
Quality by Design-Based Development of Solid Self-Emulsifying Drug Delivery System (SEDDS) as a Potential Carrier for Oral Delivery of Lysozyme

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
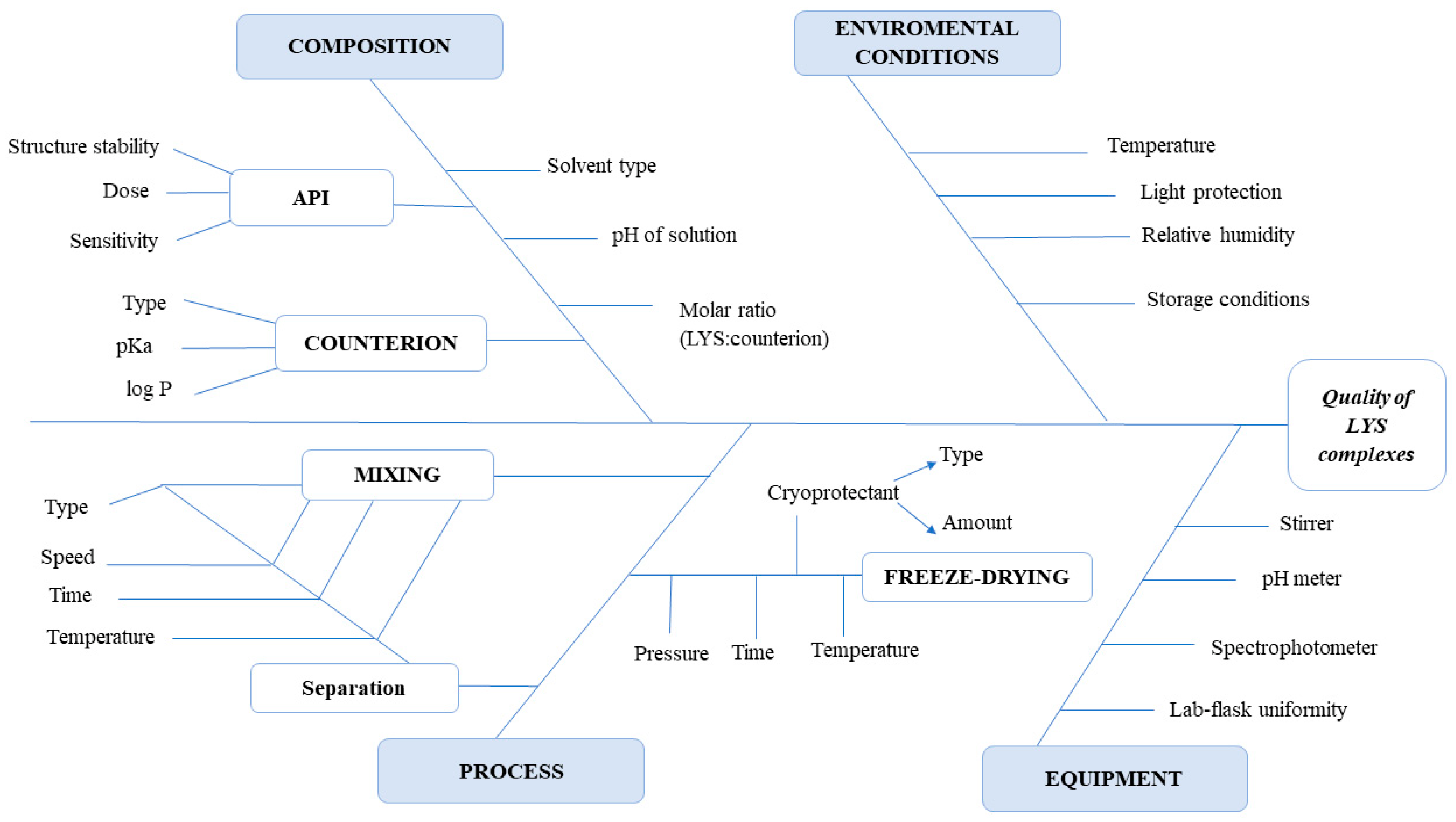
2.2.1. Quality by Design Methodology
2.2.2. Development and Characterization of HIP Complex—LYS:SDS
Hydrophobic Ion Pairing of LYS with SDS
Experimental Design—Full Factorial Design (23)
Determination of LYS Binding Efficiency
Dissociation of LYS from HIP Complex
Evaluation of LYS Enzymatic Activity after HIP
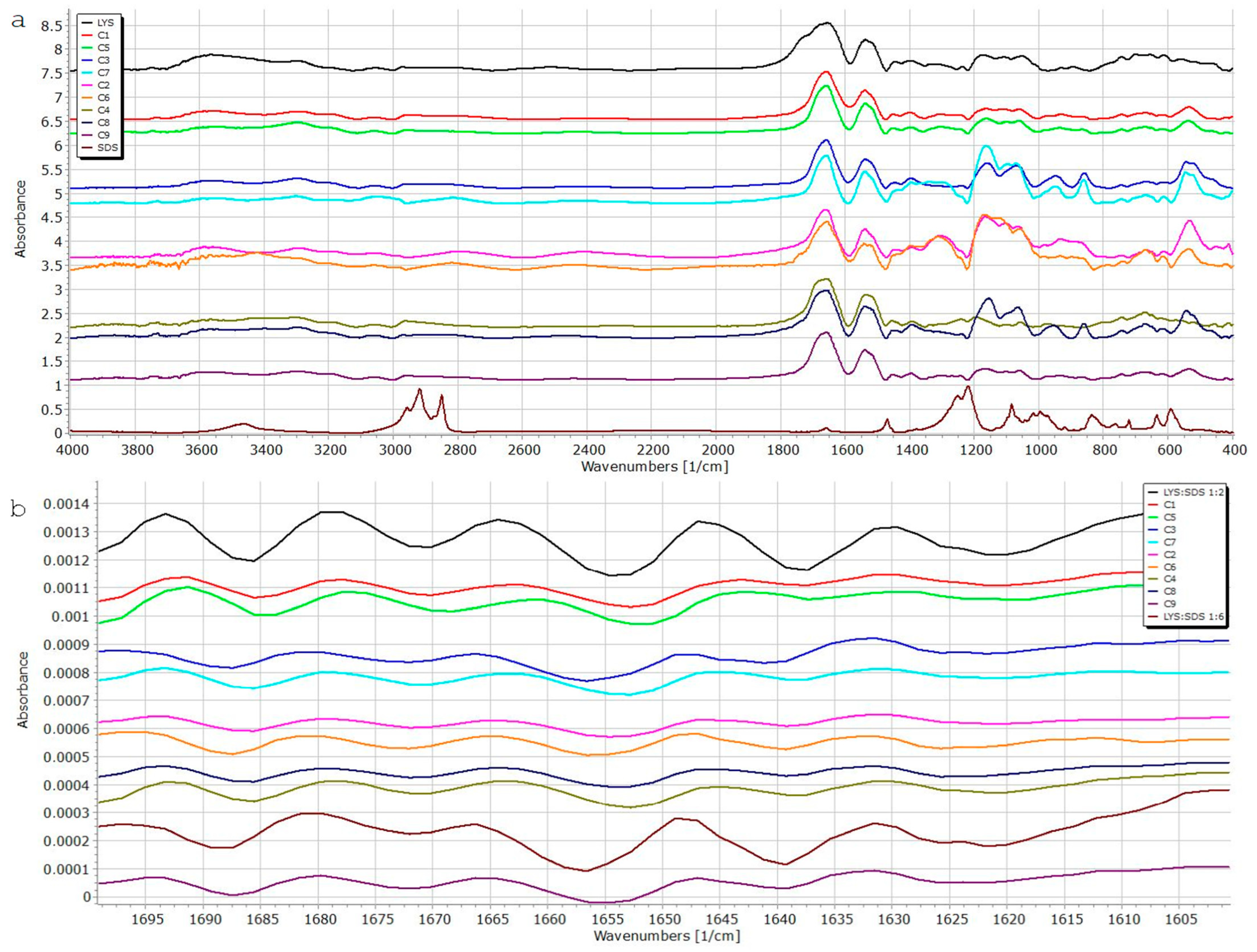
Fourier-Transform Infrared Spectroscopy (FTIR)
2.2.3. Development and Characterization of Liquid SEDDS Loaded with LYS:SDS Complex
Screening of Oils, Surfactants, and Co-Surfactants
Preparation of Liquid SEDDS According to the Mixture Design
Evaluation of Self-Emulsifying Properties
Droplet Size and Zeta Potential Measurements
Stability Studies of SEDDS
Preparation of LYS:SDS-Complex-Loaded SEDDS Formulations
Robustness to Dilution and pH Changes
Determination of log DSEDDS/release medium
2.2.4. Development and Characterization of Solid SEDDS Loaded with LYS:SDS Complex
Preparation of Solid SEDDS Containing LYS:SDS Complex
Characterization of Solid SEDDS Containing LYS:SDS—Flow Properties
Self-Emulsifying Properties of Solid SEDDS
Determination of LYS Activity after Incorporation into liquid and Solid SEDDS
2.2.5. Development and Characterization of Self-Emulsifying Tablets with LYS:SDS Complex
Compression of Tablets and Their Characterization
Dissolution Studies
3. Results and Discussion
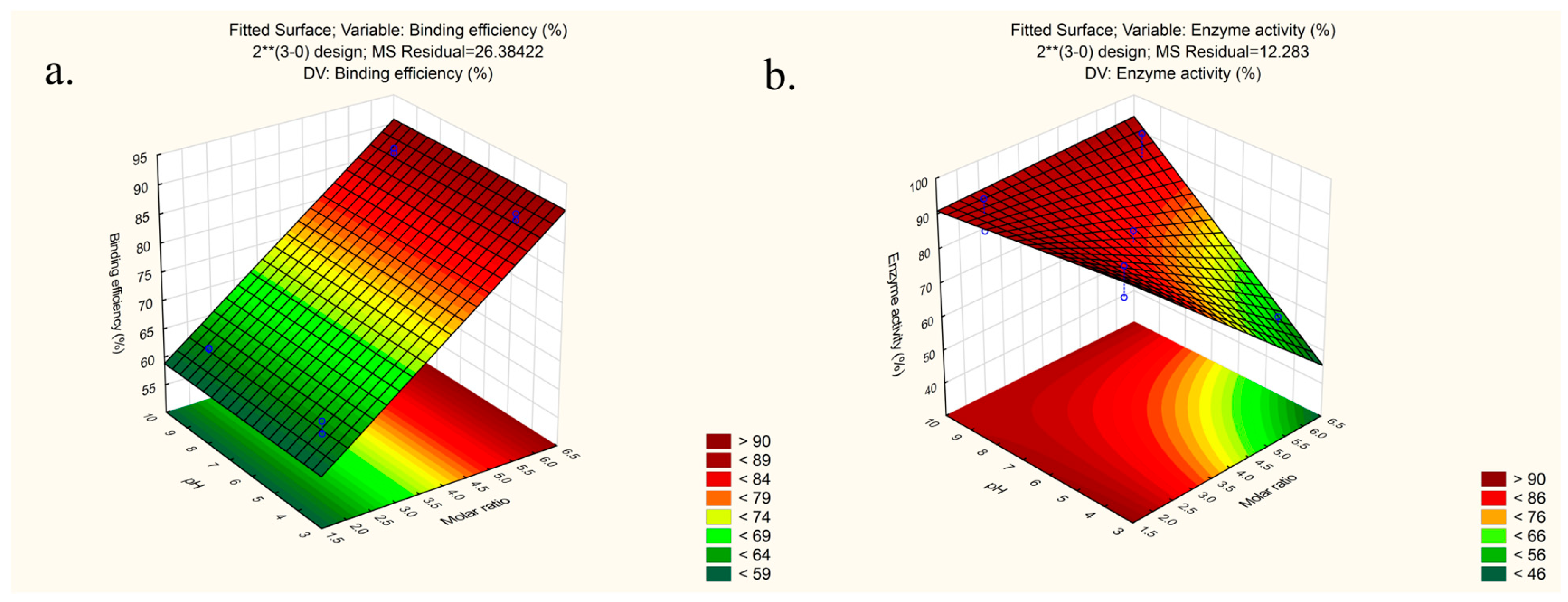
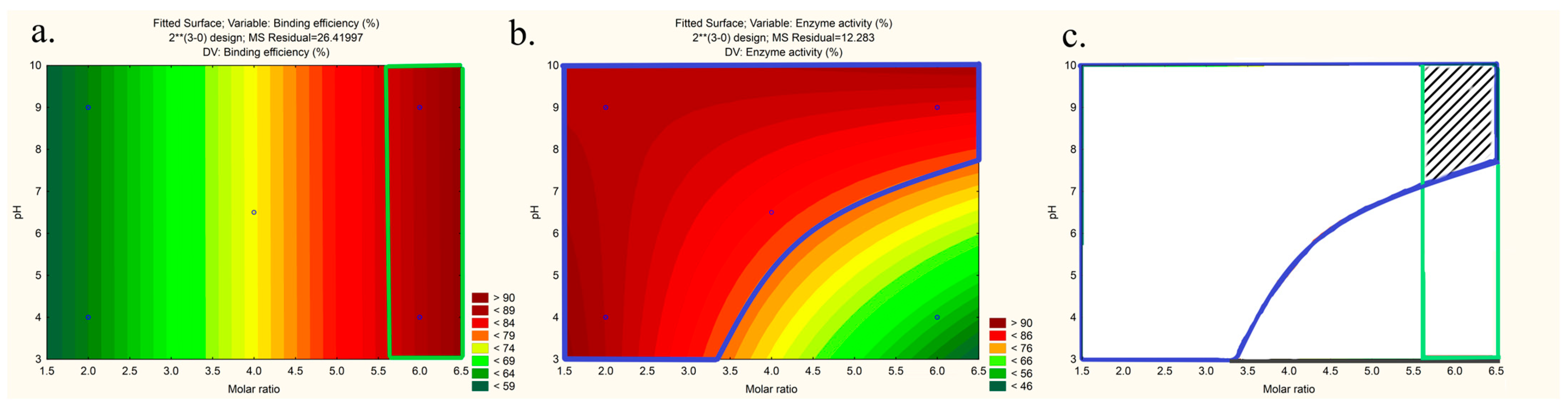
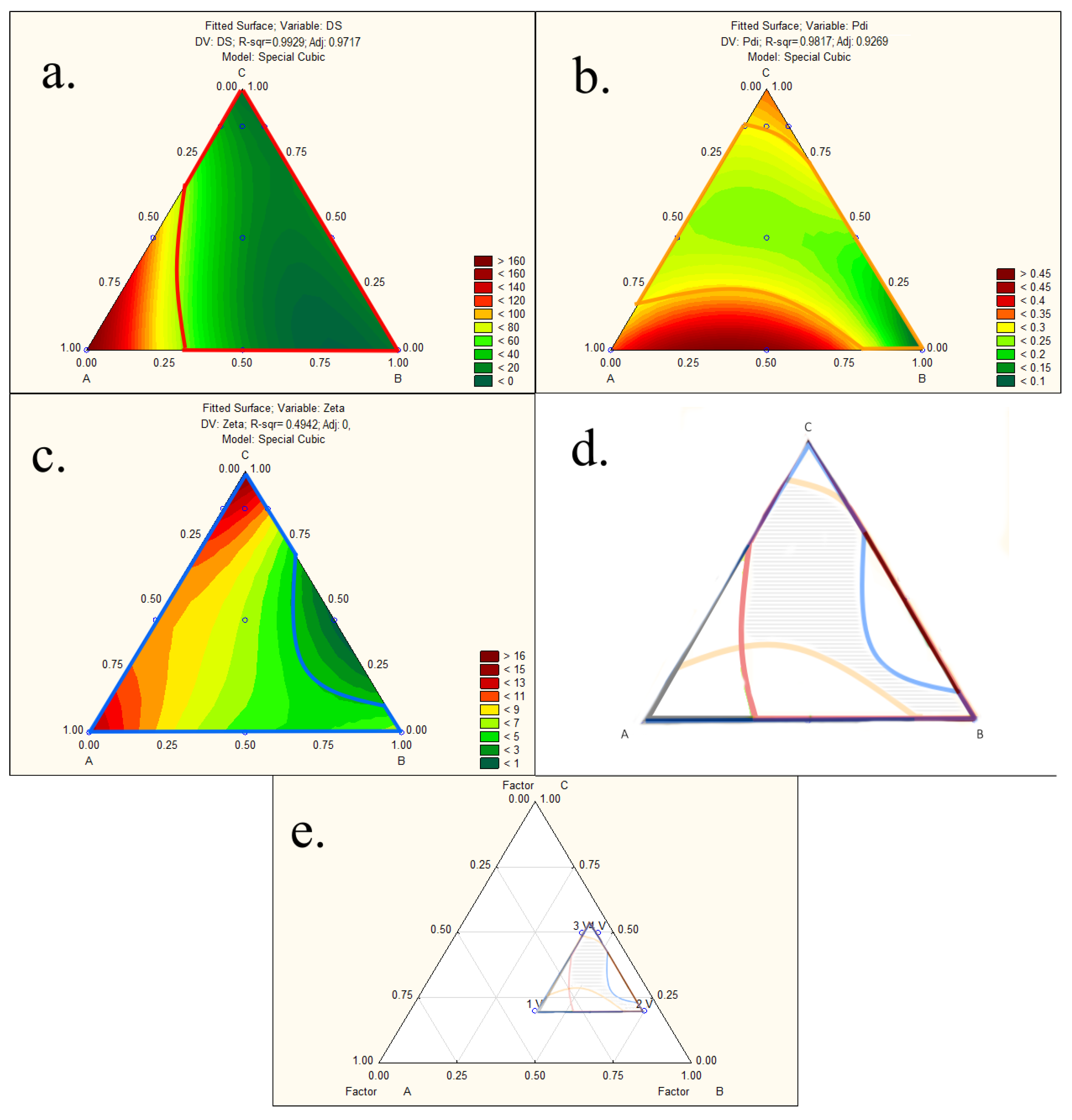
3.1. Preparation and Optimization of LYS:SDS HIP Complex
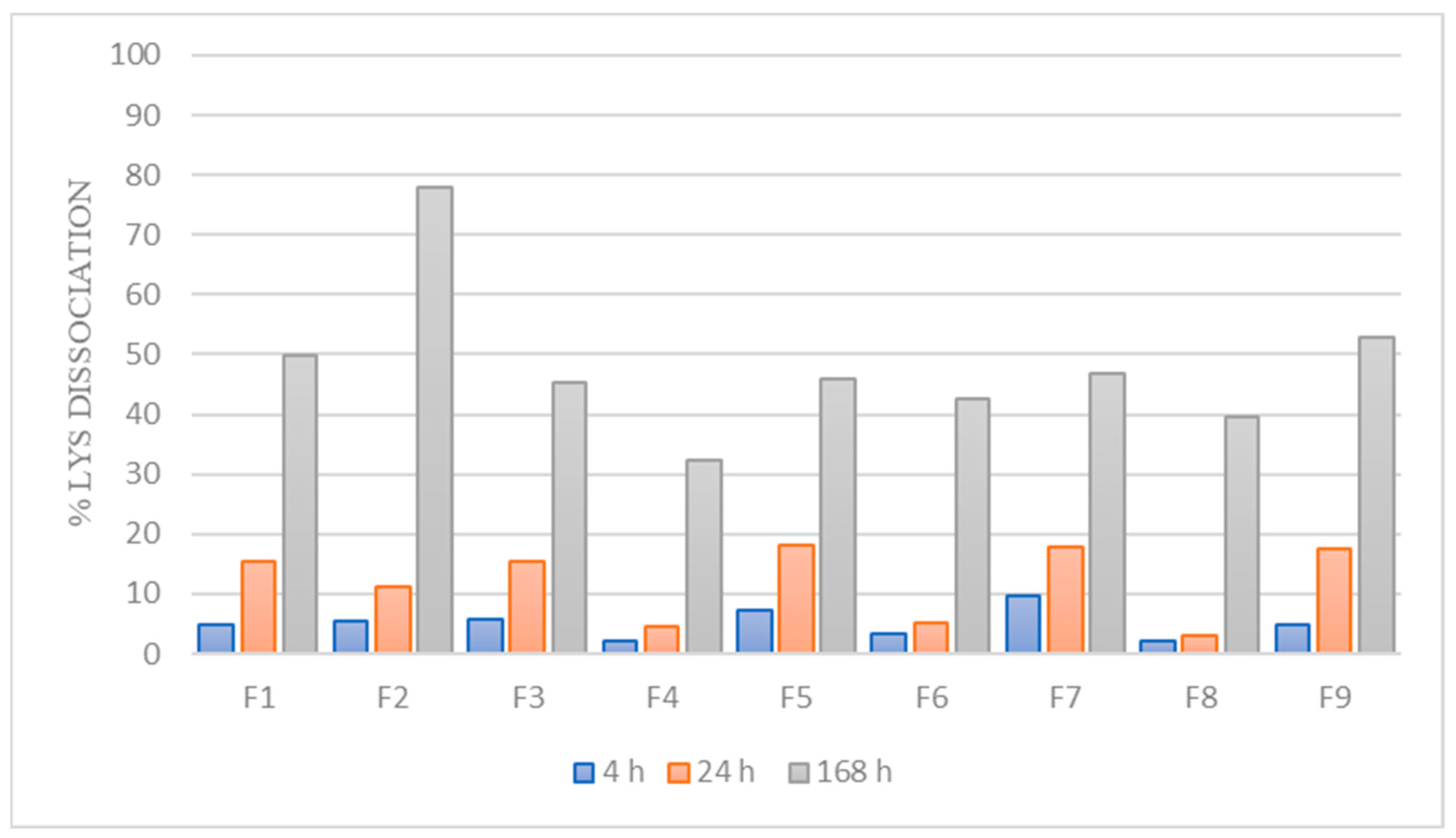
3.2. Dissociation of LYS from the HIP Complex
3.3. Fourier-Transform Infrared Spectroscopy (FT-IR)
3.4. Development and Characterization of Liquid-SEDDS Loaded with LYS:SDS Complex
3.5. Incorporation of the LYS:SDS Complex in Liquid SEDDS
3.6. Robustness to Dilution and pH Changes
3.7. Determination of log DSEDDS/release medium
3.8. Preparation of solid SEDDS Containing LYS:SDS Complex
3.9. Determination of LYS Activity after Incorporation in Liquid and Solid SEDDS
3.10. Preparation of Self-Emulsifying Tablets Containing LYS:SDS Complex
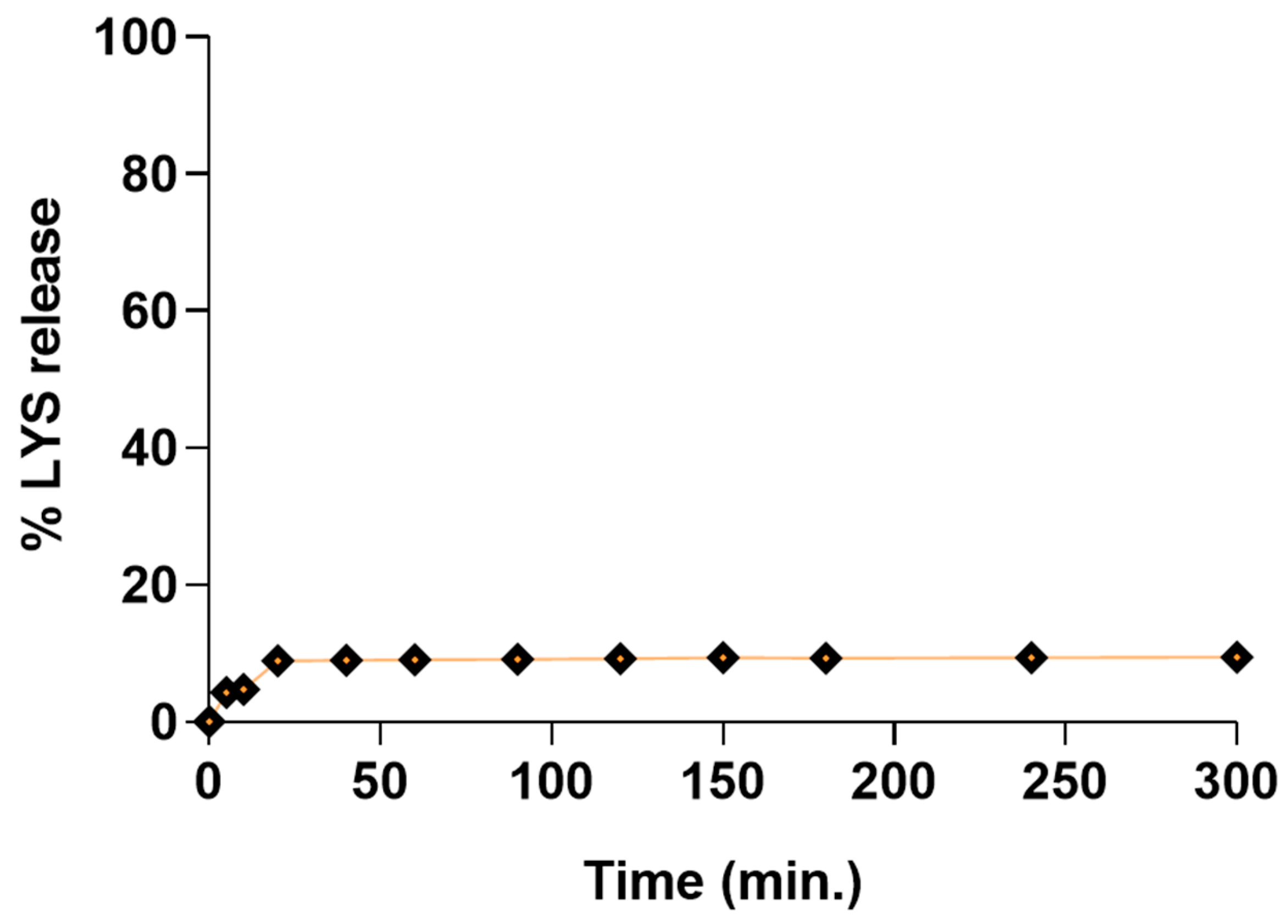
3.11. Dissolution Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brayden, D.J.; Hill, T.A.; Fairlie, D.P.; Maher, S.; Mrsny, R.J. Systemic delivery of peptides by the oral route: Formulation and medicinal chemistry approaches. Adv. Drug Deliv. Rev. 2020, 157, 2–36. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.D.; Whitehead, K.A.; Mitragotri, S. Materials for oral delivery of proteins and peptides. Nat. Rev. Mater. 2020, 5, 127–148. [Google Scholar] [CrossRef]
- Ibrahim, Y.H.-E.Y.; Regdon, G.; Hamedelniel, E.I.; Sovány, T. Review of recently used techniques and materials to improve the efficiency of orally administered proteins/peptides. DARU J. Pharm. Sci. 2020, 28, 403–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddadzadegan, S.; Dorkoosh, F.; Bernkop-Schnürch, A. Oral delivery of therapeutic peptides and proteins: Technology landscape of lipid-based nanocarriers. Adv. Drug Deliv. Rev. 2022, 182, 114097. [Google Scholar] [CrossRef]
- Pouton, C.W. Formulation of self-emulsifying drug delivery systems. Adv. Drug Deliv. Rev. 1997, 25, 47–58. [Google Scholar] [CrossRef]
- Rani, S.; Rana, R.; Saraogi, G.K.; Kumar, V.; Gupta, U. Self-Emulsifying Oral Lipid Drug Delivery Systems: Advances and Challenges. AAPS PharmSciTech 2019, 20, 129. [Google Scholar] [CrossRef]
- Tang, B.; Cheng, G.; Gu, J.-C.; Xu, C.-H. Development of solid self-emulsifying drug delivery systems: Preparation techniques and dosage forms. Drug Discov. Today 2008, 13, 606–612. [Google Scholar] [CrossRef]
- Tan, A.; Rao, S.; Prestidge, C.A. Transforming Lipid-Based Oral Drug Delivery Systems into Solid Dosage Forms: An Overview of Solid Carriers, Physicochemical Properties, and Biopharmaceutical Performance. Pharm. Res. 2013, 30, 2993–3017. [Google Scholar] [CrossRef]
- Joyce, P.; Dening, T.J.; Meola, T.R.; Schultz, H.B.; Holm, R.; Thomas, N.; Prestidge, C.A. Solidification to improve the biopharmaceutical performance of SEDDS: Opportunities and challenges. Adv. Drug Deliv. Rev. 2019, 142, 102–117. [Google Scholar] [CrossRef]
- Mahmood, A.; Bernkop-Schnürch, A. SEDDS: A game changing approach for the oral administration of hydrophilic macromolecular drugs. Adv. Drug Deliv. Rev. 2019, 142, 91–101. [Google Scholar] [CrossRef]
- Leonaviciute, G.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems in oral (poly)peptide drug delivery. Expert Opin. Drug Deliv. 2015, 12, 1703–1716. [Google Scholar] [CrossRef]
- Zupančič, O.; Bernkop-Schnürch, A. Lipophilic peptide character–What oral barriers fear the most. J. Control. Release 2017, 255, 242–257. [Google Scholar] [CrossRef]
- Ristroph, K.D.; Prud’homme, R.K. Hydrophobic ion pairing: Encapsulating small molecules, peptides, and proteins into nanocarriers. Nanoscale Adv. 2019, 1, 4207–4237. [Google Scholar] [CrossRef] [Green Version]
- Wibel, R.; Friedl, J.D.; Zaichik, S.; Bernkop-Schnürch, A. Hydrophobic ion pairing (HIP) of (poly)peptide drugs: Benefits and drawbacks of different preparation methods. Eur. J. Pharm. Biopharm. 2020, 151, 73–80. [Google Scholar] [CrossRef]
- Phan, T.N.Q.; Shahzadi, I.; Bernkop-Schnürch, A. Hydrophobic ion-pairs and lipid-based nanocarrier systems: The perfect match for delivery of BCS class 3 drugs. J. Control. Release 2019, 304, 146–155. [Google Scholar] [CrossRef]
- Ruan, J.; Liu, J.; Zhu, D.; Gong, T.; Yang, F.; Hao, X.; Zhang, Z. Preparation and evaluation of self-nanoemulsified drug delivery systems (SNEDDSs) of matrine based on drug–phospholipid complex technique. Int. J. Pharm. 2010, 386, 282–290. [Google Scholar] [CrossRef]
- Zhang, J.; Peng, Q.; Shi, S.; Zhang, Q.; Sun, X.; Gong, T.; Zhang, Z. Preparation, characterization, and in vivo evaluation of a self-nanoemulsifying drug delivery system (SNEDDS) loaded with morin-phospholipid complex. Int. J. Nanomed. 2011, 6, 3405–3414. [Google Scholar] [CrossRef] [Green Version]
- Zupančič, O.; Grieβinger, J.A.; Rohrer, J.; de Sousa, I.P.; Danninger, L.; Partenhauser, A.; Sündermann, N.E.; Laffleur, F.; Bernkop-Schnürch, A. Development, in vitro and in vivo evaluation of a self-emulsifying drug delivery system (SEDDS) for oral enoxaparin administration. Eur. J. Pharm. Biopharm. 2016, 109, 113–121. [Google Scholar] [CrossRef]
- Morgen, M.; Saxena, A.; Chen, X.-Q.; Miller, W.; Nkansah, R.; Goodwin, A.; Cape, J.; Haskell, R.; Su, C.; Gudmundsson, O.; et al. Lipophilic salts of poorly soluble compounds to enable high-dose lipidic SEDDS formulations in drug discovery. Eur. J. Pharm. Biopharm. 2017, 117, 212–223. [Google Scholar] [CrossRef]
- Zaichik, S.; Steinbring, C.; Caliskan, C.; Bernkop-Schnürch, A. Development and in vitro evaluation of a self-emulsifying drug delivery system (SEDDS) for oral vancomycin administration. Int. J. Pharm. 2019, 554, 125–133. [Google Scholar] [CrossRef]
- Asfour, M.H.; Kassem, A.A.; Salama, A.; Abd El-Alim, S.H. Hydrophobic ion pair loaded self-emulsifying drug delivery system (SEDDS): A novel oral drug delivery approach of cromolyn sodium for management of bronchial asthma. Int. J. Pharm. 2020, 585, 119494. [Google Scholar] [CrossRef] [PubMed]
- Jalil, A.; Asim, M.H.; Nazir, I.; Matuszczak, B.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems containing hydrophobic ion pairs of polymyxin B and agaric acid: A decisive strategy for enhanced antimicrobial activity. J. Mol. Liq. 2020, 311, 113298. [Google Scholar] [CrossRef]
- Bonengel, S.; Jelkmann, M.; Abdulkarim, M.; Gumbleton, M.; Reinstadler, V.; Oberacher, H.; Prüfert, F.; Bernkop-Schnürch, A. Impact of different hydrophobic ion pairs of octreotide on its oral bioavailability in pigs. J. Control. Release 2018, 273, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.N.Q.; Ismail, R.; Le-Vinh, B.; Zaichik, S.; Laffleur, F.; Bernkop-Schnürch, A. The Effect of Counterions in Hydrophobic Ion Pairs on Oral Bioavailability of Exenatide. ACS Biomater. Sci. Eng. 2020, 6, 5032–5039. [Google Scholar] [CrossRef] [PubMed]
- Leichner, C.; Menzel, C.; Laffleur, F.; Bernkop-Schnürch, A. Development and in vitro characterization of a papain loaded mucolytic self-emulsifying drug delivery system (SEDDS). Int. J. Pharm. 2017, 530, 346–353. [Google Scholar] [CrossRef]
- Zhang, Q.; He, N.; Zhang, L.; Zhu, F.; Chen, Q.; Qin, Y.; Zhang, Z.; Zhang, Q.; Wang, S.; He, Q. The In Vitro and In Vivo Study on Self-Nanoemulsifying Drug Delivery System (SNEDDS) Based on Insulin-Phospholipid Complex. J. Biomed. Nanotechnol. 2012, 8, 90–97. [Google Scholar] [CrossRef]
- Hintzen, F.; Perera, G.; Hauptstein, S.; Müller, C.; Laffleur, F.; Bernkop-Schnürch, A. In vivo evaluation of an oral self-microemulsifying drug delivery system (SMEDDS) for leuprorelin. Int. J. Pharm. 2014, 472, 20–26. [Google Scholar] [CrossRef]
- Menzel, C.; Holzeisen, T.; Laffleur, F.; Zaichik, S.; Abdulkarim, M.; Gumbleton, M.; Bernkop-Schnürch, A. In vivo evaluation of an oral self-emulsifying drug delivery system (SEDDS) for exenatide. J. Control. Release 2018, 277, 165–172. [Google Scholar] [CrossRef]
- Lupo, N.; Tkadlečková, V.N.; Jelkmann, M.; Laffleur, F.; Hetényi, G.; Kubová, K.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems: In vivo evaluation of their potential for oral vaccination. Acta Biomater. 2019, 94, 425–434. [Google Scholar] [CrossRef]
- Noh, G.; Keum, T.; Bashyal, S.; Seo, J.-E.; Shrawani, L.; Kim, J.H.; Lee, S. Recent progress in hydrophobic ion-pairing and lipid-based drug delivery systems for enhanced oral delivery of biopharmaceuticals. J. Pharm. Investig. 2021, 52, 75–93. [Google Scholar] [CrossRef]
- Gamboa, A.; Schüßler, N.; Soto-Bustamante, E.; Romero-Hasler, P.; Meinel, L.; Morales, J.O. Delivery of ionizable hydrophilic drugs based on pharmaceutical formulation of ion pairs and ionic liquids. Eur. J. Pharm. Biopharm. 2020, 156, 203–218. [Google Scholar] [CrossRef]
- Ferraboschi, P.; Ciceri, S.; Grisenti, P. Applications of Lysozyme, an Innate Immune Defense Factor, as an Alternative Antibiotic. Antibiotics 2021, 10, 1534. [Google Scholar] [CrossRef] [PubMed]
- Yuzuriha, T.; Katayama, K.; Fujita, T. Studies on biotransformation of lysozyme. I. Preparation of labeled lysozyme and its intestinal absorption. Chem. Pharm. Bull. 1975, 23, 1309–1314. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, M.; Hasegawa, S.; Yamashita, F.; Takakura, Y.; Hashida, M. Electrical charge on protein regulates its absorption from the rat small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G711–G719. [Google Scholar] [CrossRef] [Green Version]
- Hashida, S.; Ishikawa, E.; Nakamichi, N.; Sekino, H. Concentration of Egg White Lysozyme in the Serum of Healthy Subjects After Oral Administration. Clin. Exp. Pharmacol. Physiol. 2002, 29, 79–83. [Google Scholar] [CrossRef]
- European Medicines Agency. ICH Guideline Q9 on quality risk management. ICH Harmon. Tripart. Guid. 2015, 44, 1–20. [Google Scholar]
- ICH Expert Working Group. Pharmaceutical Development Q8. Harmon. Tripart. Guid. 2009, 8, 1–28. [Google Scholar]
- European Medicines Agency. ICH Guideline Q10 on Pharmaceutical Quality System; European Medicines Agency: Amsterdam, The Netherlands, 2015; Volume 44, pp. 1–20. [Google Scholar]
- Yoo, H.S.; Choi, H.K.; Park, T.G. Protein-fatty acid complex for enhanced loading and stability within biodegradable nanoparticles. J. Pharm. Sci. 2001, 90, 194–201. [Google Scholar] [CrossRef]
- Devrim, B.; Bozkır, A. Design and Evaluation of Hydrophobic Ion-Pairing Complexation of Lysozyme with Sodium Dodecyl Sulfate for Improved Encapsulation of Hydrophilic Peptides/Proteins by Lipid-Polymer Hybrid Nanoparticles. J. Nanomed. Nanotechnol. 2015, 6, 1000259. [Google Scholar]
- Pouton, C.W. Lipid formulations for oral administration of drugs: Non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur. J. Pharm. Sci. 2000, 11, S93–S98. [Google Scholar] [CrossRef]
- Mohd, A.B.; Sanka, K.; Bandi, S.; Diwan, P.V.; Shastri, N. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) for oral delivery of glimepiride: Development and antidiabetic activity in albino rabbits. Drug Deliv. 2015, 22, 499–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.; Bandopadhyay, S.; Kapil, R.; Singh, R.; Katare, O.P. Self-Emulsifying Drug Delivery Systems (SEDDS): Formulation Development, Characterization, and Applications. Crit. Rev. Ther. Drug Carr. Syst. 2009, 26, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Griesser, J.; Hetényi, G.; Moser, M.; Demarne, F.; Jannin, V.; Bernkop-Schnürch, A. Hydrophobic ion pairing: Key to highly payloaded self-emulsifying peptide drug delivery systems. Int. J. Pharm. 2017, 520, 267–274. [Google Scholar] [CrossRef]
- Bernkop-Schnürch, A.; Jalil, A. Do drug release studies from SEDDS make any sense? J. Control. Release 2018, 271, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Pallagi, E.; Ismail, R.; Paál, T.L.; Csóka, I. Initial Risk Assessment as part of the Quality by Design in peptide drug containing formulation development. Eur. J. Pharm. Sci. 2018, 122, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Kristó, K.; Manteghi, R.; Ibrahim, Y.H.-E.Y.; Ungor, D.; Csapó, E.; Berkesi, D.; Kónya, Z.; Csóka, I. Optimization of layering technique and secondary structure analysis during the formulation of nanoparticles containing lysozyme by quality by design approach. PLoS ONE 2021, 16, e0260603. [Google Scholar] [CrossRef]
- Shahzadi, I.; Nazir, I.; Phan, T.N.Q.; Bernkop-Schnürch, A. About the impact of superassociation of hydrophobic ion pairs on membrane permeability. Eur. J. Pharm. Biopharm. 2020, 151, 1–8. [Google Scholar] [CrossRef]
- Vaishya, R.D.; Mandal, A.; Gokulgandhi, M.; Patel, S.; Mitra, A.K. Reversible hydrophobic ion-paring complex strategy to minimize acylation of octreotide during long-term delivery from PLGA microparticles. Int. J. Pharm. 2015, 489, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Haris, P.I.; Chapman, D. Analysis of polypeptide and protein structures using Fourier transform infrared spectroscopy. Methods Mol. Biol. 1994, 22, 183–202. [Google Scholar] [CrossRef]
- Son, H.Y.; Chae, B.R.; Choi, J.Y.; Shin, D.J.; Goo, Y.T.; Lee, E.S.; Kang, T.H.; Kim, C.H.; Yoon, H.Y.; Choi, Y.W. Optimization of self-microemulsifying drug delivery system for phospholipid complex of telmisartan using D-optimal mixture design. PLoS ONE 2018, 13, e0208339. [Google Scholar] [CrossRef] [Green Version]
- Hetényi, G.; Griesser, J.; Moser, M.; Demarne, F.; Jannin, V.; Bernkop-Schnürch, A. Comparison of the protective effect of self-emulsifying peptide drug delivery systems towards intestinal proteases and glutathione. Int. J. Pharm. 2017, 523, 357–365. [Google Scholar] [CrossRef]
- Phan, T.N.Q.; Le-Vinh, B.; Efiana, N.A.; Bernkop-Schnürch, A. Oral self-emulsifying delivery systems for systemic administration of therapeutic proteins: Science fiction? J. Drug Target. 2019, 27, 1017–1024. [Google Scholar] [CrossRef]
- Abou Assi, R.; Abdulbaqi, I.M.; Seok Ming, T.; Siok Yee, C.; Wahab, H.A.; Asif, S.M.; Darwis, Y. Liquid and Solid Self-Emulsifying Drug Delivery Systems (SEDDs) as Carriers for the Oral Delivery of Azithromycin: Optimization, In Vitro Characterization and Stability Assessment. Pharmaceutics 2020, 12, 1052. [Google Scholar] [CrossRef]
- Vohra, A.M.; Patel, C.V.; Kumar, P.; Thakkar, H.P. Development of dual drug loaded solid self microemulsifying drug delivery system: Exploring interfacial interactions using QbD coupled risk based approach. J. Mol. Liq. 2017, 242, 1156–1168. [Google Scholar] [CrossRef]
- Sanka, K.; Suda, D.; Bakshi, V. Optimization of solid-self nanoemulsifying drug delivery system for solubility and release profile of clonazepam using simplex lattice design. J. Drug Deliv. Sci. Technol. 2016, 33, 114–124. [Google Scholar] [CrossRef]
- Li, F.; Hu, R.; Wang, B.; Gui, Y.; Cheng, G.; Gao, S.; Ye, L.; Tang, J. Self-microemulsifying drug delivery system for improving the bioavailability of huperzine A by lymphatic uptake. Acta Pharm. Sin. B 2017, 7, 353–360. [Google Scholar] [CrossRef]
- Nazir, I.; Asim, M.H.; Dizdarević, A.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems: Impact of stability of hydrophobic ion pairs on drug release. Int. J. Pharm. 2019, 561, 197–205. [Google Scholar] [CrossRef]
- Beringhs, A.O.; Minatovicz, B.C.; Zhang, G.G.Z.; Chaudhuri, B.; Lu, X. Impact of Porous Excipients on the Manufacturability and Product Performance of Solid Self-Emulsifying Drug Delivery Systems. AAPS PharmSciTech 2018, 19, 3298–3310. [Google Scholar] [CrossRef]
- Mandić, J.; Pobirk, A.Z.; Vrečer, F.; Gašperlin, M. Overview of solidification techniques for self-emulsifying drug delivery systems from industrial perspective. Int. J. Pharm. 2017, 533, 335–345. [Google Scholar] [CrossRef]
- Almeida, S.R.D.; Tippavajhala, V.K. A Rundown Through Various Methods Used in the Formulation of Solid Self-Emulsifying Drug Delivery Systems (S-SEDDS). AAPS PharmSciTech 2019, 20, 323. [Google Scholar] [CrossRef]
- Araújo, G.P.; Martins, F.T.; Taveira, S.F.; Cunha-Filho, M.; Marreto, R.N. Effects of Formulation and Manufacturing Process on Drug Release from Solid Self-emulsifying Drug Delivery Systems Prepared by High Shear Mixing. AAPS PharmSciTech 2021, 22, 254. [Google Scholar] [CrossRef] [PubMed]
- Shahzadi, I.; Dizdarević, A.; Efiana, N.A.; Matuszczak, B.; Bernkop-Schnürch, A. Trypsin decorated self-emulsifying drug delivery systems (SEDDS): Key to enhanced mucus permeation. J. Colloid Interface Sci. 2018, 531, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Etezadi, H.; Maleki, A.; Friedl, J.D.; Bernkop-Schnürch, A. Storage stability of proteins in a liquid-based formulation: Liquid vs. solid self-emulsifying drug delivery. Int. J. Pharm. 2020, 590, 119918. [Google Scholar] [CrossRef] [PubMed]
- Seljak, K.B.; Ilić, I.G.; Gašperlin, M.; Pobirk, A.Z. Self-microemulsifying tablets prepared by direct compression for improved resveratrol delivery. Int. J. Pharm. 2018, 548, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Beg, S.; Swain, S.; Singh, H.P.; Patra, C.N.; Rao, M.B. Development, Optimization, and Characterization of Solid Self-Nanoemulsifying Drug Delivery Systems of Valsartan Using Porous Carriers. AAPS PharmSciTech 2012, 13, 1416–1427. [Google Scholar] [CrossRef] [Green Version]
- Beg, S.; Katare, O.P.; Saini, S.; Garg, B.; Khurana, R.K.; Singh, B. Solid self-nanoemulsifying systems of olmesartan medoxomil: Formulation development, micromeritic characterization, in vitro and in vivo evaluation. Powder Technol. 2016, 294, 93–104. [Google Scholar] [CrossRef]
- Agarwal, V.; Siddiqui, A.; Ali, H.; Nazzal, S. Dissolution and powder flow characterization of solid self-emulsified drug delivery system (SEDDS). Int. J. Pharm. 2009, 366, 44–52. [Google Scholar] [CrossRef]
- Kostelanská, K.; Prudilová, B.B.; Holešová, S.; Vlček, J.; Vetchý, D.; Gajdziok, J. Comparative Study of Powder Carriers Physical and Structural Properties. Pharmaceutics 2022, 14, 818. [Google Scholar] [CrossRef]
- Williams, H.D.; Van Speybroeck, M.; Augustijns, P.; Porter, C.J.H. Lipid-Based Formulations Solidified Via Adsorption onto the Mesoporous Carrier Neusilin® US2: Effect of Drug Type and Formulation Composition on In Vitro Pharmaceutical Performance. J. Pharm. Sci. 2014, 103, 1734–1746. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment (C1–C9) | Molar ratio (LYS:SDS) (x1) | pH (x2) | Temperature (°C) (x3) |
|---|---|---|---|
| 1 | 1:2 (−1) | 4 (−1) | 25.0 (−1) |
| 2 | 1:6 (+1) | 4 (−1) | 25.0 (−1) |
| 3 | 1:2 (−1) | 9 (+1) | 25.0 (−1) |
| 4 | 1:6 (+1) | 9 (+1) | 25.0 (−1) |
| 5 | 1:2 (−1) | 4 (−1) | 37.0 (+1) |
| 6 | 1:6 (+1) | 4 (−1) | 37.0 (+1) |
| 7 | 1:2 (−1) | 9 (+1) | 37.0 (+1) |
| 8 | 1:6 (+1) | 9 (+1) | 37.0 (+1) |
| 9 | 1:4 (0) | 6.5 (0) | 31.0 (0) |
| Formulation | Oil | Surfactant | Co-Surfactant | Oil:Smix | S:CS |
|---|---|---|---|---|---|
| L1 | 40.0 | 40.0 | 20.0 | 1:1.5 | 2:1 |
| L2 | 5.0 | 75.0 | 20.0 | 1:19 | 4:1 |
| L3 | 10.0 | 40.0 | 50.0 | 1:9 | 1:1.3 |
| L4 | 5.0 | 45.0 | 50.0 | 1:19 | 1:1 |
| L5 | 5.0 | 60.0 | 35.0 | 1:19 | 1:1.7 |
| L6 | 25.0 | 40.0 | 35.0 | 1:3 | 1:1.4 |
| L7 | 22.5 | 57.5 | 20.0 | 1:3.4 | 1:2.9 |
| L8 | 7.5 | 42.5 | 50.0 | 1:12.3 | 1:1.2 |
| L9 | 15.0 | 50.0 | 35.0 | 1:5.7 | 1:1.4 |
| CQA | CMAs/CPPs | |||||||
|---|---|---|---|---|---|---|---|---|
| Type of Counterion | Molar Ratio (LYS:Counterion) | pH of the Solution | Ionic Strength of Solution | Mixing Type | Mixing Speed | Mixing Temperature | Centrifugation Speed | |
| Binding efficiency | High | High | High | High | Medium | Medium | Medium | Low |
| Enzyme activity | High | Medium | Medium | Medium | High | High | High | High |
| Zeta potential | High | High | High | Medium | Low | Low | Low | Low |
| Dissociation of the complex | High | High | High | High | Low | Low | Low | Low |
| Lipophilicity of the complex | High | High | Low | Low | Low | Low | Low | Low |
| HIP payload | High | High | High | Medium | Low | Low | Low | Medium |
| Sample | Binding Efficiency (%) (y1) | Enzyme Activity (%) (y2) |
|---|---|---|
| 1 | 61.77 | 87.10 |
| 2 | 88.83 | 53.70 |
| 3 | 62.77 | 86.14 |
| 4 | 88.77 | 79.25 |
| 5 | 63.93 | 95.70 |
| 6 | 87.71 | 58.95 |
| 7 | 62.94 | 95.35 |
| 8 | 87.94 | 94.34 |
| 9 | 67.92 | 85.27 |
| Responses | Check Point Formulation | Factors | Experimental Value | Predicted Value | % Bias | ||
|---|---|---|---|---|---|---|---|
| x1 | x2 | x3 | |||||
| Binding efficiency (%) | 1 | 1:6 | 9 | 25 | 88.88 ± 0.05 | 87.91 | 0.48 |
| 2 | 1:6 | 6.5 | 25 | 86.43 ± 0.77 | 87.94 | 0.755 | |
| Enzyme activity (%) | 1 | 1:6 | 9 | 25 | 77.19 ± 1.22 | 80.84 | 1.82 |
| 2 | 1:6 | 6.5 | 25 | 59.65 ± 1.61 | 66.90 | 3.62 | |
| Code | Appearance | SE Time (min) | % T | Droplet Size (nm) | PdI | Zeta Potential (mV) |
|---|---|---|---|---|---|---|
| L1 | bluish | ≤2 | 3.14 ± 0.01 | 177.20 ±1.56 | 0.348 ± 0.03 | −13.40 ± 0.20 |
| L2 | translucent | ≤1 | 99.43 ± 0.23 | 11.22 ± 0.14 | 0.095 ± 0.02 | −6.96 ± 2.81 |
| L3 | translucent | ≤1 | 98.93 ± 0.13 | 20.40 ± 0.08 | 0.289 ±0.014 | −7.39 ± 0.42 |
| L4 | translucent | ≤1 | 99.37 ± 0.12 | 14.53 ± 0.42 | 0.301 ± 0.01 | −5.70 ± 0.58 |
| L5 | translucent | ≤1 | 99.43 ±0.03 | 15.87 ± 0.67 | 0.239 ± 0.011 | −0.73 ± 0.55 |
| L6 | slightly bluish | ≤1 | 76.00 ±0.01 | 107.47 ± 1.26 | 0.250 ± 0.004 | −10.29 ± 0.64 |
| L7 | translucent | ≤1 | 99.55 ± 0.08 | 24.43 ± 0.82 | 0.481 ± 0.016 | −5.62 ± 0.47 |
| L8 | translucent | ≤1 | 99.67 ± 0.09 | 25.09 ± 1.25 | 0.336 ± 0.033 | −21.10 ± 1.95 |
| L9 | translucent | ≤1 | 99.55 ± 0.08 | 21.79 ± 0.14 | 0.239 ± 0.003 | −5.56 ± 1.62 |
| Code | Appearance | SE Time (min) | % T | Droplet Size (nm) | PdI | Zeta Potential (mV) |
|---|---|---|---|---|---|---|
| After 24 h | ||||||
| L2 | translucent | ≤20 s | 99.71 ± 0.04 | 13.02 ± 0.54 | 0.245 ± 0.008 | −4.85 ± 0.50 |
| L9 | translucent | ≤20 s | 97.98 ± 0.17 | 24.98 ± 0.35 | 0.320 ± 0.020 | −5.56 ± 1.53 |
| L10 | translucent | ≤20 s | 99.48 ± 0.11 | 14.62 ± 0.19 | 0.214 ± 0.037 | −3.09 ± 0.39 |
| After 7 days | ||||||
| L2 | translucent | ≤20 s | 99.34 ± 0.05 | 13.84 ± 0.74 | 0.218 ± 0.003 | −0.49 ± 0.41 |
| L9 | translucent | ≤20 s | 97.56 ± 0.15 | 29.08 ± 0.54 | 0.210 ± 0.005 | −9.60 ± 3.30 |
| L10 | translucent | ≤20 s | 99.45 ± 0.12 | 15.12 ± 0.23 | 0.191 ± 0.018 | −5.81 ± 3.50 |
| Code | Oil (%) | Surfactant (%) | Co- Surfactant (%) | Maximum Concentration of Dissolved Complex (%) |
|---|---|---|---|---|
| L2 | 5.0 | 75.0 | 20.0 | 4.0 |
| L9 | 15.0 | 50.0 | 35.0 | 1.0 |
| L10 | 10.0 | 62.5 | 27.5 | 2.0 |
| Medium | Droplet Size (nm) | PDI | Droplet Size (nm) | PDI |
|---|---|---|---|---|
| 1:100 | 1:1000 | |||
| DW | 13.02 ± 0.54 | 0.245 ± 0.008 | 21.31 ± 2.50 | 0.135 ± 0.007 |
| PBS pH 6.8 | 14.13 ± 0.35 | 0.223 ± 0.017 | 18.02 ± 0.94 | 0.418 ± 0.029 |
| 0.1 M HCl pH 1.4 | 13.94 ± 0.52 | 0.208 ± 0.001 | 14.83 ± 0.19 | 0.317 ± 0.032 |
| Release Medium | Log D | C Oil Droplets (%) | C in RM (%) |
|---|---|---|---|
| PBS 6.8 pH | 1.37 | 31.80 | 68.19 |
| DW | 2.72 | 91.29 | 8.71 |
| Solid Carrier (SC) | Ratio (SC-SEDDS) | Bulk Density (g/mL) | Tapped Density (g/mL) | Carr Index (%) | Hausner Ratio | Angle of Repose (°) | Flow Time (s) |
|---|---|---|---|---|---|---|---|
| Neusilin®UFL2 | 1:1 | 0.2056 | 0.2977 | 30.95 | 1.448 | 44.29 | 16 |
| 1:2 | 0.3246 | 0.4253 | 23.68 | 1.310 | 42.73 | 9 | |
| Syloid® 244P | 1:1 | 0.1363 | 0.1828 | 25.42 | 1.341 | 45.51 | 8 |
| 1:2 | 0.2953 | 0.3937 | 25.00 | 1.333 | 44.08 | 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šahinović, M.; Hassan, A.; Kristó, K.; Regdon, G., Jr.; Vranić, E.; Sovány, T. Quality by Design-Based Development of Solid Self-Emulsifying Drug Delivery System (SEDDS) as a Potential Carrier for Oral Delivery of Lysozyme. Pharmaceutics 2023, 15, 995. https://doi.org/10.3390/pharmaceutics15030995
Šahinović M, Hassan A, Kristó K, Regdon G Jr., Vranić E, Sovány T. Quality by Design-Based Development of Solid Self-Emulsifying Drug Delivery System (SEDDS) as a Potential Carrier for Oral Delivery of Lysozyme. Pharmaceutics. 2023; 15(3):995. https://doi.org/10.3390/pharmaceutics15030995
Chicago/Turabian StyleŠahinović, Merima, Alharith Hassan, Katalin Kristó, Géza Regdon, Jr., Edina Vranić, and Tamás Sovány. 2023. "Quality by Design-Based Development of Solid Self-Emulsifying Drug Delivery System (SEDDS) as a Potential Carrier for Oral Delivery of Lysozyme" Pharmaceutics 15, no. 3: 995. https://doi.org/10.3390/pharmaceutics15030995
APA StyleŠahinović, M., Hassan, A., Kristó, K., Regdon, G., Jr., Vranić, E., & Sovány, T. (2023). Quality by Design-Based Development of Solid Self-Emulsifying Drug Delivery System (SEDDS) as a Potential Carrier for Oral Delivery of Lysozyme. Pharmaceutics, 15(3), 995. https://doi.org/10.3390/pharmaceutics15030995