
Development of Small Molecules Targeting α-Synuclein Aggregation: A Promising Strategy to Treat Parkinson’s Disease




Abstract
:1. Introduction
2. Historical Overview
3. Symptomatology
4. Risk Factors and Genetics of Parkinson’s Disease
5. Molecular Mechanism Implicated in PD Development
6. Alpha-Synuclein
7. New Therapies: Modulating α-Synuclein Aggregation
7.1. Polyphenolic Scaffolds
7.2. Repositioned Compounds
7.3. Compounds Developed by Rational Design
7.4. Compounds Derived from High-Throughput Screenings
7.5. Structure-Based Strategies for Drug Discovery
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sabate, R.; Ventura, S. Cross-beta-sheet supersecondary structure in amyloid folds: Techniques for detection and characterization. Methods Mol. Biol. 2013, 932, 237–257. [Google Scholar] [CrossRef]
- Sunde, M.; Serpell, L.C.; Bartlam, M.; Fraser, P.E.; Pepys, M.B.; Blake, C.C. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 1997, 273, 729–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, P.C.; Zhou, R.; Serpell, L.C.; Riek, R.; Knowles, T.P.J.; Lashuel, H.A.; Gazit, E.; Hamley, I.W.; Davis, T.P.; Fandrich, M.; et al. Half a century of amyloids: Past, present and future. Chem. Soc. Rev. 2020, 49, 5473–5509. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.; Ventura, S. Functional Amyloids Germinate in Plants. Trends Plant Sci. 2021, 26, 7–10. [Google Scholar] [CrossRef]
- Claessen, D.; Rink, R.; de Jong, W.; Siebring, J.; de Vreugd, P.; Boersma, F.G.; Dijkhuizen, L.; Wosten, H.A. A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes Dev. 2003, 17, 1714–1726. [Google Scholar] [CrossRef] [Green Version]
- Chapman, M.R.; Robinson, L.S.; Pinkner, J.S.; Roth, R.; Heuser, J.; Hammar, M.; Normark, S.; Hultgren, S.J. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 2002, 295, 851–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, D.M.; Koulov, A.V.; Alory-Jost, C.; Marks, M.S.; Balch, W.E.; Kelly, J.W. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006, 4, e6. [Google Scholar] [CrossRef] [PubMed]
- Otzen, D.; Riek, R. Functional Amyloids. Cold Spring Harb. Perspect. Biol. 2019, 11, a033860. [Google Scholar] [CrossRef]
- Garcia-Pardo, J.; Bartolome-Nafria, A.; Chaves-Sanjuan, A.; Gil-Garcia, M.; Visentin, C.; Bolognesi, M.; Ricagno, S.; Ventura, S. Cryo-EM structure of hnRNPDL-2 fibrils, a functional amyloid associated with limb-girdle muscular dystrophy D3. Nat. Commun. 2023, 14, 239. [Google Scholar] [CrossRef]
- Berson, J.F.; Harper, D.C.; Tenza, D.; Raposo, G.; Marks, M.S. Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol. Biol. Cell 2001, 12, 3451–3464. [Google Scholar] [CrossRef] [Green Version]
- Hervas, R.; Rau, M.J.; Park, Y.; Zhang, W.; Murzin, A.G.; Fitzpatrick, J.A.J.; Scheres, S.H.W.; Si, K. Cryo-EM structure of a neuronal functional amyloid implicated in memory persistence in Drosophila. Science 2020, 367, 1230–1234. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef] [Green Version]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussbaum, R.L.; Ellis, C.E. Alzheimer’s disease and Parkinson’s disease. New Engl. J. Med. 2003, 348, 1356–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dexter, D.T.; Jenner, P. Parkinson disease: From pathology to molecular disease mechanisms. Free Radic. Biol. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1583–1590. [Google Scholar] [CrossRef]
- Goetz, C.G. The history of Parkinson’s disease: Early clinical descriptions and neurological therapies. Cold Spring Harbor. Perspect. Med. 2011, 1, a008862. [Google Scholar] [CrossRef] [Green Version]
- Duncan, G.W.; Khoo, T.K.; Yarnall, A.J.; O’Brien, J.T.; Coleman, S.Y.; Brooks, D.J.; Barker, R.A.; Burn, D.J. Health-related quality of life in early Parkinson’s disease: The impact of nonmotor symptoms. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 195–202. [Google Scholar] [CrossRef]
- Martinez-Martin, P.; Rodriguez-Blazquez, C.; Kurtis, M.M.; Chaudhuri, K.R.; Group, N.V. The impact of non-motor symptoms on health-related quality of life of patients with Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2011, 26, 399–406. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Cotzias, G.C. L-Dopa for Parkinsonism. New Engl. J. Med. 1968, 278, 630. [Google Scholar] [CrossRef] [PubMed]
- Hornykiewicz, O. L-DOPA: From a biologically inactive amino acid to a successful therapeutic agent. Amino Acids 2002, 23, 65–70. [Google Scholar] [CrossRef]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar]
- Wakabayashi, K.; Hayashi, S.; Kakita, A.; Yamada, M.; Toyoshima, Y.; Yoshimoto, M.; Takahashi, H. Accumulation of alpha-synuclein/NACP is a cytopathological feature common to Lewy body disease and multiple system atrophy. Acta Neuropathol. 1998, 96, 445–452. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. Lewy body diseases and multiple system atrophy as alpha-synucleinopathies. Mol. Psychiatry 1998, 3, 462–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. Alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkinson, J. An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236, discussion 222. [Google Scholar] [CrossRef]
- Charcot, J.-M. Leçons sur le maladies du système nerveus. In Bureaux du Progrès Medical; Oeuvres Complètes (Tome 1); Delahaye, A., Lecronsnier, E., Eds.; Bureaux du Progrès Médical: Paris, France, 1872; pp. 155–188. [Google Scholar]
- Blocq, C.; Marinescu, G. Sur un cas de tremblement parkinsonien hémiplégique symptomatique d’une tumeur du pédoncle cérébral. C. R. Cos. Biol. 1893, 5, 105–111. [Google Scholar]
- Brissaud, E. Leçons sur les Maladies Nerveuses; Masson & Associates, Inc.: Escondido, CA, USA, 1899; Volume 2. [Google Scholar]
- Trétiakoff, C.D. Contribution à L’étude de L’anatomie Pathologique du Locus Niger de Soemmering Avec Quelques Deductions Relatives A la Pathogenie des Troubles du Tonus Musculaire et de la Maladie de Parkinson; Université de Paris: Paris, France, 1919. [Google Scholar]
- Lewy, F. Zur pathologischen anatomie der paralysis agitans. Dtsch. Z. Nervenheilkd. 1912, 50, 50–55. [Google Scholar]
- Postuma, R.B.; Berg, D. Advances in markers of prodromal Parkinson disease. Nat. Rev. Neurol. 2016, 12, 622–634. [Google Scholar] [CrossRef]
- Carlsson, A.; Lindqvist, M.; Magnusson, T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature 1957, 180, 1200. [Google Scholar] [CrossRef] [PubMed]
- Ehringer, H.; Hornykiewicz, O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Klin. Wochenschr. 1960, 38, 1236–1239. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Athanassiadou, A.; Voutsinas, G.; Psiouri, L.; Leroy, E.; Polymeropoulos, M.H.; Ilias, A.; Maniatis, G.M.; Papapetropoulos, T. Genetic analysis of families with Parkinson disease that carry the Ala53Thr mutation in the gene encoding alpha-synuclein. Am. J. Hum. Genet. 1999, 65, 555–558. [Google Scholar] [CrossRef] [Green Version]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 811–813. [Google Scholar] [CrossRef]
- Kiely, A.P.; Asi, Y.T.; Kara, E.; Limousin, P.; Ling, H.; Lewis, P.; Proukakis, C.; Quinn, N.; Lees, A.J.; Hardy, J.; et al. alpha-Synucleinopathy associated with G51D SNCA mutation: A link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013, 125, 753–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, A.B.; Farrer, M.J.; Bonifati, V. The genetics of Parkinson’s disease: Progress and therapeutic implications. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Samaranch, L.; Lorenzo-Betancor, O.; Arbelo, J.M.; Ferrer, I.; Lorenzo, E.; Irigoyen, J.; Pastor, M.A.; Marrero, C.; Isla, C.; Herrera-Henriquez, J.; et al. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain A J. Neurol. 2010, 133, 1128–1142. [Google Scholar] [CrossRef] [Green Version]
- Taipa, R.; Pereira, C.; Reis, I.; Alonso, I.; Bastos-Lima, A.; Melo-Pires, M.; Magalhaes, M. DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain A J. Neurol. 2016, 139, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Schober, A. Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res. 2004, 318, 215–224. [Google Scholar] [CrossRef]
- Ko, W.K.D.; Bezard, E. Experimental animal models of Parkinson’s disease: A transition from assessing symptomatology to alpha-synuclein targeted disease modification. Exp. Neurol. 2017, 298, 172–179. [Google Scholar] [CrossRef]
- Koprich, J.B.; Kalia, L.V.; Brotchie, J.M. Animal models of alpha-synucleinopathy for Parkinson disease drug development. Nat. Rev. Neurosci. 2017, 18, 515–529. [Google Scholar] [CrossRef]
- Lazaro, D.F.; Pavlou, M.A.S.; Outeiro, T.F. Cellular models as tools for the study of the role of alpha-synuclein in Parkinson’s disease. Exp. Neurol. 2017, 298, 162–171. [Google Scholar] [CrossRef]
- Marmion, D.J.; Kordower, J.H. alpha-Synuclein nonhuman primate models of Parkinson’s disease. J. Neural Transm. 2018, 125, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Trigo-Damas, I.; Del Rey, N.L.; Blesa, J. Novel models for Parkinson’s disease and their impact on future drug discovery. Expert Opin. Drug Discov. 2018, 13, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E.; et al. Targeting alpha-synuclein for treatment of Parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef] [Green Version]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coune, P.G.; Schneider, B.L.; Aebischer, P. Parkinson’s disease: Gene therapies. Cold Spring Harb. Perspect. Med. 2012, 2, a009431. [Google Scholar] [CrossRef]
- Tolosa, E.; Wenning, G.; Poewe, W. The diagnosis of Parkinson’s disease. Lancet Neurol. 2006, 5, 75–86. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Marder, K.S.; Tang, M.X.; Mejia-Santana, H.; Rosado, L.; Louis, E.D.; Comella, C.L.; Colcher, A.; Siderowf, A.D.; Jennings, D.; Nance, M.A.; et al. Predictors of parkin mutations in early-onset Parkinson disease: The consortium on risk for early-onset Parkinson disease study. Arch. Neurol. 2010, 67, 731–738. [Google Scholar] [CrossRef]
- Pont-Sunyer, C.; Hotter, A.; Gaig, C.; Seppi, K.; Compta, Y.; Katzenschlager, R.; Mas, N.; Hofeneder, D.; Brucke, T.; Bayes, A.; et al. The onset of nonmotor symptoms in Parkinson’s disease (the ONSET PD study). Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 229–237. [Google Scholar] [CrossRef]
- Schrag, A.; Sauerbier, A.; Chaudhuri, K.R. New clinical trials for nonmotor manifestations of Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 1490–1504. [Google Scholar] [CrossRef]
- Hinnell, C.; Hurt, C.S.; Landau, S.; Brown, R.G.; Samuel, M.; Group, P.-P.S. Nonmotor versus motor symptoms: How much do they matter to health status in Parkinson’s disease? Mov. Disord. Off. J. Mov. Disord. Soc. 2012, 27, 236–241. [Google Scholar] [CrossRef]
- Storch, A.; Schneider, C.B.; Wolz, M.; Sturwald, Y.; Nebe, A.; Odin, P.; Mahler, A.; Fuchs, G.; Jost, W.H.; Chaudhuri, K.R.; et al. Nonmotor fluctuations in Parkinson disease: Severity and correlation with motor complications. Neurology 2013, 80, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Weerkamp, N.J.; Tissingh, G.; Poels, P.J.; Zuidema, S.U.; Munneke, M.; Koopmans, R.T.; Bloem, B.R. Nonmotor symptoms in nursing home residents with Parkinson’s disease: Prevalence and effect on quality of life. J. Am. Geriatr. Soc. 2013, 61, 1714–1721. [Google Scholar] [CrossRef]
- Kalia, L.V.; Brotchie, J.M.; Fox, S.H. Novel nondopaminergic targets for motor features of Parkinson’s disease: Review of recent trials. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Karachi, C.; Grabli, D.; Bernard, F.A.; Tande, D.; Wattiez, N.; Belaid, H.; Bardinet, E.; Prigent, A.; Nothacker, H.P.; Hunot, S.; et al. Cholinergic mesencephalic neurons are involved in gait and postural disorders in Parkinson disease. J. Clin. Investig. 2010, 120, 2745–2754. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, E.C.; Graybiel, A.M.; Duyckaerts, C.; Javoy-Agid, F. Neuronal loss in the pedunculopontine tegmental nucleus in Parkinson disease and in progressive supranuclear palsy. Proc. Natl. Acad. Sci. USA 1987, 84, 5976–5980. [Google Scholar] [CrossRef] [Green Version]
- Seppi, K.; Weintraub, D.; Coelho, M.; Perez-Lloret, S.; Fox, S.H.; Katzenschlager, R.; Hametner, E.M.; Poewe, W.; Rascol, O.; Goetz, C.G.; et al. The Movement Disorder Society Evidence-Based Medicine Review Update: Treatments for the non-motor symptoms of Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2011, 26 (Suppl. S3), S42–S80. [Google Scholar] [CrossRef] [PubMed]
- Connolly, B.; Fox, S.H. Treatment of cognitive, psychiatric, and affective disorders associated with Parkinson’s disease. Neurotherapeutics 2014, 11, 78–91. [Google Scholar] [CrossRef] [Green Version]
- Mahlknecht, P.; Seppi, K.; Poewe, W. The Concept of Prodromal Parkinson’s Disease. J. Park. Dis. 2015, 5, 681–697. [Google Scholar] [CrossRef] [Green Version]
- Salat, D.; Noyce, A.J.; Schrag, A.; Tolosa, E. Challenges of modifying disease progression in prediagnostic Parkinson’s disease. Lancet Neurol. 2016, 15, 637–648. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.S.; Williams, D.R.; Gallagher, D.A.; Massey, L.A.; Silveira-Moriyama, L.; Lees, A.J. Nonmotor symptoms as presenting complaints in Parkinson’s disease: A clinicopathological study. Mov. Disord. Off. J. Mov. Disord. Soc. 2008, 23, 101–106. [Google Scholar] [CrossRef]
- Hely, M.A.; Reid, W.G.; Adena, M.A.; Halliday, G.M.; Morris, J.G. The Sydney multicenter study of Parkinson’s disease: The inevitability of dementia at 20 years. Mov. Disord. Off. J. Mov. Disord. Soc. 2008, 23, 837–844. [Google Scholar] [CrossRef]
- Hely, M.A.; Morris, J.G.; Reid, W.G.; Trafficante, R. Sydney Multicenter Study of Parkinson’s disease: Non-L-dopa-responsive problems dominate at 15 years. Mov. Disord. Off. J. Mov. Disord. Soc. 2005, 20, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Noyce, A.J.; Bestwick, J.P.; Silveira-Moriyama, L.; Hawkes, C.H.; Giovannoni, G.; Lees, A.J.; Schrag, A. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann. Neurol. 2012, 72, 893–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadimitriou, D.; Antonelou, R.; Miligkos, M.; Maniati, M.; Papagiannakis, N.; Bostantjopoulou, S.; Leonardos, A.; Koros, C.; Simitsi, A.; Papageorgiou, S.G.; et al. Motor and Nonmotor Features of Carriers of the p.A53T Alpha-Synuclein Mutation: A Longitudinal Study. Mov. Disord. Off. J. Mov. Disord. Soc. 2016, 31, 1226–1230. [Google Scholar] [CrossRef]
- Koros, C.; Simitsi, A.; Stefanis, L. Genetics of Parkinson’s Disease: Genotype-Phenotype Correlations. Int. Rev. Neurobiol. 2017, 132, 197–231. [Google Scholar] [CrossRef]
- Somme, J.H.; Gomez-Esteban, J.C.; Molano, A.; Tijero, B.; Lezcano, E.; Zarranz, J.J. Initial neuropsychological impairments in patients with the E46K mutation of the alpha-synuclein gene (PARK 1). J. Neurol. Sci. 2011, 310, 86–89. [Google Scholar] [CrossRef]
- Ghosh, D.; Sahay, S.; Ranjan, P.; Salot, S.; Mohite, G.M.; Singh, P.K.; Dwivedi, S.; Carvalho, E.; Banerjee, R.; Kumar, A.; et al. The newly discovered Parkinson’s disease associated Finnish mutation (A53E) attenuates alpha-synuclein aggregation and membrane binding. Biochemistry 2014, 53, 6419–6421. [Google Scholar] [CrossRef]
- Kiely, A.P.; Ling, H.; Asi, Y.T.; Kara, E.; Proukakis, C.; Schapira, A.H.; Morris, H.R.; Roberts, H.C.; Lubbe, S.; Limousin, P.; et al. Distinct clinical and neuropathological features of G51D SNCA mutation cases compared with SNCA duplication and H50Q mutation. Mol. Neurodegener. 2015, 10, 41. [Google Scholar] [CrossRef] [Green Version]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honore, A.; Rozas, N.; Pieri, L.; Madiona, K.; Durr, A.; Melki, R.; et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Tokutake, T.; Ishikawa, A.; Yoshimura, N.; Miyashita, A.; Kuwano, R.; Nishizawa, M.; Ikeuchi, T. Clinical and neuroimaging features of patient with early-onset Parkinson’s disease with dementia carrying SNCA p. G51D mutation. Park. Relat. Disord. 2014, 20, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Martikainen, M.H.; Paivarinta, M.; Hietala, M.; Kaasinen, V. Clinical and imaging findings in Parkinson disease associated with the A53E SNCA mutation. Neurol. Genet. 2015, 1, e27. [Google Scholar] [CrossRef]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Poyhonen, M.; Paetau, A. Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.T.; Mahul-Mellier, A.L.; Hegde, R.N.; Riviere, G.; Moons, R.; Ibanez de Opakua, A.; Magalhaes, P.; Rostami, I.; Donzelli, S.; Sobott, F.; et al. A NAC domain mutation (E83Q) unlocks the pathogenicity of human alpha-synuclein and recapitulates its pathological diversity. Sci. Adv. 2022, 8, eabn0044. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Koros, C.; Strohaker, T.; Schulte, C.; Bozi, M.; Varvaresos, S.; Ibanez de Opakua, A.; Simitsi, A.M.; Bougea, A.; Voumvourakis, K.; et al. A Novel SNCA A30G Mutation Causes Familial Parkinson’s Disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 1624–1633. [Google Scholar] [CrossRef]
- Lee, S.; Imai, Y.; Gehrke, S.; Liu, S.; Lu, B. The synaptic function of LRRK2. Biochem. Soc. Trans. 2012, 40, 1047–1051. [Google Scholar] [CrossRef] [Green Version]
- Rassu, M.; Del Giudice, M.G.; Sanna, S.; Taymans, J.M.; Morari, M.; Brugnoli, A.; Frassineti, M.; Masala, A.; Esposito, S.; Galioto, M.; et al. Role of LRRK2 in the regulation of dopamine receptor trafficking. PLoS ONE 2017, 12, e0179082. [Google Scholar] [CrossRef] [Green Version]
- Paisan-Ruiz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; Lopez de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Zabetian, C.P.; Samii, A.; Mosley, A.D.; Roberts, J.W.; Leis, B.C.; Yearout, D.; Raskind, W.H.; Griffith, A. A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology 2005, 65, 741–744. [Google Scholar] [CrossRef]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. S1), 216–231. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R. Cellular effects of LRRK2 mutations. Biochem. Soc. Trans. 2012, 40, 1070–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lill, C.M. Genetics of Parkinson’s disease. Mol. Cell. Probes 2016, 30, 386–396. [Google Scholar] [CrossRef]
- Thaler, A.; Gurevich, T.; Bar Shira, A.; Gana Weisz, M.; Ash, E.; Shiner, T.; Orr-Urtreger, A.; Giladi, N.; Mirelman, A. A “dose” effect of mutations in the GBA gene on Parkinson’s disease phenotype. Park. Relat. Disord. 2017, 36, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Montfort, M.; Chabas, A.; Vilageliu, L.; Grinberg, D. Functional analysis of 13 GBA mutant alleles identified in Gaucher disease patients: Pathogenic changes and “modifier” polymorphisms. Hum. Mutat. 2004, 23, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef] [Green Version]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hallett, P.; Isacson, O. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. New Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Sardi, S.P.; Clarke, J.; Viel, C.; Chan, M.; Tamsett, T.J.; Treleaven, C.M.; Bu, J.; Sweet, L.; Passini, M.A.; Dodge, J.C.; et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl. Acad. Sci. USA 2013, 110, 3537–3542. [Google Scholar] [CrossRef] [Green Version]
- Sardi, S.P.; Cheng, S.H.; Shihabuddin, L.S. Gaucher-related synucleinopathies: The examination of sporadic neurodegeneration from a rare (disease) angle. Prog. Neurobiol. 2015, 125, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Riley, B.E.; Lougheed, J.C.; Callaway, K.; Velasquez, M.; Brecht, E.; Nguyen, L.; Shaler, T.; Walker, D.; Yang, Y.; Regnstrom, K.; et al. Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat. Commun. 2013, 4, 1982. [Google Scholar] [CrossRef] [Green Version]
- Smith, W.W.; Pei, Z.; Jiang, H.; Moore, D.J.; Liang, Y.; West, A.B.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 18676–18681. [Google Scholar] [CrossRef] [Green Version]
- van der Merwe, C.; Jalali Sefid Dashti, Z.; Christoffels, A.; Loos, B.; Bardien, S. Evidence for a common biological pathway linking three Parkinson’s disease-causing genes: Parkin, PINK1 and DJ-1. Eur. J. Neurosci. 2015, 41, 1113–1125. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef] [Green Version]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Di Nottia, M.; Masciullo, M.; Verrigni, D.; Petrillo, S.; Modoni, A.; Rizzo, V.; Di Giuda, D.; Rizza, T.; Niceta, M.; Torraco, A.; et al. DJ-1 modulates mitochondrial response to oxidative stress: Clues from a novel diagnosis of PARK7. Clin. Genet. 2017, 92, 18–25. [Google Scholar] [CrossRef]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef] [Green Version]
- McCoy, M.K.; Cookson, M.R. Mitochondrial quality control and dynamics in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 869–882. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Abou-Sleiman, P.M.; Muqit, M.M.; McDonald, N.Q.; Yang, Y.X.; Gandhi, S.; Healy, D.G.; Harvey, K.; Harvey, R.J.; Deas, E.; Bhatia, K.; et al. A heterozygous effect for PINK1 mutations in Parkinson’s disease? Ann. Neurol. 2006, 60, 414–419. [Google Scholar] [CrossRef]
- Kim, C.; Lee, S.J. Controlling the mass action of alpha-synuclein in Parkinson’s disease. J. Neurochem. 2008, 107, 303–316. [Google Scholar] [CrossRef]
- Melki, R. Role of Different Alpha-Synuclein Strains in Synucleinopathies, Similarities with other Neurodegenerative Diseases. J. Park. Dis. 2015, 5, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. alpha-Synuclein and protein degradation systems: A reciprocal relationship. Mol. Neurobiol. 2013, 47, 537–551. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Angot, E.; Steiner, J.A.; Hansen, C.; Li, J.Y.; Brundin, P. Are synucleinopathies prion-like disorders? Lancet Neurol. 2010, 9, 1128–1138. [Google Scholar] [CrossRef]
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 2010, 11, 301–307. [Google Scholar] [CrossRef] [Green Version]
- George, S.; Rey, N.L.; Reichenbach, N.; Steiner, J.A.; Brundin, P. alpha-Synuclein: The long distance runner. Brain Pathol. 2013, 23, 350–357. [Google Scholar] [CrossRef]
- Brundin, P.; Li, J.Y.; Holton, J.L.; Lindvall, O.; Revesz, T. Research in motion: The enigma of Parkinson’s disease pathology spread. Nat. Rev. Neurosci. 2008, 9, 741–745. [Google Scholar] [CrossRef]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 2009, 4, e5515. [Google Scholar] [CrossRef] [Green Version]
- Volpicelli-Daley, L.A.; Abdelmotilib, H.; Liu, Z.; Stoyka, L.; Daher, J.P.; Milnerwood, A.J.; Unni, V.K.; Hirst, W.D.; Yue, Z.; Zhao, H.T.; et al. G2019S-LRRK2 Expression Augments alpha-Synuclein Sequestration into Inclusions in Neurons. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 7415–7427. [Google Scholar] [CrossRef]
- Fernandes, H.J.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular alpha-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.L.; Erion, J.R.; Tian, Y.; Liu, W.; Yin, D.M.; Ye, J.; Tang, B.; Mei, L.; Xiong, W.C. VPS35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for alpha-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 10613–10628. [Google Scholar] [CrossRef] [Green Version]
- Steele, J.W.; Ju, S.; Lachenmayer, M.L.; Liken, J.; Stock, A.; Kim, S.H.; Delgado, L.M.; Alfaro, I.E.; Bernales, S.; Verdile, G.; et al. Latrepirdine stimulates autophagy and reduces accumulation of alpha-synuclein in cells and in mouse brain. Mol. Psychiatry 2013, 18, 882–888. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [Green Version]
- Emmanouilidou, E.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 2010, 31, 953–968. [Google Scholar] [CrossRef]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [Green Version]
- Eschbach, J.; von Einem, B.; Muller, K.; Bayer, H.; Scheffold, A.; Morrison, B.E.; Rudolph, K.L.; Thal, D.R.; Witting, A.; Weydt, P.; et al. Mutual exacerbation of peroxisome proliferator-activated receptor gamma coactivator 1alpha deregulation and alpha-synuclein oligomerization. Ann. Neurol. 2015, 77, 15–32. [Google Scholar] [CrossRef] [Green Version]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Bolam, J.P.; Pissadaki, E.K. Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov. Disord. Off. J. Mov. Disord. Soc. 2012, 27, 1478–1483. [Google Scholar] [CrossRef] [Green Version]
- Pissadaki, E.K.; Bolam, J.P. The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in Parkinson’s disease. Front. Comput. Neurosci. 2013, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Surmeier, D.J.; Guzman, J.N.; Sanchez-Padilla, J.; Schumacker, P.T. The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in Parkinson’s disease. Neuroscience 2011, 198, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Surmeier, D.J.; Schumacker, P.T.; Guzman, J.D.; Ilijic, E.; Yang, B.; Zampese, E. Calcium and Parkinson’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 1013–1019. [Google Scholar] [CrossRef] [Green Version]
- Moehle, M.S.; West, A.B. M1 and M2 immune activation in Parkinson’s Disease: Foe and ally? Neuroscience 2015, 302, 59–73. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Gao, H.M.; Kotzbauer, P.T.; Uryu, K.; Leight, S.; Trojanowski, J.Q.; Lee, V.M. Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 7687–7698. [Google Scholar] [CrossRef] [Green Version]
- Jao, C.C.; Hegde, B.G.; Chen, J.; Haworth, I.S.; Langen, R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. USA 2008, 105, 19666–19671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of alpha-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Wang, L.; Bao, H.; Premi, S.; Das, U.; Chapman, E.R.; Roy, S. Functional cooperation of alpha-synuclein and VAMP2 in synaptic vesicle recycling. Proc. Natl. Acad. Sci. USA 2019, 116, 11113–11115. [Google Scholar] [CrossRef] [Green Version]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Burre, J.; Sharma, M.; Sudhof, T.C. alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283. [Google Scholar] [CrossRef] [Green Version]
- Hallacli, E.; Kayatekin, C.; Nazeen, S.; Wang, X.H.; Sheinkopf, Z.; Sathyakumar, S.; Sarkar, S.; Jiang, X.; Dong, X.; Di Maio, R.; et al. The Parkinson’s disease protein alpha-synuclein is a modulator of processing bodies and mRNA stability. Cell 2022, 185, 2035–2056.e33. [Google Scholar] [CrossRef]
- Schaser, A.J.; Osterberg, V.R.; Dent, S.E.; Stackhouse, T.L.; Wakeham, C.M.; Boutros, S.W.; Weston, L.J.; Owen, N.; Weissman, T.A.; Luna, E.; et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 2019, 9, 10919. [Google Scholar] [CrossRef] [Green Version]
- Fusco, G.; De Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G. Direct observation of the three regions in alpha-synuclein that determine its membrane-bound behaviour. Nat. Commun. 2014, 5, 3827. [Google Scholar] [CrossRef] [Green Version]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human alpha-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Li, J.; Fink, A.L. The association of alpha-synuclein with membranes affects bilayer structure, stability, and fibril formation. J. Biol. Chem. 2003, 278, 40186–40197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvagnion, C.; Brown, J.W.; Ouberai, M.M.; Flagmeier, P.; Vendruscolo, M.; Buell, A.K.; Sparr, E.; Dobson, C.M. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 7065–7070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiechle, M.; Grozdanov, V.; Danzer, K.M. The Role of Lipids in the Initiation of alpha-Synuclein Misfolding. Front. Cell Dev. Biol. 2020, 8, 562241. [Google Scholar] [CrossRef]
- O’Leary, E.I.; Lee, J.C. Interplay between alpha-synuclein amyloid formation and membrane structure. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Doherty, C.P.A.; Ulamec, S.M.; Maya-Martinez, R.; Good, S.C.; Makepeace, J.; Khan, G.N.; van Oosten-Hawle, P.; Radford, S.E.; Brockwell, D.J. A short motif in the N-terminal region of alpha-synuclein is critical for both aggregation and function. Nat. Struct. Mol. Biol. 2020, 27, 249–259. [Google Scholar] [CrossRef]
- McGlinchey, R.P.; Ni, X.; Shadish, J.A.; Jiang, J.; Lee, J.C. The N terminus of alpha-synuclein dictates fibril formation. Proc. Natl. Acad. Sci. USA 2021, 118, e2023487118. [Google Scholar] [CrossRef]
- Buratti, F.A.; Boeffinger, N.; Garro, H.A.; Flores, J.S.; Hita, F.J.; Goncalves, P.D.C.; Copello, F.D.R.; Lizarraga, L.; Rossetti, G.; Carloni, P.; et al. Aromaticity at position 39 in alpha-synuclein: A modulator of amyloid fibril assembly and membrane-bound conformations. Protein Sci. 2022, 31, e4360. [Google Scholar] [CrossRef]
- Ueda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, J.A.; Ivanova, M.I.; Sawaya, M.R.; Cascio, D.; Reyes, F.E.; Shi, D.; Sangwan, S.; Guenther, E.L.; Johnson, L.M.; Zhang, M.; et al. Structure of the toxic core of alpha-synuclein from invisible crystals. Nature 2015, 525, 486–490. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Ferreira, R.; Taylor, N.M.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM structure of alpha-synuclein fibrils. eLife 2018, 7. [Google Scholar] [CrossRef]
- Chakraborty, R.; Chattopadhyay, K. Cryo-Electron Microscopy Uncovers Key Residues within the Core of Alpha-Synuclein Fibrils. ACS Chem. Neurosci. 2019, 10, 1135–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izawa, Y.; Tateno, H.; Kameda, H.; Hirakawa, K.; Hato, K.; Yagi, H.; Hongo, K.; Mizobata, T.; Kawata, Y. Role of C-terminal negative charges and tyrosine residues in fibril formation of alpha-synuclein. Brain Behav. 2012, 2, 595–605. [Google Scholar] [CrossRef]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef]
- Gillam, J.E.; MacPhee, C.E. Modelling amyloid fibril formation kinetics: Mechanisms of nucleation and growth. J. Phys. Condens. Matter Inst. Phys. J. 2013, 25, 373101. [Google Scholar] [CrossRef]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [Green Version]
- Invernizzi, G.; Papaleo, E.; Sabate, R.; Ventura, S. Protein aggregation: Mechanisms and functional consequences. Int. J. Biochem. Cell Biol. 2012, 44, 1541–1554. [Google Scholar] [CrossRef] [PubMed]
- Marvian, A.T.; Koss, D.J.; Aliakbari, F.; Morshedi, D.; Outeiro, T.F. In vitro models of synucleinopathies: Informing on molecular mechanisms and protective strategies. J. Neurochem. 2019, 150, 535–565. [Google Scholar] [CrossRef] [Green Version]
- Rutherford, N.J.; Moore, B.D.; Golde, T.E.; Giasson, B.I. Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of alpha-synuclein. J. Neurochem. 2014, 131, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J. Biol. Chem. 2001, 276, 10737–10744. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, R.; Lund, M.; Sparr, E.; Linse, S. Anomalous Salt Dependence Reveals an Interplay of Attractive and Repulsive Electrostatic Interactions in α-synuclein Fibril Formation. QRB Discov. 2020, 1, e2. [Google Scholar] [CrossRef]
- Ziaunys, M.; Sakalauskas, A.; Mikalauskaite, K.; Smirnovas, V. Polymorphism of Alpha-Synuclein Amyloid Fibrils Depends on Ionic Strength and Protein Concentration. Int. J. Mol. Sci. 2021, 22, 12382. [Google Scholar] [CrossRef]
- de Oliveira, G.A.P.; Silva, J.L. Alpha-synuclein stepwise aggregation reveals features of an early onset mutation in Parkinson’s disease. Commun. Biol. 2019, 2, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buell, A.K.; Galvagnion, C.; Gaspar, R.; Sparr, E.; Vendruscolo, M.; Knowles, T.P.; Linse, S.; Dobson, C.M. Solution conditions determine the relative importance of nucleation and growth processes in alpha-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2014, 111, 7671–7676. [Google Scholar] [CrossRef] [Green Version]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Bockmann, A.; Meier, B.H.; et al. Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef] [Green Version]
- Pena-Diaz, S.; Pujols, J.; Vasili, E.; Pinheiro, F.; Santos, J.; Manglano-Artunedo, Z.; Outeiro, T.F.; Ventura, S. The small aromatic compound SynuClean-D inhibits the aggregation and seeded polymerization of multiple alpha-synuclein strains. J. Biol. Chem. 2022, 298, 101902. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Heikenwalder, M.; Polymenidou, M. Insights into prion strains and neurotoxicity. Nat. Rev. Mol. Cell Biol. 2007, 8, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Behbahanipour, M.; Garcia-Pardo, J.; Ventura, S. Decoding the role of coiled-coil motifs in human prion-like proteins. Prion 2021, 15, 143–154. [Google Scholar] [CrossRef]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, T.R.; Holmes, B.B.; Furman, J.L.; Dhavale, D.D.; Su, B.W.; Song, E.S.; Cairns, N.J.; Kotzbauer, P.T.; Diamond, M.I. Parkinson’s disease and multiple system atrophy have distinct alpha-synuclein seed characteristics. J. Biol. Chem. 2019, 294, 1045–1058. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Ferreira, R.; Taylor, N.M.; Arteni, A.A.; Kumari, P.; Mona, D.; Ringler, P.; Britschgi, M.; Lauer, M.E.; Makky, A.; Verasdonck, J.; et al. Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. eLife 2019, 8, e48907. [Google Scholar] [CrossRef]
- Boyer, D.R.; Li, B.; Sun, C.; Fan, W.; Sawaya, M.R.; Jiang, L.; Eisenberg, D.S. Structures of fibrils formed by alpha-synuclein hereditary disease mutant H50Q reveal new polymorphs. Nat. Struct. Mol. Biol. 2019, 26, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of full-length alpha-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 2018, 9, 3609. [Google Scholar] [CrossRef] [PubMed]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of alpha-synuclein filaments from multiple system atrophy. Nature 2020, 585, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating alpha-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; So, R.W.L.; Lau, H.H.C.; Sang, J.C.; Ruiz-Riquelme, A.; Fleck, S.C.; Stuart, E.; Menon, S.; Visanji, N.P.; Meisl, G.; et al. alpha-Synuclein strains target distinct brain regions and cell types. Nat. Neurosci. 2020, 23, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. alpha-Synuclein aggregation nucleates through liquid-liquid phase separation. Nat. Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef]
- Hardenberg, M.C.; Sinnige, T.; Casford, S.; Dada, S.T.; Poudel, C.; Robinson, E.A.; Fuxreiter, M.; Kaminksi, C.F.; Kaminski Schierle, G.S.; Nollen, E.A.A.; et al. Observation of an alpha-synuclein liquid droplet state and its maturation into Lewy body-like assemblies. J. Mol. Cell Biol. 2021, 13, 282–294. [Google Scholar] [CrossRef]
- Takamuku, M.; Sugishita, T.; Tamaki, H.; Dong, L.; So, M.; Fujiwara, T.; Matsuki, Y. Evolution of alpha-synuclein conformation ensemble toward amyloid fibril via liquid-liquid phase separation (LLPS) as investigated by dynamic nuclear polarization-enhanced solid-state MAS NMR. Neurochem. Int. 2022, 157, 105345. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Julicher, F.; Hyman, A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef]
- Li, X.H.; Chavali, P.L.; Pancsa, R.; Chavali, S.; Babu, M.M. Function and Regulation of Phase-Separated Biological Condensates. Biochemistry 2018, 57, 2452–2461. [Google Scholar] [CrossRef]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, J.; Weber, S.C.; Vaidya, N.; Haataja, M.; Brangwynne, C.P. RNA transcription modulates phase transition-driven nuclear body assembly. Proc. Natl. Acad. Sci. USA 2015, 112, E5237–E5245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Banjade, S.; Cheng, H.C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodruff, J.B.; Ferreira Gomes, B.; Widlund, P.O.; Mahamid, J.; Honigmann, A.; Hyman, A.A. The Centrosome Is a Selective Condensate that Nucleates Microtubules by Concentrating Tubulin. Cell 2017, 169, 1066–1077.e1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawner, A.S.; Ray, S.; Yadav, P.; Mukherjee, S.; Panigrahi, R.; Poudyal, M.; Patel, K.; Ghosh, D.; Kummerant, E.; Kumar, A.; et al. Modulating alpha-Synuclein Liquid-Liquid Phase Separation. Biochemistry 2021, 60, 3676–3696. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Xu, B.; Liu, Y. Calcium promotes alpha-synuclein liquid-liquid phase separation to accelerate amyloid aggregation. Biochem. Biophys. Res. Commun. 2022, 603, 13–20. [Google Scholar] [CrossRef]
- Xu, B.; Huang, S.; Liu, Y.; Wan, C.; Gu, Y.; Wang, D.; Yu, H. Manganese promotes alpha-synuclein amyloid aggregation through the induction of protein phase transition. J. Biol. Chem. 2022, 298, 101469. [Google Scholar] [CrossRef]
- Oliva, R.; Mukherjee, S.K.; Ostermeier, L.; Pazurek, L.A.; Kriegler, S.; Bader, V.; Prumbaum, D.; Raunser, S.; Winklhofer, K.F.; Tatzelt, J.; et al. Remodeling of the Fibrillation Pathway of alpha-Synuclein by Interaction with Antimicrobial Peptide LL-III. Chemistry 2021, 27, 11845–11851. [Google Scholar] [CrossRef]
- Agarwal, A.; Arora, L.; Rai, S.K.; Avni, A.; Mukhopadhyay, S. Spatiotemporal modulations in heterotypic condensates of prion and alpha-synuclein control phase transitions and amyloid conversion. Nat. Commun. 2022, 13, 1154. [Google Scholar] [CrossRef]
- Siegert, A.; Rankovic, M.; Favretto, F.; Ukmar-Godec, T.; Strohaker, T.; Becker, S.; Zweckstetter, M. Interplay between tau and alpha-synuclein liquid-liquid phase separation. Protein Sci. 2021, 30, 1326–1336. [Google Scholar] [CrossRef]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by alpha-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremades, N.; Cohen, S.I.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campioni, S.; Mannini, B.; Zampagni, M.; Pensalfini, A.; Parrini, C.; Evangelisti, E.; Relini, A.; Stefani, M.; Dobson, C.M.; Cecchi, C.; et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010, 6, 140–147. [Google Scholar] [CrossRef]
- Cascella, R.; Chen, S.W.; Bigi, A.; Camino, J.D.; Xu, C.K.; Dobson, C.M.; Chiti, F.; Cremades, N.; Cecchi, C. The release of toxic oligomers from alpha-synuclein fibrils induces dysfunction in neuronal cells. Nat. Commun. 2021, 12, 1814. [Google Scholar] [CrossRef] [PubMed]
- Gadhe, L.; Sakunthala, A.; Mukherjee, S.; Gahlot, N.; Bera, R.; Sawner, A.S.; Kadu, P.; Maji, S.K. Intermediates of alpha-synuclein aggregation: Implications in Parkinson’s disease pathogenesis. Biophys. Chem. 2022, 281, 106736. [Google Scholar] [CrossRef]
- Cascella, R.; Bigi, A.; Cremades, N.; Cecchi, C. Effects of oligomer toxicity, fibril toxicity and fibril spreading in synucleinopathies. Cell. Mol. Life Sci. 2022, 79, 174. [Google Scholar] [CrossRef]
- Hansen, C.; Angot, E.; Bergstrom, A.L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Rietdijk, C.D.; Perez-Pardo, P.; Garssen, J.; van Wezel, R.J.; Kraneveld, A.D. Exploring Braak’s Hypothesis of Parkinson’s Disease. Front. Neurol. 2017, 8, 37. [Google Scholar] [CrossRef]
- Alba, R.; Bosch, A.; Chillon, M. Gutless adenovirus: Last-generation adenovirus for gene therapy. Gene Ther. 2005, 12 (Suppl. S1), S18–S27. [Google Scholar] [CrossRef] [Green Version]
- Froelich, S.; Tai, A.; Wang, P. Lentiviral vectors for immune cells targeting. Immunopharmacol. Immunotoxicol. 2010, 32, 208–218. [Google Scholar] [CrossRef]
- Kotterman, M.A.; Schaffer, D.V. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet 2014, 15, 445–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeWitt, P.A.; Rezai, A.R.; Leehey, M.A.; Ojemann, S.G.; Flaherty, A.W.; Eskandar, E.N.; Kostyk, S.K.; Thomas, K.; Sarkar, A.; Siddiqui, M.S.; et al. AAV2-GAD gene therapy for advanced Parkinson’s disease: A double-blind, sham-surgery controlled, randomised trial. Lancet Neurol. 2011, 10, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, S.; Wang, L.; Ikeguchi, K.; Fujimoto, K.; Nakano, I.; Ozawa, K. Recombinant adeno-associated viral vectors bring gene therapy for Parkinson’s disease closer to reality. J. Neurol. 2002, 249 (Suppl. S2), II36–II40. [Google Scholar] [CrossRef]
- Jarraya, B.; Boulet, S.; Ralph, G.S.; Jan, C.; Bonvento, G.; Azzouz, M.; Miskin, J.E.; Shin, M.; Delzescaux, T.; Drouot, X.; et al. Dopamine gene therapy for Parkinson’s disease in a nonhuman primate without associated dyskinesia. Sci. Transl. Med. 2009, 1, 2ra4. [Google Scholar] [CrossRef]
- Marks, W.J., Jr.; Bartus, R.T.; Siffert, J.; Davis, C.S.; Lozano, A.; Boulis, N.; Vitek, J.; Stacy, M.; Turner, D.; Verhagen, L.; et al. Gene delivery of AAV2-neurturin for Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol. 2010, 9, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, A.; Kirik, D.; Rosenblad, C.; Georgievska, B.; Lundberg, C.; Mandel, R.J. Towards a neuroprotective gene therapy for Parkinson’s disease: Use of adenovirus, AAV and lentivirus vectors for gene transfer of GDNF to the nigrostriatal system in the rat Parkinson model. Brain Res. 2000, 886, 82–98. [Google Scholar] [CrossRef]
- Tian, Y.Y.; Tang, C.J.; Wang, J.N.; Feng, Y.; Chen, X.W.; Wang, L.; Qiao, X.; Sun, S.G. Favorable effects of VEGF gene transfer on a rat model of Parkinson disease using adeno-associated viral vectors. Neurosci. Lett. 2007, 421, 239–244. [Google Scholar] [CrossRef]
- Oh, S.M.; Chang, M.Y.; Song, J.J.; Rhee, Y.H.; Joe, E.H.; Lee, H.S.; Yi, S.H.; Lee, S.H. Combined Nurr1 and Foxa2 roles in the therapy of Parkinson’s disease. EMBO Mol. Med. 2015, 7, 510–525. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.U.; Bilal, M.; Shah, J.A.; Kaushik, A.; Teissedre, P.L.; Kujawska, M. CRISPR-Cas9-Based Technology and Its Relevance to Gene Editing in Parkinson’s Disease. Pharmaceutics 2022, 14, 1252. [Google Scholar] [CrossRef]
- Lindvall, O.; Brundin, P.; Widner, H.; Rehncrona, S.; Gustavii, B.; Frackowiak, R.; Leenders, K.L.; Sawle, G.; Rothwell, J.C.; Marsden, C.D.; et al. Grafts of fetal dopamine neurons survive and improve motor function in Parkinson’s disease. Science 1990, 247, 574–577. [Google Scholar] [CrossRef]
- Piccini, P.; Brooks, D.J.; Bjorklund, A.; Gunn, R.N.; Grasby, P.M.; Rimoldi, O.; Brundin, P.; Hagell, P.; Rehncrona, S.; Widner, H.; et al. Dopamine release from nigral transplants visualized in vivo in a Parkinson’s patient. Nat. Neurosci. 1999, 2, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A.; Drouin-Ouellet, J.; Parmar, M. Cell-based therapies for Parkinson disease-past insights and future potential. Nat. Rev. Neurol. 2015, 11, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Petit, G.H.; Olsson, T.T.; Brundin, P. The future of cell therapies and brain repair: Parkinson’s disease leads the way. Neuropathol. Appl. Neurobiol. 2014, 40, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Hauser, R.A. alpha-Synuclein in Parkinson’s disease: Getting to the core of the matter. Lancet Neurol. 2015, 14, 785–786. [Google Scholar] [CrossRef]
- Kingwell, K. Zeroing in on neurodegenerative alpha-synuclein. Nat. Rev. Drug Discov. 2017, 16, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Mikitsh, J.L.; Chacko, A.M. Pathways for small molecule delivery to the central nervous system across the blood-brain barrier. Perspect. Medicin. Chem. 2014, 6, 11–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhu, M.; Manning-Bog, A.B.; Di Monte, D.A.; Fink, A.L. Dopamine and L-dopa disaggregate amyloid fibrils: Implications for Parkinson’s and Alzheimer’s disease. FASEB J. 2004, 18, 962–964. [Google Scholar] [CrossRef] [PubMed]
- Li, H.T.; Lin, D.H.; Luo, X.Y.; Zhang, F.; Ji, L.N.; Du, H.N.; Song, G.Q.; Hu, J.; Zhou, J.W.; Hu, H.Y. Inhibition of alpha-synuclein fibrillization by dopamine analogs via reaction with the amino groups of alpha-synuclein. Implication for dopaminergic neurodegeneration. FEBS J. 2005, 272, 3661–3672. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.L.; Pham, C.L.; Galatis, D.; Fodero-Tavoletti, M.T.; Perez, K.; Hill, A.F.; Masters, C.L.; Ali, F.E.; Barnham, K.J.; Cappai, R. Formation of dopamine-mediated alpha-synuclein-soluble oligomers requires methionine oxidation. Free Radic. Biol. Med. 2009, 46, 1328–1337. [Google Scholar] [CrossRef]
- Uversky, V.N.; Yamin, G.; Souillac, P.O.; Goers, J.; Glaser, C.B.; Fink, A.L. Methionine oxidation inhibits fibrillation of human alpha-synuclein in vitro. FEBS Lett. 2002, 517, 239–244. [Google Scholar] [CrossRef] [Green Version]
- Norris, E.H.; Giasson, B.I.; Hodara, R.; Xu, S.; Trojanowski, J.Q.; Ischiropoulos, H.; Lee, V.M. Reversible inhibition of alpha-synuclein fibrillization by dopaminochrome-mediated conformational alterations. J. Biol. Chem. 2005, 280, 21212–21219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latawiec, D.; Herrera, F.; Bek, A.; Losasso, V.; Candotti, M.; Benetti, F.; Carlino, E.; Kranjc, A.; Lazzarino, M.; Gustincich, S.; et al. Modulation of alpha-synuclein aggregation by dopamine analogs. PLoS ONE 2010, 5, e9234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, F.E.; Chesi, A.; Paleologou, K.E.; Schmid, A.; Munoz, A.; Vendruscolo, M.; Gustincich, S.; Lashuel, H.A.; Carloni, P. Inhibition of alpha-synuclein fibrillization by dopamine is mediated by interactions with five C-terminal residues and with E83 in the NAC region. PLoS ONE 2008, 3, e3394. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Armakola, M.; Dumoulin, M.; Parastatidis, I.; Ischiropoulos, H. Cellular oligomerization of alpha-synuclein is determined by the interaction of oxidized catechols with a C-terminal sequence. J. Biol. Chem. 2007, 282, 31621–31630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dibenedetto, D.; Rossetti, G.; Caliandro, R.; Carloni, P. A molecular dynamics simulation-based interpretation of nuclear magnetic resonance multidimensional heteronuclear spectra of alpha-synuclein.dopamine adducts. Biochemistry 2013, 52, 6672–6683. [Google Scholar] [CrossRef]
- Mor, D.E.; Tsika, E.; Mazzulli, J.R.; Gould, N.S.; Kim, H.; Daniels, M.J.; Doshi, S.; Gupta, P.; Grossman, J.L.; Tan, V.X.; et al. Dopamine induces soluble alpha-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 2017, 20, 1560–1568. [Google Scholar] [CrossRef] [Green Version]
- Mor, D.E.; Daniels, M.J.; Ischiropoulos, H. The usual suspects, dopamine and alpha-synuclein, conspire to cause neurodegeneration. Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 167–179. [Google Scholar] [CrossRef]
- Ono, K.; Yamada, M. Antioxidant compounds have potent anti-fibrillogenic and fibril-destabilizing effects for alpha-synuclein fibrils in vitro. J. Neurochem. 2006, 97, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Suzuki, N.; Taniguchi, S.; Oikawa, T.; Nonaka, T.; Iwatsubo, T.; Hisanaga, S.; Goedert, M.; Hasegawa, M. Small molecule inhibitors of alpha-synuclein filament assembly. Biochemistry 2006, 45, 6085–6094. [Google Scholar] [CrossRef]
- Dhouafli, Z.; Cuanalo-Contreras, K.; Hayouni, E.A.; Mays, C.E.; Soto, C.; Moreno-Gonzalez, I. Inhibition of protein misfolding and aggregation by natural phenolic compounds. Cell. Mol. Life Sci. 2018, 75, 3521–3538. [Google Scholar] [CrossRef]
- Hornedo-Ortega, R.; Alvarez-Fernandez, M.A.; Cerezo, A.B.; Richard, T.; Troncoso, A.M.A.; Garcia-Parrilla, M.A.C. Protocatechuic Acid: Inhibition of Fibril Formation, Destabilization of Preformed Fibrils of Amyloid-beta and alpha-Synuclein, and Neuroprotection. J. Agric. Food Chem. 2016, 64, 7722–7732. [Google Scholar] [CrossRef] [PubMed]
- Pandey, N.; Strider, J.; Nolan, W.C.; Yan, S.X.; Galvin, J.E. Curcumin inhibits aggregation of alpha-synuclein. Acta Neuropathol. 2008, 115, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Rajamani, S.; Kaylor, J.; Han, S.; Zhou, F.; Fink, A.L. The flavonoid baicalein inhibits fibrillation of alpha-synuclein and disaggregates existing fibrils. J. Biol. Chem. 2004, 279, 26846–26857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazili, N.A.; Naeem, A. Anti-fibrillation potency of caffeic acid against an antidepressant induced fibrillogenesis of human alpha-synuclein: Implications for Parkinson’s disease. Biochimie 2015, 108, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, B.; Lapidus, L.J. Curcumin prevents aggregation in alpha-synuclein by increasing reconfiguration rate. J. Biol. Chem. 2012, 287, 9193–9199. [Google Scholar] [CrossRef] [Green Version]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef]
- Lorenzen, N.; Nielsen, S.B.; Yoshimura, Y.; Vad, B.S.; Andersen, C.B.; Betzer, C.; Kaspersen, J.D.; Christiansen, G.; Pedersen, J.S.; Jensen, P.H.; et al. How epigallocatechin gallate can inhibit alpha-synuclein oligomer toxicity in vitro. J. Biol. Chem. 2014, 289, 21299–21310. [Google Scholar] [CrossRef] [Green Version]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.K.; Kotia, V.; Ghosh, D.; Mohite, G.M.; Kumar, A.; Maji, S.K. Curcumin modulates alpha-synuclein aggregation and toxicity. ACS Chem. Neurosci. 2013, 4, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Chen, J.; Liu, Y. Curcumin Interacts with alpha-Synuclein Condensates To Inhibit Amyloid Aggregation under Phase Separation. ACS Omega 2022, 7, 30281–30290. [Google Scholar] [CrossRef]
- Xu, B.; Mo, X.; Chen, J.; Yu, H.; Liu, Y. Myricetin Inhibits alpha-Synuclein Amyloid Aggregation by Delaying the Liquid-to-Solid Phase Transition. Chembiochem 2022, 23, e202200216. [Google Scholar] [CrossRef] [PubMed]
- Ardah, M.T.; Paleologou, K.E.; Lv, G.; Abul Khair, S.B.; Kazim, A.S.; Minhas, S.T.; Al-Tel, T.H.; Al-Hayani, A.A.; Haque, M.E.; Eliezer, D.; et al. Structure activity relationship of phenolic acid inhibitors of alpha-synuclein fibril formation and toxicity. Front. Aging Neurosci. 2014, 6, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kujawska, M.; Jodynis-Liebert, J. Polyphenols in Parkinson’s Disease: A Systematic Review of In Vivo Studies. Nutrients 2018, 10, 642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Q.; Uversky, V.N.; Huang, M.; Kang, H.; Xu, F.; Liu, X.; Lian, L.; Liang, Q.; Jiang, H.; Liu, A.; et al. Baicalein inhibits alpha-synuclein oligomer formation and prevents progression of alpha-synuclein accumulation in a rotenone mouse model of Parkinson’s disease. Biochim. Biophys. Acta 2016, 1862, 1883–1890. [Google Scholar] [CrossRef]
- Sharma, N.; Nehru, B. Curcumin affords neuroprotection and inhibits alpha-synuclein aggregation in lipopolysaccharide-induced Parkinson’s disease model. Inflammopharmacology 2018, 26, 349–360. [Google Scholar] [CrossRef]
- Ogawa, N.; Mizukawa, K.; Hirose, Y.; Kajita, S.; Ohara, S.; Watanabe, Y. MPTP-induced parkinsonian model in mice: Biochemistry, pharmacology and behavior. Eur. Neurol. 1987, 26 (Suppl. S1), 16–23. [Google Scholar] [CrossRef]
- Jeon, B.S.; Jackson-Lewis, V.; Burke, R.E. 6-Hydroxydopamine lesion of the rat substantia nigra: Time course and morphology of cell death. Neurodegeneration 1995, 4, 131–137. [Google Scholar] [CrossRef]
- Pan-Montojo, F.; Anichtchik, O.; Dening, Y.; Knels, L.; Pursche, S.; Jung, R.; Jackson, S.; Gille, G.; Spillantini, M.G.; Reichmann, H.; et al. Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS ONE 2010, 5, e8762. [Google Scholar] [CrossRef] [Green Version]
- Vajragupta, O.; Boonchoong, P.; Watanabe, H.; Tohda, M.; Kummasud, N.; Sumanont, Y. Manganese complexes of curcumin and its derivatives: Evaluation for the radical scavenging ability and neuroprotective activity. Free Radic. Biol. Med. 2003, 35, 1632–1644. [Google Scholar] [CrossRef]
- Rajeswari, A.; Sabesan, M. Inhibition of monoamine oxidase-B by the polyphenolic compound, curcumin and its metabolite tetrahydrocurcumin, in a model of Parkinson’s disease induced by MPTP neurodegeneration in mice. Inflammopharmacology 2008, 16, 96–99. [Google Scholar] [CrossRef]
- Nagarajan, S.; Chellappan, D.R.; Chinnaswamy, P.; Thulasingam, S. Ferulic acid pretreatment mitigates MPTP-induced motor impairment and histopathological alterations in C57BL/6 mice. Pharm. Biol. 2015, 53, 1591–1601. [Google Scholar] [CrossRef]
- Lv, C.; Hong, T.; Yang, Z.; Zhang, Y.; Wang, L.; Dong, M.; Zhao, J.; Mu, J.; Meng, Y. Effect of Quercetin in the 1-Methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-Induced Mouse Model of Parkinson’s Disease. Evid. Based Complement. Alternat. Med. 2012, 2012, 928643. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; He, G.R.; Sun, L.; Lan, X.; Shi, L.L.; Xuan, Z.H.; Du, G.H. Assessment of the treatment effect of baicalein on a model of Parkinsonian tremor and elucidation of the mechanism. Life Sci. 2012, 91, 5–13. [Google Scholar] [CrossRef]
- Li, S.; Pu, X.P. Neuroprotective effect of kaempferol against a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mouse model of Parkinson’s disease. Biol. Pharm. Bull. 2011, 34, 1291–1296. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Park, H.R.; Ji, S.T.; Lee, Y.; Lee, J. Baicalein attenuates astroglial activation in the 1-methyl-4-phenyl-1,2,3,4-tetrahydropyridine-induced Parkinson’s disease model by downregulating the activations of nuclear factor-kappaB, ERK, and JNK. J. Neurosci. Res. 2014, 92, 130–139. [Google Scholar] [CrossRef]
- Chen, M.; Wang, T.; Yue, F.; Li, X.; Wang, P.; Li, Y.; Chan, P.; Yu, S. Tea polyphenols alleviate motor impairments, dopaminergic neuronal injury, and cerebral alpha-synuclein aggregation in MPTP-intoxicated parkinsonian monkeys. Neuroscience 2015, 286, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, K.J.; Osterberg, V.R.; Meshul, C.K.; Soumyanath, A.; Unni, V.K. Curcumin Treatment Improves Motor Behavior in alpha-Synuclein Transgenic Mice. PLoS ONE 2015, 10, e0128510. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kumar, R.; Kumari, M.; Kumari, R.; Saha, S.; Bhavesh, N.S.; Maiti, T.K. Ellagic Acid Inhibits alpha-Synuclein Aggregation at Multiple Stages and Reduces Its Cytotoxicity. ACS Chem. Neurosci. 2021, 12, 1919–1930. [Google Scholar] [CrossRef] [PubMed]
- Ardah, M.T.; Eid, N.; Kitada, T.; Haque, M.E. Ellagic Acid Prevents alpha-Synuclein Aggregation and Protects SH-SY5Y Cells from Aggregated alpha-Synuclein-Induced Toxicity via Suppression of Apoptosis and Activation of Autophagy. Int. J. Mol. Sci. 2021, 22, 13398. [Google Scholar] [CrossRef]
- Chen, M.; Liu, A.; Ouyang, Y.; Huang, Y.; Chao, X.; Pi, R. Fasudil and its analogs: A new powerful weapon in the long war against central nervous system disorders? Expert Opin. Investig. Drugs 2013, 22, 537–550. [Google Scholar] [CrossRef]
- He, Q.; Li, Y.H.; Guo, S.S.; Wang, Y.; Lin, W.; Zhang, Q.; Wang, J.; Ma, C.G.; Xiao, B.G. Inhibition of Rho-kinase by Fasudil protects dopamine neurons and attenuates inflammatory response in an intranasal lipopolysaccharide-mediated Parkinson’s model. Eur. J. Neurosci. 2016, 43, 41–52. [Google Scholar] [CrossRef]
- Tatenhorst, L.; Eckermann, K.; Dambeck, V.; Fonseca-Ornelas, L.; Walle, H.; Lopes da Fonseca, T.; Koch, J.C.; Becker, S.; Tonges, L.; Bahr, M.; et al. Fasudil attenuates aggregation of alpha-synuclein in models of Parkinson’s disease. Acta Neuropathol. Commun. 2016, 4, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robustelli, P.; Ibanez-de-Opakua, A.; Campbell-Bezat, C.; Giordanetto, F.; Becker, S.; Zweckstetter, M.; Pan, A.C.; Shaw, D.E. Molecular Basis of Small-Molecule Binding to alpha-Synuclein. J. Am. Chem. Soc. 2022, 144, 2501–2510. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koprich, J.B.; Johnston, T.H.; Reyes, M.G.; Sun, X.; Brotchie, J.M. Expression of human A53T alpha-synuclein in the rat substantia nigra using a novel AAV1/2 vector produces a rapidly evolving pathology with protein aggregation, dystrophic neurite architecture and nigrostriatal degeneration with potential to model the pathology of Parkinson’s disease. Mol. Neurodegener. 2010, 5, 43. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.J.; Bu, L.L.; Shen, C.; Ge, J.J.; He, S.J.; Yu, H.L.; Tang, Y.L.; Jue, Z.; Sun, Y.M.; Yu, W.B.; et al. Fasudil Promotes alpha-Synuclein Clearance in an AAV-Mediated alpha-Synuclein Rat Model of Parkinson’s Disease by Autophagy Activation. J. Park. Dis. 2020, 10, 969–979. [Google Scholar] [CrossRef]
- Wood, J.G.; Mirra, S.S.; Pollock, N.J.; Binder, L.I. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc. Natl. Acad. Sci. USA 1986, 83, 4040–4043. [Google Scholar] [CrossRef] [Green Version]
- Delacourte, A.; Defossez, A. Alzheimer’s disease: Tau proteins, the promoting factors of microtubule assembly, are major components of paired helical filaments. J. Neurol. Sci. 1986, 76, 173–186. [Google Scholar] [CrossRef]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.; Roth, M.; Harrington, C.R. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. USA 1996, 93, 11213–11218. [Google Scholar] [CrossRef] [Green Version]
- Wischik, C.M.; Staff, R.T.; Wischik, D.J.; Bentham, P.; Murray, A.D.; Storey, J.M.; Kook, K.A.; Harrington, C.R. Tau aggregation inhibitor therapy: An exploratory phase 2 study in mild or moderate Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 705–720. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, S.; Feldman, H.H.; Schneider, L.S.; Wilcock, G.K.; Frisoni, G.B.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Wischik, D.J.; et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: A randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 2016, 388, 2873–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcock, G.K.; Gauthier, S.; Frisoni, G.B.; Jia, J.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Schelter, B.O.; Wischik, D.J.; et al. Potential of Low Dose Leuco-Methylthioninium Bis(Hydromethanesulphonate) (LMTM) Monotherapy for Treatment of Mild Alzheimer’s Disease: Cohort Analysis as Modified Primary Outcome in a Phase III Clinical Trial. J. Alzheimers Dis. 2018, 61, 435–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, K.; Frahm, S.; Horsley, D.; Rickard, J.E.; Melis, V.; Goatman, E.A.; Magbagbeolu, M.; Douglas, M.; Leith, M.G.; Baddeley, T.C.; et al. A Protein Aggregation Inhibitor, Leuco-Methylthioninium Bis(Hydromethanesulfonate), Decreases alpha-Synuclein Inclusions in a Transgenic Mouse Model of Synucleinopathy. Front. Mol. Neurosci. 2017, 10, 447. [Google Scholar] [CrossRef] [PubMed]
- Frahm, S.; Melis, V.; Horsley, D.; Rickard, J.E.; Riedel, G.; Fadda, P.; Scherma, M.; Harrington, C.R.; Wischik, C.M.; Theuring, F.; et al. Alpha-Synuclein transgenic mice, h-alpha-SynL62, display alpha-Syn aggregation and a dopaminergic phenotype reminiscent of Parkinson’s disease. Behav. Brain Res. 2018, 339, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Brycki, B.; Koenig, H.; Pospieszny, T. Quaternary Alkylammonium Conjugates of Steroids: Synthesis, Molecular Structure, and Biological Studies. Molecules 2015, 20, 20887–20900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.S.; Wehrli, S.; Roder, H.; Rogers, M.; Forrest, J.N., Jr.; McCrimmon, D.; Zasloff, M. Squalamine: An aminosterol antibiotic from the shark. Proc. Natl. Acad. Sci. USA 1993, 90, 1354–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khelaifia, S.; Drancourt, M. Susceptibility of archaea to antimicrobial agents: Applications to clinical microbiology. Clin. Microbiol. Infect. 2012, 18, 841–848. [Google Scholar] [CrossRef] [Green Version]
- Cushnie, T.P.; Cushnie, B.; Lamb, A.J. Alkaloids: An overview of their antibacterial, antibiotic-enhancing and antivirulence activities. Int. J. Antimicrob. Agents 2014, 44, 377–386. [Google Scholar] [CrossRef]
- Schlottmann, P.G.; Alezzandrini, A.A.; Zas, M.; Rodriguez, F.J.; Luna, J.D.; Wu, L. New Treatment Modalities for Neovascular Age-Related Macular Degeneration. Asia Pac. J. Ophthalmol. 2017, 6, 514–519. [Google Scholar] [CrossRef]
- Perni, M.; Galvagnion, C.; Maltsev, A.; Meisl, G.; Muller, M.B.; Challa, P.K.; Kirkegaard, J.B.; Flagmeier, P.; Cohen, S.I.; Cascella, R.; et al. A natural product inhibits the initiation of alpha-synuclein aggregation and suppresses its toxicity. Proc. Natl. Acad. Sci. USA 2017, 114, E1009–E1017. [Google Scholar] [CrossRef] [Green Version]
- Yeung, T.; Gilbert, G.E.; Shi, J.; Silvius, J.; Kapus, A.; Grinstein, S. Membrane phosphatidylserine regulates surface charge and protein localization. Science 2008, 319, 210–213. [Google Scholar] [CrossRef]
- Sumioka, A.; Yan, D.; Tomita, S. TARP phosphorylation regulates synaptic AMPA receptors through lipid bilayers. Neuron 2010, 66, 755–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, R.T.; Jaumouille, V.; Yeung, T.; Furuya, W.; Peltekova, I.; Boucher, A.; Zasloff, M.; Orlowski, J.; Grinstein, S. Membrane surface charge dictates the structure and function of the epithelial Na+/H+ exchanger. EMBO J. 2011, 30, 679–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvagnion, C.; Buell, A.K.; Meisl, G.; Michaels, T.C.; Vendruscolo, M.; Knowles, T.P.; Dobson, C.M. Lipid vesicles trigger alpha-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 2015, 11, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Limbocker, R.; Staats, R.; Chia, S.; Ruggeri, F.S.; Mannini, B.; Xu, C.K.; Perni, M.; Cascella, R.; Bigi, A.; Sasser, L.R.; et al. Squalamine and Its Derivatives Modulate the Aggregation of Amyloid-beta and alpha-Synuclein and Suppress the Toxicity of Their Oligomers. Front. Neurosci. 2021, 15, 680026. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Maguire-Nguyen, K.K.; Rando, T.A.; Zasloff, M.A.; Strange, K.B.; Yin, V.P. The protein tyrosine phosphatase 1B inhibitor MSI-1436 stimulates regeneration of heart and multiple other tissues. NPJ Regen. Med. 2017, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Perni, M.; Flagmeier, P.; Limbocker, R.; Cascella, R.; Aprile, F.A.; Galvagnion, C.; Heller, G.T.; Meisl, G.; Chen, S.W.; Kumita, J.R.; et al. Multistep Inhibition of alpha-Synuclein Aggregation and Toxicity in vitro and in vivo by Trodusquemine. ACS Chem. Biol. 2018, 13, 2308–2319. [Google Scholar] [CrossRef] [PubMed]
- Limbocker, R.; Mannini, B.; Ruggeri, F.S.; Cascella, R.; Xu, C.K.; Perni, M.; Chia, S.; Chen, S.W.; Habchi, J.; Bigi, A.; et al. Trodusquemine displaces protein misfolded oligomers from cell membranes and abrogates their cytotoxicity through a generic mechanism. Commun. Biol. 2020, 3, 435. [Google Scholar] [CrossRef] [PubMed]
- Flagmeier, P.; Meisl, G.; Vendruscolo, M.; Knowles, T.P.; Dobson, C.M.; Buell, A.K.; Galvagnion, C. Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of alpha-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2016, 113, 10328–10333. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.W.; Buell, A.K.; Michaels, T.C.; Meisl, G.; Carozza, J.; Flagmeier, P.; Vendruscolo, M.; Knowles, T.P.; Dobson, C.M.; Galvagnion, C. Beta-Synuclein suppresses both the initiation and amplification steps of alpha-synuclein aggregation via competitive binding to surfaces. Sci. Rep. 2016, 6, 36010. [Google Scholar] [CrossRef]
- Chen, S.W.; Drakulic, S.; Deas, E.; Ouberai, M.; Aprile, F.A.; Arranz, R.; Ness, S.; Roodveldt, C.; Guilliams, T.; De-Genst, E.J.; et al. Structural characterization of toxic oligomers that are kinetically trapped during alpha-synuclein fibril formation. Proc. Natl. Acad. Sci. USA 2015, 112, E1994–E2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelisti, E.; Cascella, R.; Becatti, M.; Marrazza, G.; Dobson, C.M.; Chiti, F.; Stefani, M.; Cecchi, C. Binding affinity of amyloid oligomers to cellular membranes is a generic indicator of cellular dysfunction in protein misfolding diseases. Sci. Rep. 2016, 6, 32721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhudesai, S.; Sinha, S.; Attar, A.; Kotagiri, A.; Fitzmaurice, A.G.; Lakshmanan, R.; Ivanova, M.I.; Loo, J.A.; Klarner, F.G.; Schrader, T.; et al. A novel “molecular tweezer” inhibitor of alpha-synuclein neurotoxicity in vitro and in vivo. Neurotherapeutics 2012, 9, 464–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, S.; Safaie, B.M.; Wongkongkathep, P.; Ivanova, M.I.; Attar, A.; Klarner, F.G.; Schrader, T.; Loo, J.A.; Bitan, G.; Lapidus, L.J. Molecular basis for preventing alpha-synuclein aggregation by a molecular tweezer. J. Biol. Chem. 2014, 289, 10727–10737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rink, E.; Wullimann, M.F. The teleostean (zebrafish) dopaminergic system ascending to the subpallium (striatum) is located in the basal diencephalon (posterior tuberculum). Brain Res. 2001, 889, 316–330. [Google Scholar] [CrossRef]
- Chesselet, M.F.; Richter, F.; Zhu, C.; Magen, I.; Watson, M.B.; Subramaniam, S.R. A progressive mouse model of Parkinson’s disease: The Thy1-aSyn (“Line 61”) mice. Neurotherapeutics 2012, 9, 297–314. [Google Scholar] [CrossRef] [Green Version]
- Richter, F.; Subramaniam, S.R.; Magen, I.; Lee, P.; Hayes, J.; Attar, A.; Zhu, C.; Franich, N.R.; Bove, N.; De La Rosa, K.; et al. A Molecular Tweezer Ameliorates Motor Deficits in Mice Overexpressing alpha-Synuclein. Neurotherapeutics 2017, 14, 1107–1119. [Google Scholar] [CrossRef] [Green Version]
- Janezic, S.; Threlfell, S.; Dodson, P.D.; Dowie, M.J.; Taylor, T.N.; Potgieter, D.; Parkkinen, L.; Senior, S.L.; Anwar, S.; Ryan, B.; et al. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc. Natl. Acad. Sci. USA 2013, 110, E4016–E4025. [Google Scholar] [CrossRef] [Green Version]
- Refolo, V.; Bez, F.; Polissidis, A.; Kuzdas-Wood, D.; Sturm, E.; Kamaratou, M.; Poewe, W.; Stefanis, L.; Angela Cenci, M.; Romero-Ramos, M.; et al. Progressive striatonigral degeneration in a transgenic mouse model of multiple system atrophy: Translational implications for interventional therapies. Acta Neuropathol. Commun. 2018, 6, 2. [Google Scholar] [CrossRef]
- Bengoa-Vergniory, N.; Faggiani, E.; Ramos-Gonzalez, P.; Kirkiz, E.; Connor-Robson, N.; Brown, L.V.; Siddique, I.; Li, Z.; Vingill, S.; Cioroch, M.; et al. CLR01 protects dopaminergic neurons in vitro and in mouse models of Parkinson’s disease. Nat. Commun. 2020, 11, 4885. [Google Scholar] [CrossRef]
- Herrera-Vaquero, M.; Bouquio, D.; Kallab, M.; Biggs, K.; Nair, G.; Ochoa, J.; Heras-Garvin, A.; Heid, C.; Hadrovic, I.; Poewe, W.; et al. The molecular tweezer CLR01 reduces aggregated, pathologic, and seeding-competent alpha-synuclein in experimental multiple system atrophy. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 165513. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Lopes, D.H.; Du, Z.; Pang, E.S.; Shanmugam, A.; Lomakin, A.; Talbiersky, P.; Tennstaedt, A.; McDaniel, K.; Bakshi, R.; et al. Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J. Am. Chem. Soc. 2011, 133, 16958–16969. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, N.; Pereira-Henriques, A.; Attar, A.; Klarner, F.G.; Schrader, T.; Bitan, G.; Gales, L.; Saraiva, M.J.; Almeida, M.R. Molecular tweezers targeting transthyretin amyloidosis. Neurotherapeutics 2014, 11, 450–461. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Sharikov, Y.; Miller, M.A.; Masliah, E. Mechanism of alpha-synuclein oligomerization and membrane interaction: Theoretical approach to unstructured proteins studies. Nanomedicine 2008, 4, 350–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsigelny, I.F.; Sharikov, Y.; Wrasidlo, W.; Gonzalez, T.; Desplats, P.A.; Crews, L.; Spencer, B.; Masliah, E. Role of alpha-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 2012, 279, 1000–1013. [Google Scholar] [CrossRef] [Green Version]
- Wrasidlo, W.; Tsigelny, I.F.; Price, D.L.; Dutta, G.; Rockenstein, E.; Schwarz, T.C.; Ledolter, K.; Bonhaus, D.; Paulino, A.; Eleuteri, S.; et al. A de novo compound targeting alpha-synuclein improves deficits in models of Parkinson’s disease. Brain A J. Neurol. 2016, 139, 3217–3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, D.L.; Koike, M.A.; Khan, A.; Wrasidlo, W.; Rockenstein, E.; Masliah, E.; Bonhaus, D. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci. Rep. 2018, 8, 16165. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Johnson, R.; Wittmer, C.; Maile, M.; Tatsukawa, K.; Wong, J.L.; Gill, M.B.; Stocking, E.M.; Natala, S.R.; Paulino, A.D.; et al. NPT520-34 improves neuropathology and motor deficits in a transgenic mouse model of Parkinson’s disease. Brain A J. Neurol. 2021, 144, 3692–3709. [Google Scholar] [CrossRef]
- Maculins, T.; Garcia-Pardo, J.; Skenderovic, A.; Gebel, J.; Putyrski, M.; Vorobyov, A.; Busse, P.; Varga, G.; Kuzikov, M.; Zaliani, A.; et al. Discovery of Protein-Protein Interaction Inhibitors by Integrating Protein Engineering and Chemical Screening Platforms. Cell Chem. Biol. 2020, 27, 1441–1451.e1447. [Google Scholar] [CrossRef]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013, 125, 795–813. [Google Scholar] [CrossRef] [Green Version]
- Kurnik, M.; Sahin, C.; Andersen, C.B.; Lorenzen, N.; Giehm, L.; Mohammad-Beigi, H.; Jessen, C.M.; Pedersen, J.S.; Christiansen, G.; Petersen, S.V.; et al. Potent alpha-Synuclein Aggregation Inhibitors, Identified by High-Throughput Screening, Mainly Target the Monomeric State. Cell Chem. Biol. 2018, 25, 1389–1402.e1389. [Google Scholar] [CrossRef]
- Hideshima, M.; Kimura, Y.; Aguirre, C.; Kakuda, K.; Takeuchi, T.; Choong, C.J.; Doi, J.; Nabekura, K.; Yamaguchi, K.; Nakajima, K.; et al. Two-step screening method to identify alpha-synuclein aggregation inhibitors for Parkinson’s disease. Sci. Rep. 2022, 12, 351. [Google Scholar] [CrossRef]
- Staats, R.; Michaels, T.C.T.; Flagmeier, P.; Chia, S.A.; Horne, R.I.; Habchi, J.; Linse, S.; Knowles, T.P.J.; Dobson, C.M.; Vendruscolo, M. Screening of small molecules using the inhibition of oligomer formation in alpha-synuclein aggregation as a selection parameter. Commun. Chem. 2020, 3, 191. [Google Scholar] [CrossRef] [PubMed]
- Antonschmidt, L.; Matthes, D.; Dervisoglu, R.; Frieg, B.; Dienemann, C.; Leonov, A.; Nimerovsky, E.; Sant, V.; Ryazanov, S.; Giese, A.; et al. The clinical drug candidate anle138b binds in a cavity of lipidic alpha-synuclein fibrils. Nat. Commun. 2022, 13, 5385. [Google Scholar] [CrossRef]
- Levin, J.; Schmidt, F.; Boehm, C.; Prix, C.; Botzel, K.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A. The oligomer modulator anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset. Acta Neuropathol. 2014, 127, 779–780. [Google Scholar] [CrossRef] [Green Version]
- Heras-Garvin, A.; Weckbecker, D.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A.; Wenning, G.K.; Stefanova, N. Anle138b modulates alpha-synuclein oligomerization and prevents motor decline and neurodegeneration in a mouse model of multiple system atrophy. Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Fellner, L.; Kuzdas-Wood, D.; Levin, J.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A.; Wenning, G.K.; Stefanova, N. Anle138b Partly Ameliorates Motor Deficits Despite Failure of Neuroprotection in a Model of Advanced Multiple System Atrophy. Front. Neurosci. 2016, 10, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, J.; Sing, N.; Melbourne, S.; Morgan, A.; Mariner, C.; Spillantini, M.G.; Wegrzynowicz, M.; Dalley, J.W.; Langer, S.; Ryazanov, S.; et al. Safety, tolerability and pharmacokinetics of the oligomer modulator anle138b with exposure levels sufficient for therapeutic efficacy in a murine Parkinson model: A randomised, double-blind, placebo-controlled phase 1a trial. EBioMedicine 2022, 80, 104021. [Google Scholar] [CrossRef]
- Pujols, J.; Pena-Diaz, S.; Conde-Gimenez, M.; Pinheiro, F.; Navarro, S.; Sancho, J.; Ventura, S. High-Throughput Screening Methodology to Identify Alpha-Synuclein Aggregation Inhibitors. Int. J. Mol. Sci. 2017, 18, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujols, J.; Pena-Diaz, S.; Lazaro, D.F.; Peccati, F.; Pinheiro, F.; Gonzalez, D.; Carija, A.; Navarro, S.; Conde-Gimenez, M.; Garcia, J.; et al. Small molecule inhibits alpha-synuclein aggregation, disrupts amyloid fibrils, and prevents degeneration of dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2018, 115, 10481–10486. [Google Scholar] [CrossRef] [Green Version]
- Pena-Diaz, S.; Pujols, J.; Conde-Gimenez, M.; Carija, A.; Dalfo, E.; Garcia, J.; Navarro, S.; Pinheiro, F.; Santos, J.; Salvatella, X.; et al. ZPD-2, a Small Compound That Inhibits alpha-Synuclein Amyloid Aggregation and Its Seeded Polymerization. Front. Mol. Neurosci. 2019, 12, 306. [Google Scholar] [CrossRef] [PubMed]
- Pena-Diaz, S.; Pujols, J.; Pinheiro, F.; Santos, J.; Pallares, I.; Navarro, S.; Conde-Gimenez, M.; Garcia, J.; Salvatella, X.; Dalfo, E.; et al. Inhibition of alpha-Synuclein Aggregation and Mature Fibril Disassembling With a Minimalistic Compound, ZPDm. Front. Bioeng. Biotechnol. 2020, 8, 588947. [Google Scholar] [CrossRef] [PubMed]
- van Ham, T.J.; Thijssen, K.L.; Breitling, R.; Hofstra, R.M.; Plasterk, R.H.; Nollen, E.A.C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008, 4, e1000027. [Google Scholar] [CrossRef]
- Hamamichi, S.; Rivas, R.N.; Knight, A.L.; Cao, S.; Caldwell, K.A.; Caldwell, G.A. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc. Natl. Acad. Sci. USA 2008, 105, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Harrington, A.J.; Yacoubian, T.A.; Slone, S.R.; Caldwell, K.A.; Caldwell, G.A. Functional analysis of VPS41-mediated neuroprotection in Caenorhabditis elegans and mammalian models of Parkinson’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 2142–2153. [Google Scholar] [CrossRef] [Green Version]
- Pena, D.S.; Ventura, S. One ring is sufficient to inhibit alpha-synuclein aggregation. Neural Regen. Res. 2022, 17, 508–511. [Google Scholar] [CrossRef]
- Mahia, A.; Pena-Diaz, S.; Navarro, S.; Jose Galano-Frutos, J.; Pallares, I.; Pujols, J.; Diaz-de-Villegas, M.D.; Galvez, J.A.; Ventura, S.; Sancho, J. Design, synthesis and structure-activity evaluation of novel 2-pyridone-based inhibitors of alpha-synuclein aggregation with potentially improved BBB permeability. Bioorg. Chem. 2021, 117, 105472. [Google Scholar] [CrossRef] [PubMed]
- De Luca, L.; Vittorio, S.; Pena-Diaz, S.; Pitasi, G.; Fornt-Sune, M.; Bucolo, F.; Ventura, S.; Gitto, R. Ligand-Based Discovery of a Small Molecule as Inhibitor of alpha-Synuclein Amyloid Formation. Int. J. Mol. Sci. 2022, 23, 14844. [Google Scholar] [CrossRef]
- Gitto, R.; Vittorio, S.; Bucolo, F.; Pena-Diaz, S.; Siracusa, R.; Cuzzocrea, S.; Ventura, S.; Di Paola, R.; De Luca, L. Discovery of Neuroprotective Agents Based on a 5-(4-Pyridinyl)-1,2,4-triazole Scaffold. ACS Chem. Neurosci. 2022, 13, 581–586. [Google Scholar] [CrossRef]
- Braun, A.R.; Liao, E.E.; Horvath, M.; Kalra, P.; Acosta, K.; Young, M.C.; Kochen, N.N.; Lo, C.H.; Brown, R.; Evans, M.D.; et al. Potent inhibitors of toxic alpha-synuclein identified via cellular time-resolved FRET biosensors. NPJ Park. Dis. 2021, 7, 52. [Google Scholar] [CrossRef]
- Wang, Q.; Yao, S.; Yang, Z.X.; Zhou, C.; Zhang, Y.; Zhang, Y.; Zhang, L.; Li, J.T.; Xu, Z.J.; Zhu, W.L.; et al. Pharmacological characterization of the small molecule 03A10 as an inhibitor of alpha-synuclein aggregation for Parkinson’s disease treatment. Acta Pharmacol. Sin. 2023. [Google Scholar] [CrossRef] [PubMed]
- Priss, A.; Afitska, K.; Galkin, M.; Yushchenko, D.A.; Shvadchak, V.V. Rationally Designed Protein-Based Inhibitor of alpha-Synuclein Fibrillization in Cells. J. Med. Chem. 2021, 64, 6827–6837. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hu, J.; Zhang, G.; Qin, L.; Wen, H.; Tang, Y. Pharmacophore modeling and 3D-QSAR study for the design of novel alpha-synuclein aggregation inhibitors. J. Mol. Model. 2021, 27, 260. [Google Scholar] [CrossRef]
- Sahihi, M.; Gaci, F.; Navizet, I. Identification of new alpha-synuclein fibrillogenesis inhibitor using in silico structure-based virtual screening. J. Mol. Graph. Model. 2021, 108, 108010. [Google Scholar] [CrossRef]
- Chia, S.; Faidon Brotzakis, Z.; Horne, R.I.; Possenti, A.; Mannini, B.; Cataldi, R.; Nowinska, M.; Staats, R.; Linse, S.; Knowles, T.P.J.; et al. Structure-Based Discovery of Small-Molecule Inhibitors of the Autocatalytic Proliferation of alpha-Synuclein Aggregates. Mol. Pharm. 2023, 20, 183–193. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
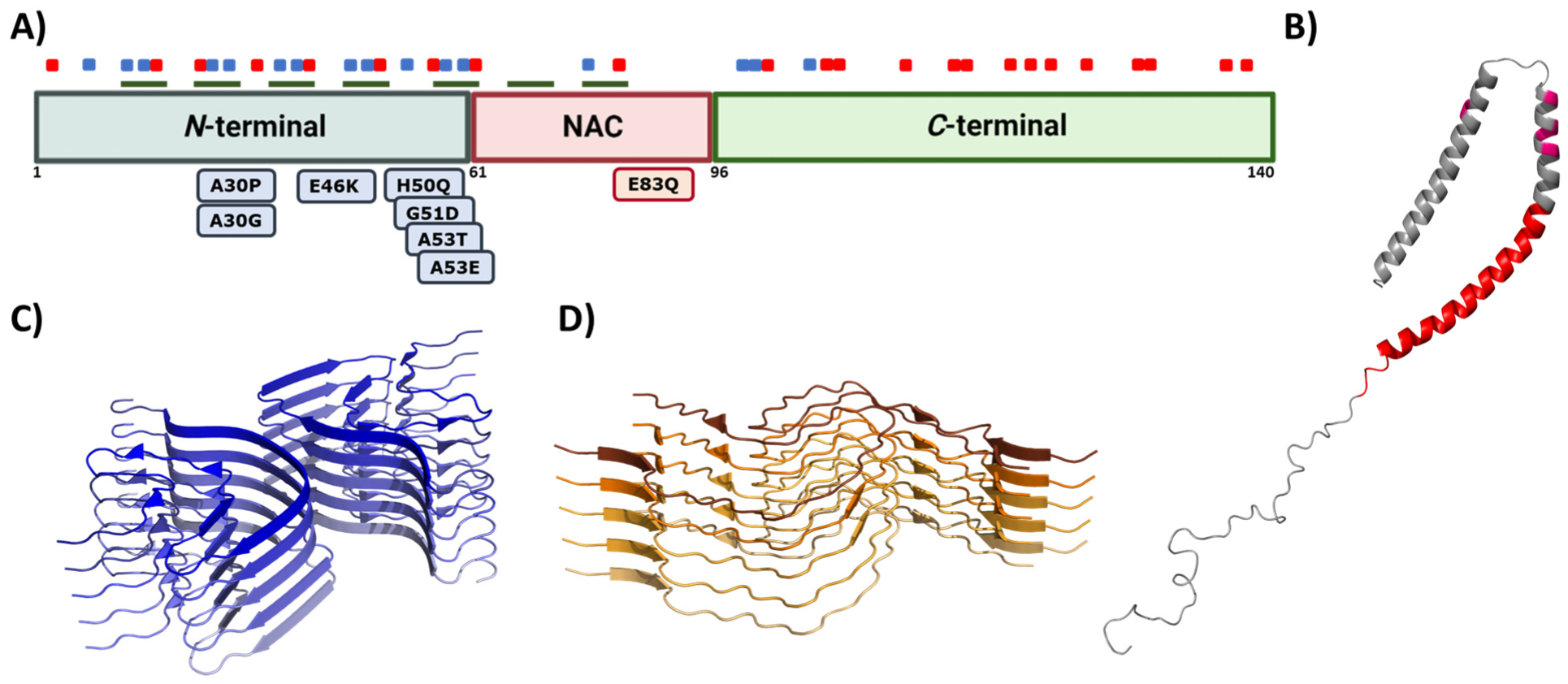{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Role in PD |
|---|---|
| SNCA (α-Synuclein) | Protein aggregation Prion-like transmission Synaptic function and dopamine transmission |
| GBA (Glucocerebrosidase) | Lysosome mediated autophagy pathway |
| LRRK2 | Neurite structure Protein and membrane trafficking Lysosome-mediated autophagy pathway Synaptic function and dopamine transmission |
| MAPT (Tau) | Protein aggregation Neurite structure |
| VPS35 | Protein and membrane trafficking Lysosome-mediated autophagy pathway |
| DNAJC13 (REM-8) | Protein and membrane trafficking Lysosome-mediated autophagy pathway |
| GAK | Protein and membrane trafficking |
| RAB7L1 | Protein and membrane trafficking |
| RAB39B | Protein and membrane trafficking |
| Parkin | Ubiquitin-mediated proteasome Mitochondrial dysfunction and mitophagy |
| FBX07 | Ubiquitin-mediated proteasome |
| SCA3 (Ataxin-3) | Ubiquitin-mediated proteasome |
| PINK1 | Mitochondrial dysfunction and mitophagy |
| DJ-1 | Mitochondrial dysfunction and mitophagy |
| CHCHD2 | Mitochondrial dysfunction and mitophagy |
| POLG1 | Mitochondrial dysfunction and mitophagy |
| SREVF1 | Mitochondrial dysfunction and mitophagy |
| ATP12A2 | Lysosome-mediated autophagy pathway |
| SCARB2 (LIMP-2) | Lysosome-mediated autophagy pathway |
| SYNJ1 (Synaptojanin 1) | Synaptic function and dopamine transmission |
| GCH1 | Synaptic function and dopamine transmission |
| STX1B (Syntaxin-1B) | Synaptic function and dopamine transmission |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peña-Díaz, S.; García-Pardo, J.; Ventura, S. Development of Small Molecules Targeting α-Synuclein Aggregation: A Promising Strategy to Treat Parkinson’s Disease. Pharmaceutics 2023, 15, 839. https://doi.org/10.3390/pharmaceutics15030839
Peña-Díaz S, García-Pardo J, Ventura S. Development of Small Molecules Targeting α-Synuclein Aggregation: A Promising Strategy to Treat Parkinson’s Disease. Pharmaceutics. 2023; 15(3):839. https://doi.org/10.3390/pharmaceutics15030839
Chicago/Turabian StylePeña-Díaz, Samuel, Javier García-Pardo, and Salvador Ventura. 2023. "Development of Small Molecules Targeting α-Synuclein Aggregation: A Promising Strategy to Treat Parkinson’s Disease" Pharmaceutics 15, no. 3: 839. https://doi.org/10.3390/pharmaceutics15030839
APA StylePeña-Díaz, S., García-Pardo, J., & Ventura, S. (2023). Development of Small Molecules Targeting α-Synuclein Aggregation: A Promising Strategy to Treat Parkinson’s Disease. Pharmaceutics, 15(3), 839. https://doi.org/10.3390/pharmaceutics15030839