J. Cell. Physiol. 2019, 234, 5700–5721. [Google Scholar] [CrossRef]
- Feng, L.; Dong, Z.; Tao, D.; Zhang, Y.; Liu, Z. The acidic tumor microenvironment: A target for smart cancer nano-theranostics. Natl. Sci. Rev. 2018, 5, 269–286. [Google Scholar] [CrossRef] [Green Version]
- Mavrogiorgis, D.; Bilalis, P.; Karatzas, A.; Skoulas, D.; Fotinogiannopoulou, G.; Iatrou, H. Controlled polymerization of histidine and synthesis of well-defined stimuli responsive polymers. Elucidation of the structure–aggregation relationship of this highly multifunctional material. Polym. Chem. 2014, 5, 6256–6278. [Google Scholar] [CrossRef]
- Bilalis, P.; Tziveleka, L.-A.; Varlas, S.; Iatrou, H. pH-Sensitive nanogates based on poly (l-histidine) for controlled drug release from mesoporous silica nanoparticles. Polym. Chem. 2016, 7, 1475–1485. [Google Scholar] [CrossRef]
- Karatzas, A.; Haataja, J.S.; Skoulas, D.; Bilalis, P.; Varlas, S.; Apostolidi, P.; Sofianopoulou, S.; Stratikos, E.; Houbenov, N.; Ikkala, O. Marcromolecular Architecture and Encapsulation of the Anticancer Drug Everolimus Control the Self-Assembly of Amphiphilic Polypeptide-Containing Hybrids. Biomacromolecules 2019, 20, 4546–4562. [Google Scholar] [CrossRef]
- Li, B.; Xu, Q.; Li, X.; Zhang, P.; Zhao, X.; Wang, Y. Redox-responsive hyaluronic acid nanogels for hyperthermia-assisted chemotherapy to overcome multidrug resistance. Carbohydr. Polym. 2019, 203, 378–385. [Google Scholar] [CrossRef]
- Elzes, M.R.; Akeroyd, N.; Engbersen, J.F.; Paulusse, J.M. Disulfide-functional poly (amido amine) s with tunable degradability for gene delivery. J. Control. Release 2016, 244, 357–365. [Google Scholar] [CrossRef]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione levels in human tumors. Biomarkers 2012, 17, 671–691. [Google Scholar] [CrossRef] [Green Version]
- Perry, R.R.; Mazetta, J.; Levin, M.; Barranco, S.C. Glutathione levels and variability in breast tumors and normal tissue. Cancer 1993, 72, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Feng, F.; Meng, F.; Deng, C.; Feijen, J.; Zhong, Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J. Control. Release 2011, 152, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhao, L.; Xu, X.; Bertrand, N.; Choi, W.I.; Yameen, B.; Shi, J.; Shah, V.; Mulvale, M.; MacLean, J.L. Hydrophobic cysteine poly (disulfide)-based redox-hypersensitive nanoparticle platform for cancer theranostics. Angew. Chem. 2015, 127, 9350–9355. [Google Scholar] [CrossRef]
- Wang, L.; You, X.; Lou, Q.; He, S.; Zhang, J.; Dai, C.; Zhao, M.; Zhao, M.; Hu, H.; Wu, J. Cysteine-based redox-responsive nanoparticles for small-molecule agent delivery. Biomater. Sci. 2019, 7, 4218–4229. [Google Scholar] [CrossRef] [PubMed]
- Huo, M.; Yuan, J.; Tao, L.; Wei, Y. Redox-responsive polymers for drug delivery: From molecular design to applications. Polym. Chem. 2014, 5, 1519–1528. [Google Scholar] [CrossRef]
- Gyarmati, B.; Némethy, Á.; Szilágyi, A. Reversible disulphide formation in polymer networks: A versatile functional group from synthesis to applications. Eur. Polym. J. 2013, 49, 1268–1286. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Luo, G.-F.; Liu, Y.; Li, C.; Cheng, S.-X.; Zhuo, R.-X.; Zhang, X.-Z. Redox-sensitive shell cross-linked PEG–polypeptide hybrid micelles for controlled drug release. Polym. Chem. 2012, 3, 1084–1090. [Google Scholar] [CrossRef]
- Wu, X.; Zhou, L.; Su, Y.; Dong, C.-M. Plasmonic, targeted, and dual drugs-loaded polypeptide composite nanoparticles for synergistic cocktail chemotherapy with photothermal therapy. Biomacromolecules 2016, 17, 2489–2501. [Google Scholar] [CrossRef]
- Wu, X.; Zhou, L.; Su, Y.; Dong, C.-M. An autoreduction method to prepare plasmonic gold-embedded polypeptide micelles for synergistic chemo-photothermal therapy. J. Mater. Chem. B 2016, 4, 2142–2152. [Google Scholar] [CrossRef]
- Bilalis, P.; Varlas, S.; Kiafa, A.; Velentzas, A.; Stravopodis, D.; Iatrou, H. Preparation of hybrid triple-stimuli responsive nanogels based on poly (L-histidine). J. Polym. Sci. Part A Polym. Chem. 2016, 54, 1278–1288. [Google Scholar] [CrossRef]
- Lee, J.H.; Orfanou, K.; Driva, P.; Iatrou, H.; Hadjichristidis, N.; Lohse, D.J. Linear and Nonlinear Rheology of Dendritic Star Polymers: Experiment. Macromolecules 2008, 41, 9165–9178. [Google Scholar] [CrossRef]
- Junnila, S.; Houbenov, N.; Karatzas, A.; Hadjichristidis, N.; Hirao, A.; Iatrou, H.; Ikkala, O. Side-Chain-Controlled Self-Assembly of Polystyrene-Polypeptide Miktoarm Star Copolymers. Macromolecules 2012, 45, 2850–2856. [Google Scholar] [CrossRef]
- Yamauchi, K.; Akasaka, S.; Hasegawa, H.; Iatrou, H.; Hadjichristidis, N. Blends of a 3-miktoarm star terpolymer (3 mu-ISD) of isoprene (I), styrene (S), and dimethylsiloxane (D) with PS and PDMS. Effect on microdomain morphology and grain size. Macromolecules 2005, 38, 8022–8027. [Google Scholar] [CrossRef]
- Gitsas, A.; Floudas, G.; Mondeshki, M.; Lieberwirth, I.; Spiess, H.W.; Iatrou, H.; Hadjichristidis, N.; Hirao, A. Hierarchical Self-Assembly and Dynamics of a Miktoarm Star chimera Composed of Poly(gamma-benzyl-L-glutamate), Polystyrene, and Polyisoprene. Macromolecules 2010, 43, 1874–1881. [Google Scholar] [CrossRef]
- Houbenov, N.; Haataja, J.S.; Iatrou, H.; Hadjichristidis, N.; Ruokolainen, J.; Faul, C.F.J.; Ikkala, O. Self-Assembled Polymeric Supramolecular Frameworks. Angew. Chem. Int. Ed. 2011, 50, 2516–2520. [Google Scholar] [CrossRef]
- Habraken, G.J.; Koning, C.E.; Heuts, J.P.; Heise, A. Thiol chemistry on well-defined synthetic polypeptides. Chem. Commun. 2009, 24, 3612–3614. [Google Scholar] [CrossRef]
- Fetsch, C.; Grossmann, A.; Holz, L.; Nawroth, J.F.; Luxenhofer, R. Polypeptoids from N-substituted glycine N-carboxyanhydrides: Hydrophilic, hydrophobic, and amphiphilic polymers with poisson distribution. Macromolecules 2011, 44, 6746–6758. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Sakellariou, G. Synthesis of well-defined polypeptide-based materials via the ring-opening polymerization of α-amino acid N-carboxyanhydrides. Chem. Rev. 2009, 109, 5528–5578. [Google Scholar] [CrossRef]
- Deming, T.J. Living polymerization of α-amino acid-N-carboxyanhydrides. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 3011–3018. [Google Scholar] [CrossRef]
- Kricheldorf, H.R. Polypeptides and 100 years of chemistry of α-amino acid N-carboxyanhydrides. Angew. Chem. Int. Ed. 2006, 45, 5752–5784. [Google Scholar] [CrossRef]
- Kataoka, K.; Matsumoto, T.; Yokoyama, M.; Okano, T.; Sakurai, Y.; Fukushima, S.; Okamoto, K.; Kwon, G.S. Doxorubicin-loaded poly (ethylene glycol)–poly (β-benzyl-l-aspartate) copolymer micelles: Their pharmaceutical characteristics and biological significance. J. Control. Release 2000, 64, 143–153. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. JNCI J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Sereti, E.; Tsimplouli, C.; Kalaitsidou, E.; Sakellaridis, N.; Dimas, K. Study of the Relationship between Sigma Receptor Expression Levels and Some Common Sigma Ligand Activity in Cancer Using Human Cancer Cell Lines of the NCI-60 Cell Line Panel. Biomedicines 2021, 9, 38. [Google Scholar] [CrossRef]
- Kamposioras, K.; Tsimplouli, C.; Verbeke, C.; Anthoney, A.; Daoukopoulou, A.; Papandreou, C.N.; Sakellaridis, N.; Vassilopoulos, G.; Potamianos, S.P.; Liakouli, V.; et al. Silencing of caveolin-1 in fibroblasts as opposed to epithelial tumor cells results in increased tumor growth rate and chemoresistance in a human pancreatic cancer model. Int. J. Oncol. 2019, 54, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Iatrou, H.; Dimas, K.; Gkikas, M.; Tsimblouli, C.; Sofianopoulou, S. Polymersomes from Polypeptide Containing Triblock Co- and Terpolymers for Drug Delivery against Pancreatic Cancer: Asymmetry of the External Hydrophilic Blocks. Macromol. Biosci. 2014, 14, 1222–1238. [Google Scholar] [CrossRef]
- Greenfield, N.J. Analysis of Circular Dichroism Data. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2004; Volume 383, pp. 282–317. [Google Scholar]
- Bilalis, P.; Skoulas, D.; Karatzas, A.; Marakis, J.; Stamogiannos, A.; Tsimblouli, C.; Sereti, E.; Stratikos, E.; Dimas, K.; Vlassopoulos, D. Self-healing pH-and enzyme stimuli-responsive hydrogels for targeted delivery of gemcitabine to treat pancreatic cancer. Biomacromolecules 2018, 19, 3840–3852. [Google Scholar] [CrossRef]
- Ulkoski, D.; Scholz, C. Synthesis and application of aurophilic poly (cysteine) and poly (cysteine)-containing copolymers. Polymers 2017, 9, 500. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, N.A.; Noronha, M.A.; Câmara, M.C.C.; Kurnik, I.S.; Feng, C.; Araujo, V.H.S.; Santos, J.H.P.M.; Feitosa, V.; Molino, J.V.D.; Rangel-Yagui, C.O.; et al. Doxorubicin nanoformulations on therapy against cancer: An overview from the last 10 years. Mater. Sci. Eng. C 2021, 133, 112623. [Google Scholar] [CrossRef]
- Al-malky, H.S.; Al Harthi, S.E.; Osman, A.-M.M. Major obstacles to doxorubicin therapy: Cardiotoxicity and drug resistance. J. Oncol. Pharm. Pract. 2020, 26, 434–444. [Google Scholar] [CrossRef]
- Shafei, A.; El-Bakly, W.; Sobhy, A.; Wagdy, O.; Reda, A.; Aboelenin, O.; Marzouk, A.; El Habak, K.; Mostafa, R.; Ali, M.A.; et al. A review on the efficacy and toxicity of different doxorubicin nanoparticles for targeted therapy in metastatic breast cancer. Biomed. Pharmacother. 2017, 95, 1209–1218. [Google Scholar] [CrossRef]
- Aliferis, T.; Iatrou, H.; Hadjichristidis, N. Living polypeptides. Biomacromolecules 2004, 5, 1653–1656. [Google Scholar] [CrossRef] [PubMed]
- Pickel, D.L.; Politakos, N.; Avgeropoulos, A.; Messman, J.M. A mechanistic study of α-(amino acid)-N-carboxyanhydride polymerization: Comparing initiation and termination events in high-vacuum and traditional polymerization techniques. Macromolecules 2009, 42, 7781–7788. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pispas, S.; Pitsikalis, M. Anionic polymerization: High vacuum techniques. J. Polym. Sci. A Polym. Chem. 2000, 38, 3211–3234. [Google Scholar] [CrossRef]
- Uhrig, D.; Mays, J.W. Experimental techniques in high–vacuum anionic polymerization. J. Polym. Sci. A Polym. Chem. 2005, 43, 6179–6222. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Hirao, A. Anionic Polymerization; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Mn PEO | Mn P(Cys)x-P(His)y | I |
|---|---|---|---|
| ×10−3 (g mol–1) a | ×10−3(g mol–1) b | ( ) b | |
| PCys5-PHis | 10.0 | 5.9 | 1.16 |
| PCys10-PHis | 10.0 | 5.9 | 1.11 |
| PHis-PCys5 | 10.0 | 6.0 | 1.17 |
| PHis-PCys10 | 10.0 | 5.7 | 1.21 |
| PCys5COPHis | 10.0 | 6.1 | 1.18 |
| PCys10COPHis | 10.0 | 5.8 | 1.15 |
| POLYMER | Rg (nm) | Rh (Dh) a (nm) | Rg/Rh | Diameter by DLS (nm) | PDI by DLS | Average Core Diameter by TEM (nm) | Zeta Potential (mV) pH/GSH | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 7.4 | 6.5 | 5 | 6.5/GSH 10 mM | 5/GSH 10 mM | |||||||
| PCys5-PHis | 128 | 125 (250) | 1.1 | A.31 B.250 | 0.294 | 205 | +2.4 | +3.2 | −3.0 | −0.3 | +7.8 |
| PCys10-PHis | - | - | - | - | - | - | - | −0.6 | +5.1 | +0.4 | +1.4 |
| PHis-PCys5 | 134 | 122 (245) | 1.1 | A.32 B.215 | 0.344 | 52 | −0.1 | 0.0 | − | +3.3 | − |
| PHis-PCys10 | 150 | 107 (215) | 1.4 | A.30 B.200 | 0.284 | 45 | +3.3 | −3.5 | +0.4 | −0.3 | +0.5 |
| PCys5COPHis | 131 | 100 (200) | 1.3 | A.18 B.256 | 0.268 | 194 | +3.1 | −0.3 | +1.3 | +3.4 | +3.8 |
| PCys10COPHis | 87 | 128 (256) | 1.2 | A.31 B.250 | 0.294 | 35, 158 | +2.4 | +3.2 | −3.0 | −0.3 | +7.8 |
| POLYMER | Diameter by DLS (nm) | PDI by DLS | Average Core Diameter by TEM (nm) | Zeta Potential (mV) | EE (%) | LC (%) |
|---|---|---|---|---|---|---|
| PCys5-PHis | 204 | 0.227 | 120 | −7.6 | 13.6 | 8.5 |
| PCys10-PHis | 193 | 0.136 | 108 | −8.1 | 19.8 | 9.7 |
| PHis-PCys5 | 159 | 0.137 | 15 | 1.1 | 19.4 | 9.5 |
| PHis-PCys10 | 208 | 0.196 | 23 | 0.88 | 15.2 | 7.9 |
| PCys5COPHis | 118 | 0.146 | 100 | 4.3 | 20.3 | 10.0 |
| PCys10COPHis | 154 | 0.164 | 130 | 1.4 | 23.0 | 12.0 |
| Cancer Cell Line | DOXO_PEO-b-P(Cys)5-b-P(His)40 | DOXO_PEO-b-P(His) | DOXO_PEO-b-P(His)40-b-P(Cys)5 | DOXO_PEO-b-[P(Cys)10-co-P(His)35 | DOXIL | DOX | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GI50 | TGI | LC50 | GI50 | TGI | LC50 | GI50 | TGI | LC50 | GI50 | TGI | LC50 | GI50 | TGI | LC50 | GI50 | TGI | LC50 | |
| T47D | 0.3 | 0.9 | >10 | 0.4 | 0.8 | >10 | 0.4 | 0.8 | >10 | 0.2 | 0.8 | >10 | >10 | >10 | >10 | 0.4 | 0.9 | >10 |
| MCF7 | 0.5 | 4.5 | >10 | 0.3 | 1.0 | >10 | 0.7 | 5.4 | >10 | 0.8 | 5.4 | >10 | >10 | >10 | >10 | 0.5 | 4.7 | >10 |
| MB231 | 0.6 | 6.5 | >10 | 0.4 | 1.0 | >10 | 0.4 | 7.3 | >10 | 0.6 | 7.2 | >10 | >10 | >10 | >10 | 0.5 | 6.3 | >10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stavroulaki, D.; Kyroglou, I.; Skourtis, D.; Athanasiou, V.; Thimi, P.; Sofianopoulou, S.; Kazaryan, D.; Fragouli, P.G.; Labrianidou, A.; Dimas, K.; et al. Influence of the Topology of Poly(L-Cysteine) on the Self-Assembly, Encapsulation and Release Profile of Doxorubicin on Dual-Responsive Hybrid Polypeptides. Pharmaceutics 2023, 15, 790. https://doi.org/10.3390/pharmaceutics15030790
Stavroulaki D, Kyroglou I, Skourtis D, Athanasiou V, Thimi P, Sofianopoulou S, Kazaryan D, Fragouli PG, Labrianidou A, Dimas K, et al. Influence of the Topology of Poly(L-Cysteine) on the Self-Assembly, Encapsulation and Release Profile of Doxorubicin on Dual-Responsive Hybrid Polypeptides. Pharmaceutics. 2023; 15(3):790. https://doi.org/10.3390/pharmaceutics15030790
Chicago/Turabian StyleStavroulaki, Dimitra, Iro Kyroglou, Dimitrios Skourtis, Varvara Athanasiou, Pandora Thimi, Sosanna Sofianopoulou, Diana Kazaryan, Panagiota G. Fragouli, Andromahi Labrianidou, Konstantinos Dimas, and et al. 2023. "Influence of the Topology of Poly(L-Cysteine) on the Self-Assembly, Encapsulation and Release Profile of Doxorubicin on Dual-Responsive Hybrid Polypeptides" Pharmaceutics 15, no. 3: 790. https://doi.org/10.3390/pharmaceutics15030790
APA StyleStavroulaki, D., Kyroglou, I., Skourtis, D., Athanasiou, V., Thimi, P., Sofianopoulou, S., Kazaryan, D., Fragouli, P. G., Labrianidou, A., Dimas, K., Patias, G., Haddleton, D. M., & Iatrou, H. (2023). Influence of the Topology of Poly(L-Cysteine) on the Self-Assembly, Encapsulation and Release Profile of Doxorubicin on Dual-Responsive Hybrid Polypeptides. Pharmaceutics, 15(3), 790. https://doi.org/10.3390/pharmaceutics15030790