
Neonatal Pharmacokinetics and Biodistribution of Polymeric Nanoparticles and Effect of Surfactant

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Care and Ethics Statement
2.2. Polymer Activation and Labeling
2.3. Nanoparticle Preparation and Characterization
2.4. Transmission Electron Microscopy Method
2.5. Nanoparticle Administration and Tissue Extraction
2.6. High-Performance Liquid Chromatography (HPLC) Method
2.7. Tissue Processing for PK and Organ-Level Biodistribution Analysis
2.8. Immunohistochemistry
2.9. Statistical Analysis
3. Results
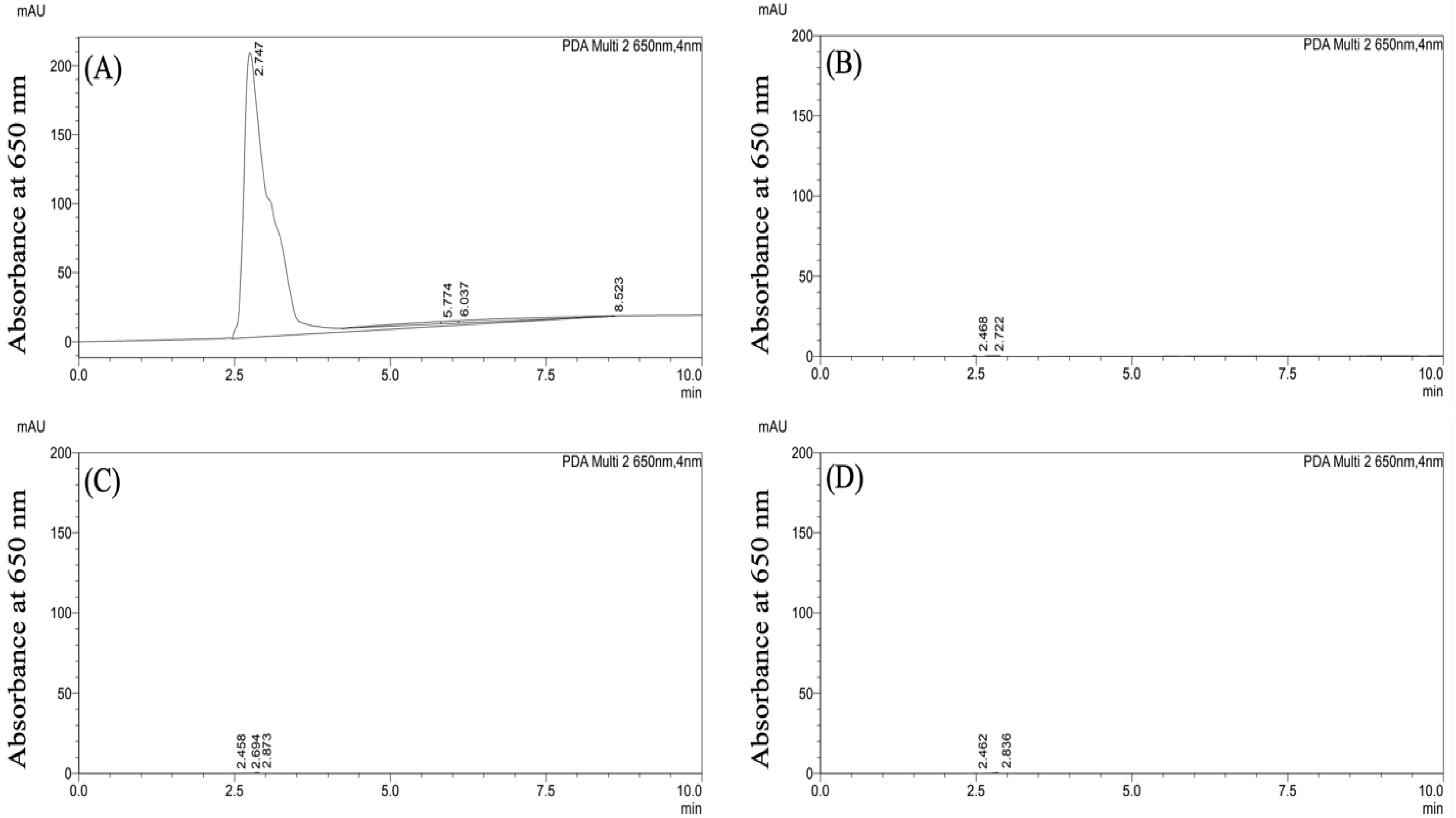
3.1. Characterization and Stability of CF647-Labeled Nanoparticles
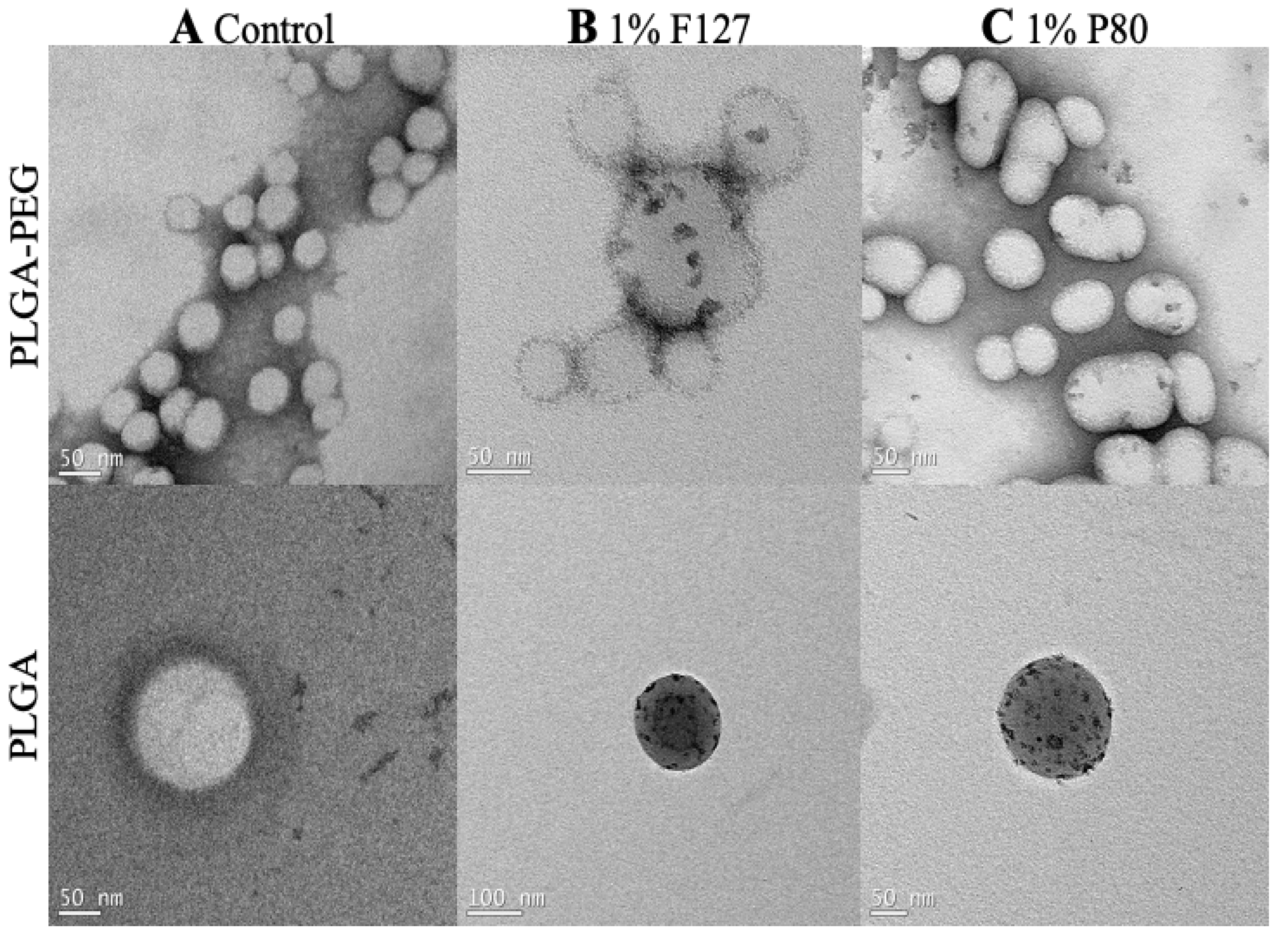
3.2. Characterization of Nanoparticles on TEM
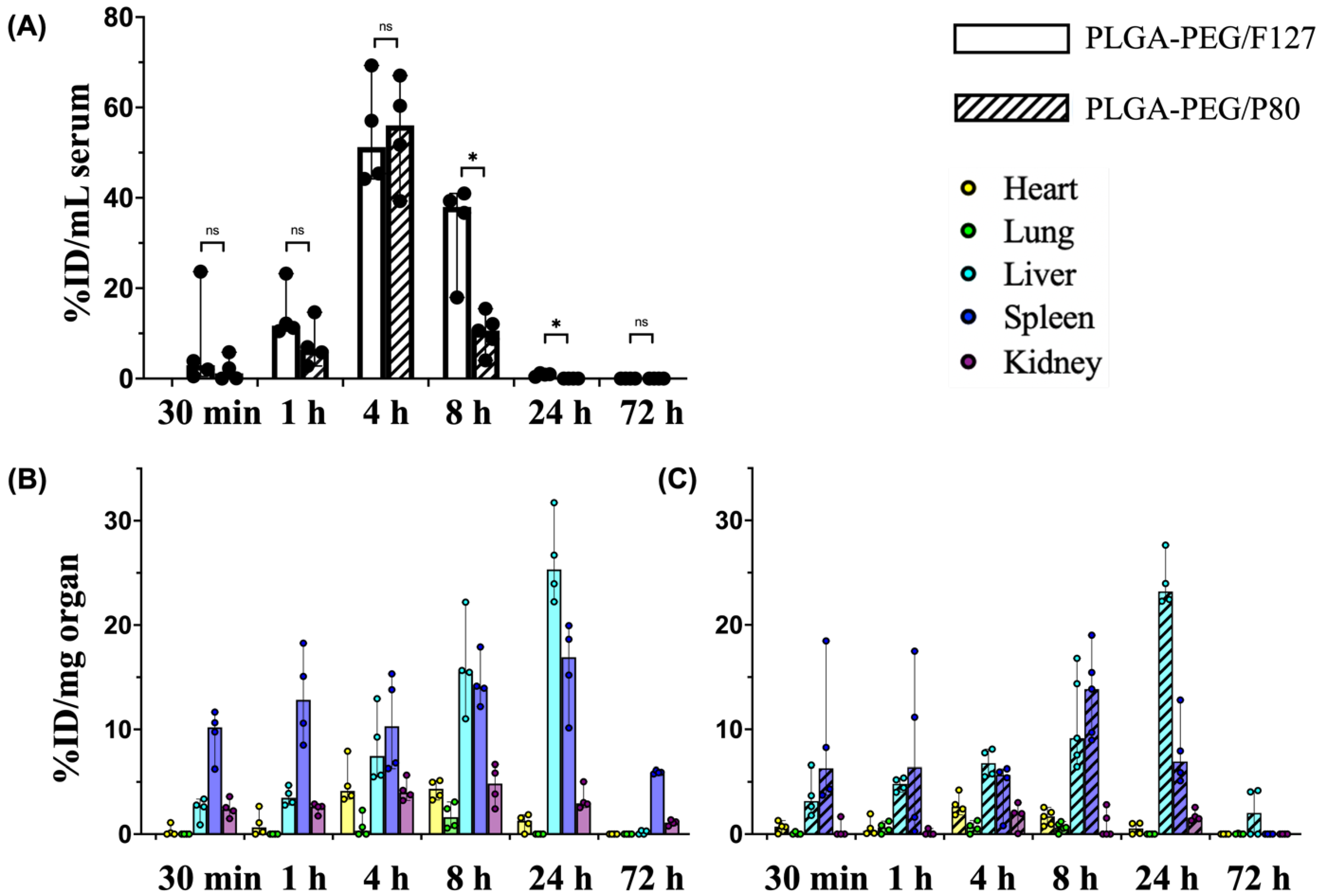
3.3. PK and Biodistribution of PLGA-PEG in Term-Equivalent Rats
3.4. Tissue-Level Biodistribution
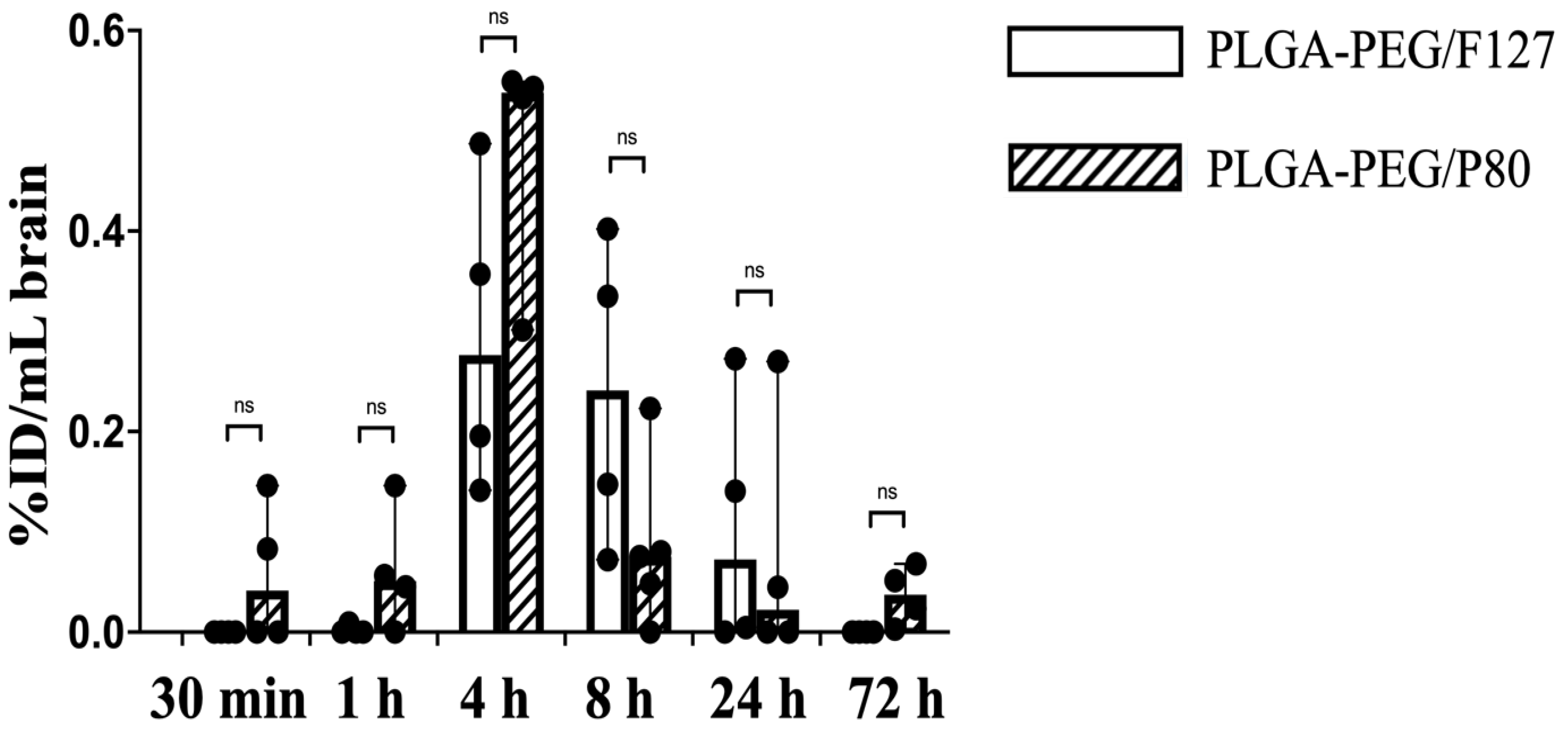
3.5. PLGA-PEG PK and Biodistribution in the Term-Equivalent Brain
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FDA. Pediatric Study Plans: Content of and Process for Submitting Initial Pediatric Study Plans and Amended Initial Pediatric Study Plans; FDA: White Oak, MD, USA, 2020. [Google Scholar]
- Giacoia, G.P.; Taylor-Zapata, P.; Zajicek, A. Eunice Kennedy Shriver National Institute of Child Health and Human Development Pediatrics Formulation Initiative: Proceedings from the Second Workshop on Pediatric Formulations. Clin. Ther. 2012, 34, S1–S10. [Google Scholar] [CrossRef]
- Nieto Gonzalez, N.; Obinu, A.; Rassu, G.; Giunchedi, P.; Gavini, E. Polymeric and Lipid Nanoparticles: Which Applications in Pediatrics? Pharmaceutics 2021, 13, 670. [Google Scholar] [CrossRef] [PubMed]
- Penkov, D.; Tomasi, P.; Eichler, I.; Murphy, D.; Yao, L.P.; Temeck, J. Pediatric Medicine Development: An Overview and Comparison of Regulatory Processes in the European Union and United States. Ther. Innov. Regul. Sci. 2017, 51, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.Y.; Rutka, J.T.; Chan, W.C. Nanomedicine. N. Engl. J. Med. 2010, 363, 2434–2443. [Google Scholar] [CrossRef] [Green Version]
- Freitas, R.A., Jr. What is nanomedicine? Nanomedicine 2005, 1, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Xu, X.; Barwe, S.P.; Yang, X.; Czymmek, K.; Waldman, S.A.; Mason, R.W.; Jia, X.; Rajasekaran, A.K. Dexamethasone-loaded block copolymer nanoparticles induce leukemia cell death and enhance therapeutic efficacy: A novel application in pediatric nanomedicine. Mol. Pharm. 2013, 10, 2199–2210. [Google Scholar] [CrossRef]
- Chiappetta, D.A.; Hocht, C.; Taira, C.; Sosnik, A. Efavirenz-loaded polymeric micelles for pediatric anti-HIV pharmacotherapy with significantly higher oral bioavailability [corrected]. Nanomedicine 2010, 5, 11–23. [Google Scholar] [CrossRef]
- Choi, J.; Rui, Y.; Kim, J.; Gorelick, N.; Wilson, D.R.; Kozielski, K.; Mangraviti, A.; Sankey, E.; Brem, H.; Tyler, B.; et al. Nonviral polymeric nanoparticles for gene therapy in pediatric CNS malignancies. Nanomedicine 2020, 23, 102115. [Google Scholar] [CrossRef]
- Belayneh, A.; Tadese, E.; Molla, F. Safety and Biopharmaceutical Challenges of Excipients in Off-Label Pediatric Formulations. Int. J. Gen. Med. 2020, 13, 1051–1066. [Google Scholar] [CrossRef]
- Yellepeddi, V.K.; Joseph, A.; Nance, E. Pharmacokinetics of nanotechnology-based formulations in pediatric populations. Adv. Drug Deliv. Rev. 2019, 151-152, 44–55. [Google Scholar] [CrossRef]
- Frattarelli, D.A.; Galinkin, J.L.; Green, T.P.; Johnson, T.D.; Neville, K.A.; Paul, I.M.; Van Den Anker, J.N. American Academy of Pediatrics Committee on Drugs. Off-label use of drugs in children. Pediatrics 2014, 133, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.S.; Hall, M.; Goodman, D.M.; Feuer, P.; Sharma, V.; Fargason, C., Jr.; Hyman, D.; Jenkins, K.; White, M.L.; Levy, F.H.; et al. Off-label drug use in hospitalized children. Arch. Pediatr. Adolesc. Med. 2007, 161, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Pratico, A.D.; Longo, L.; Mansueto, S.; Gozzo, L.; Barberi, I.; Tiralongo, V.; Salvo, V.; Falsaperla, R.; Vitaliti, G.; La Rosa, M.; et al. Off-Label Use of Drugs and Adverse Drug Reactions in Pediatric Units: A Prospective, Multicenter Study. Curr. Drug Saf. 2018, 13, 200–207. [Google Scholar] [CrossRef]
- Allen, H.C.; Garbe, M.C.; Lees, J.; Aziz, N.; Chaaban, H.; Miller, J.L.; Johnson, P.; DeLeon, S. Off-Label Medication use in Children, More Common than We Think: A Systematic Review of the Literature. J. Okla. State Med. Assoc. 2018, 111, 776–783. [Google Scholar] [PubMed]
- Amin, F.U.; Shah, S.A.; Badshah, H.; Khan, M.; Kim, M.O. Anthocyanins encapsulated by PLGA@PEG nanoparticles potentially improved its free radical scavenging capabilities via p38/JNK pathway against Abeta1-42-induced oxidative stress. J. Nanobiotechnol. 2017, 15, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saneja, A.; Kumar, R.; Mintoo, M.J.; Dubey, R.D.; Sangwan, P.L.; Mondhe, D.M.; Panda, A.K.; Gupta, P.N. Gemcitabine and betulinic acid co-encapsulated PLGA-PEG polymer nanoparticles for improved efficacy of cancer chemotherapy. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 98, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Khalil, N.M.; do Nascimento, T.C.; Casa, D.M.; Dalmolin, L.F.; de Mattos, A.C.; Hoss, I.; Romano, M.A.; Mainardes, R.M. Pharmacokinetics of curcumin-loaded PLGA and PLGA-PEG blend nanoparticles after oral administration in rats. Colloids Surf. B Biointerfaces 2013, 101, 353–360. [Google Scholar] [CrossRef]
- Operti, M.C.; Bernhardt, A.; Grimm, S.; Engel, A.; Figdor, C.G.; Tagit, O. PLGA-based nanomedicines manufacturing: Technologies overview and challenges in industrial scale-up. Int. J. Pharm. 2021, 605, 120807. [Google Scholar] [CrossRef]
- NCT05456022. Available online: https://clinicaltrials.gov (accessed on 4 April 2023).
- Joseph, A.; Wood, T.; Chen, C.C.; Corry, K.; Snyder, J.M.; Juul, S.E.; Parikh, P.; Nance, E. Curcumin-loaded polymeric nanoparticles for neuro-protection in neonatal rats with hypoxic-ischemic encephalopathy. Nano Res. 2018, 11, 5670–5688. [Google Scholar] [CrossRef]
- Araujo, L.; Lobenberg, R.; Kreuter, J. Influence of the surfactant concentration on the body distribution of nanoparticles. J. Drug Target 1999, 6, 373–385. [Google Scholar] [CrossRef]
- Troster, S.D.; Muller, U.; Kreuter, J. Modification of the Body Distribution of Poly(Methyl Methacrylate) Nanoparticles in Rats by Coating with Surfactants. Int. J. Pharm. 1990, 61, 85–100. [Google Scholar] [CrossRef]
- Joseph, A.; Simo, G.M.; Gao, T.; Alhindi, N.; Xu, N.; Graham, D.J.; Gamble, L.J.; Nance, E. Surfactants influence polymer nanoparticle fate within the brain. Biomaterials 2021, 277, 121086. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, T.; Itaya, M.; Burdeos, G.C.; Nakagawa, K.; Miyazawa, T. A Critical Review of the Use of Surfactant-Coated Nanoparticles in Nanomedicine and Food Nanotechnology. Int. J. Nanomed. 2021, 16, 3937–3999. [Google Scholar] [CrossRef] [PubMed]
- Cortes, H.; Hernandez-Parra, H.; Bernal-Chavez, S.A.; Prado-Audelo, M.L.D.; Caballero-Floran, I.H.; Borbolla-Jimenez, F.V.; Gonzalez-Torres, M.; Magana, J.J.; Leyva-Gomez, G. Non-Ionic Surfactants for Stabilization of Polymeric Nanoparticles for Biomedical Uses. Materials 2021, 14, 3197. [Google Scholar] [CrossRef]
- Kreuter, J.; Shamenkov, D.; Petrov, V.; Ramge, P.; Cychutek, K.; Koch-Brandt, C.; Alyautdin, R. Apolipoprotein-mediated transport of nanoparticle-bound drugs across the blood-brain barrier. J. Drug Target 2002, 10, 317–325. [Google Scholar] [CrossRef]
- Wunsch, A.; Mulac, D.; Langer, K. Lipoprotein imitating nanoparticles: Lecithin coating binds ApoE and mediates non-lysosomal uptake leading to transcytosis over the blood-brain barrier. Int. J. Pharm. 2020, 589, 119821. [Google Scholar] [CrossRef]
- Li, Y.; Wu, M.; Zhang, N.; Tang, C.; Jiang, P.; Liu, X.; Yan, F.; Zheng, H. Mechanisms of enhanced antiglioma efficacy of polysorbate 80-modified paclitaxel-loaded PLGA nanoparticles by focused ultrasound. J. Cell. Mol. Med. 2018, 22, 4171–4182. [Google Scholar] [CrossRef]
- Portioli, C.; Bovi, M.; Benati, D.; Donini, M.; Perduca, M.; Romeo, A.; Dusi, S.; Monaco, H.L.; Bentivoglio, M. Novel functionalization strategies of polymeric nanoparticles as carriers for brain medications. J. Biomed. Mater. Res. A 2017, 105, 847–858. [Google Scholar] [CrossRef]
- Wagner, S.; Zensi, A.; Wien, S.L.; Tschickardt, S.E.; Maier, W.; Vogel, T.; Worek, F.; Pietrzik, C.U.; Kreuter, J.; von Briesen, H. Uptake mechanism of ApoE-modified nanoparticles on brain capillary endothelial cells as a blood-brain barrier model. PLoS ONE 2012, 7, e32568. [Google Scholar] [CrossRef]
- Rafiei, P.; Haddadi, A. Docetaxel-loaded PLGA and PLGA-PEG nanoparticles for intravenous application: Pharmacokinetics and biodistribution profile. Int. J. Nanomed. 2017, 12, 935–947. [Google Scholar] [CrossRef] [Green Version]
- Malatesta, M. Transmission Electron Microscopy as a Powerful Tool to Investigate the Interaction of Nanoparticles with Subcellular Structures. Int. J. Mol. Sci. 2021, 22, 12789. [Google Scholar] [CrossRef]
- Douglas-Escobar, M.; Weiss, M.D. Hypoxic-ischemic encephalopathy: A review for the clinician. JAMA Pediatr. 2015, 169, 397–403. [Google Scholar] [CrossRef]
- Pressler, R.M.; Boylan, G.B.; Marlow, N.; Blennow, M.; Chiron, C.; Cross, J.H.; de Vries, L.S.; Hallberg, B.; Hellstrom-Westas, L.; Jullien, V.; et al. Bumetanide for the treatment of seizures in newborn babies with hypoxic ischaemic encephalopathy (NEMO): An open-label, dose finding, and feasibility phase 1/2 trial. Lancet Neurol. 2015, 14, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr. Res. 2001, 50, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Joseph, A.; Nyambura, C.W.; Bondurant, D.; Corry, K.; Beebout, D.; Wood, T.R.; Pfaendtner, J.; Nance, E. Formulation and Efficacy of Catalase-Loaded Nanoparticles for the Treatment of Neonatal Hypoxic-Ischemic Encephalopathy. Pharmaceutics 2021, 13, 1131. [Google Scholar] [CrossRef] [PubMed]
- Tahara, K.; Miyazaki, Y.; Kawashima, Y.; Kreuter, J.; Yamamoto, H. Brain targeting with surface-modified poly(D,L-lactic-co-glycolic acid) nanoparticles delivered via carotid artery administration. Eur. J. Pharm. Biopharm. 2011, 77, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Bhawana; Basniwal, R.K.; Buttar, H.S.; Jain, V.K.; Jain, N. Curcumin nanoparticles: Preparation, characterization, and antimicrobial study. J. Agric. Food Chem. 2011, 59, 2056–2061. [Google Scholar] [CrossRef] [PubMed]
- Li, S.D.; Huang, L. Pharmacokinetics and biodistribution of nanoparticles. Mol. Pharm. 2008, 5, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Mori, A.; Huang, L. Role of liposome size and RES blockade in controlling biodistribution and tumor uptake of GM1-containing liposomes. Biochim. Biophys. Acta 1992, 1104, 95–101. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, S.P.H.; Mohammadpour, R.; Ghandehari, H. In vitro and in vivo evaluation of degradation, toxicity, biodistribution, and clearance of silica nanoparticles as a function of size, porosity, density, and composition. J. Control. Release 2019, 311, 1–15. [Google Scholar] [CrossRef]
- Li, X.M.; Hu, Z.P.; Ma, J.L.; Wang, X.Y.; Zhang, Y.P.; Wang, W.; Yuan, Z. The systematic evaluation of size-dependent toxicity and multi-time biodistribution of gold nanoparticles. Colloids Surf. B 2018, 167, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Al Shoyaib, A.; Archie, S.R.; Karamyan, V.T. Intraperitoneal Route of Drug Administration: Should it Be Used in Experimental Animal Studies? Pharm. Res. 2019, 37, 12. [Google Scholar] [CrossRef]
- Tao, X.; Mao, S.; Zhang, Q.; Yu, H.; Li, Y.; He, X.; Yang, S.; Zhang, Z.; Yi, Z.; Song, Y.; et al. Brain-Targeted Polysorbate 80-Emulsified Donepezil Drug-Loaded Nanoparticles for Neuroprotection. Nanoscale Res. Lett. 2021, 16, 132. [Google Scholar] [CrossRef]
- Scheuplein, R.; Charnley, G.; Dourson, M. Differential sensitivity of children and adults to chemical toxicity. I. Biological basis. Regul. Toxicol. Pharmacol. 2002, 35, 429–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.A.; Xin, X.; Liu, C.; Liu, Y.H.; Duan, H.X.; Qi, L.L.; Zhang, Y.Y.; Zhao, H.M.; Chen, L.Q.; Jin, M.J.; et al. Novel brain-targeted nanomicelles for anti-glioma therapy mediated by the ApoE-enriched protein corona in vivo. J. Nanobiotechnol. 2021, 19, 453. [Google Scholar] [CrossRef]
- Panagi, Z.; Beletsi, A.; Evangelatos, G.; Livaniou, E.; Ithakissios, D.S.; Avgoustakis, K. Effect of dose on the biodistribution and pharmacokinetics of PLGA and PLGA-mPEG nanoparticles. Int. J. Pharm. 2001, 221, 143–152. [Google Scholar] [CrossRef]
- Martinez, M.N.; Amidon, G.L. A mechanistic approach to understanding the factors affecting drug absorption: A review of fundamentals. J. Clin. Pharmacol. 2002, 42, 620–643. [Google Scholar] [CrossRef] [Green Version]
- Micossi, P.; Cristallo, M.; Librenti, M.C.; Petrella, G.; Galimberti, G.; Melandri, M.; Monti, L.; Spotti, D.; Scavini, M.; Di Carlo, V.; et al. Free-insulin profiles after intraperitoneal, intramuscular, and subcutaneous insulin administration. Diabetes Care 1986, 9, 575–578. [Google Scholar] [CrossRef]
- Costanzo, M.; Carton, F.; Marengo, A.; Berlier, G.; Stella, B.; Arpicco, S.; Malatesta, M. Fluorescence and electron microscopy to visualize the intracellular fate of nanoparticles for drug delivery. Eur. J. Histochem. 2016, 60, 2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinals, R.L.; Yang, D.; Rosenberg, D.J.; Chaudhary, T.; Crothers, A.R.; Iavarone, A.T.; Hammel, M.; Landry, M.P. Quantitative Protein Corona Composition and Dynamics on Carbon Nanotubes in Biological Environments. Angew. Chem. Int. Ed. Engl. 2020, 59, 23668–23677. [Google Scholar] [CrossRef] [PubMed]
- Tekie, F.S.M.; Hajiramezanali, M.; Geramifar, P.; Raoufi, M.; Dinarvand, R.; Soleimani, M.; Atyabi, F. Controlling evolution of protein corona: A prosperous approach to improve chitosan-based nanoparticle biodistribution and half-life. Sci. Rep. 2020, 10, 9664. [Google Scholar] [CrossRef]
- Bertrand, N.; Grenier, P.; Mahmoudi, M.; Lima, E.M.; Appel, E.A.; Dormont, F.; Lim, J.M.; Karnik, R.; Langer, R.; Farokhzad, O.C. Mechanistic understanding of in vivo protein corona formation on polymeric nanoparticles and impact on pharmacokinetics. Nat. Commun. 2017, 8, 777. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Size ± SEM (nm) | PDI ± SEM | Zeta Potential ± SEM (mV) |
|---|---|---|---|
| PLGA-PEG/F127 | 60.2 ± 0.8 | 0.2 ± 0.01 | −2.6 ± 0.3 |
| PLGA-PEG/P80 | 66.2 ± 1.4 | 0.2 ± 0.01 | −2.0 ± 0.3 |
| Formulation | PK Parameter | |
|---|---|---|
| PLGA-PEG/F127 | Tmax (h) | 4 |
| Cmax (mg/mL) | 2.1 ± 0.4 | |
| Vd (mL) | 1.4 ± 0.4 | |
| Vss (mL) | 1.2 ± 1.1 | |
| Clearance (mL/h) | 0.17 ± 0.03 | |
| T1/2 (h) | 5.9 ± 2.1 | |
| AUC (h-mg/mL) | 23.1 ± 3.8 | |
| MRT (h) | 7.2 ± 1.7 | |
| PLGA-PEG/P80 | Tmax (h) | 4 |
| Cmax (mg/mL) | 2.3 ± 0.6 | |
| Vd (mL) | 0.74 ± 0.3 | |
| Vss (mL) | 1.6 ± 1.6 | |
| Clearance (mL/h) | 0.31 ± 0.06 | |
| T1/2 (h) | 1.7 ± 0.7 | |
| AUC (h-mg/mL) | 12.4 ± 2.3 | |
| MRT (h) | 5.2 ± 1.5 | |
| Organ | ||||||
|---|---|---|---|---|---|---|
| Formulation | PK Parameter | Heart | Lung | Liver | Spleen | Kidney |
| PLGA-PEG/F127 | Tmax (h) | 4 | 8 | 24 | 24 | 8 |
| Cmax (mg/mL) | 0.19 ± 0.06 | 0.069 ± 0.04 | 1.2 ± 0.1 | 0.70 ± 0.2 | 0.19 ± 0.07 | |
| Vd (mL) | 13.1 ± 6.9 | 591.8 ± 531.4 | 0.77 ± 0.1 | 5.3 ± 1.2 | 22.7 ± 6.4 | |
| Vss (mL) | 13.0 ± 15.5 | 33.5 ± 53.0 | 1.8 ± 1.2 | 2.9 ± 2.3 | 10.9 ± 9.7 | |
| Clearance (mL/h) | 1.0 ± 0.2 | 4.6 ± 2.3 | 0.082 ± 0.07 | 0.11 ± 0.2 | 0.47 ± 0.08 | |
| T1/2 (h) | 9.4 ± 2.4 | 89.3 ± 21.0 | 6.5 ± 0.5 | 33.5 ± 2.9 | 33.4 ± 6.7 | |
| AUC (h-mg/mL) | 4.0 ± 1.0 | 0.84 ± 0.4 | 47.0 ± 4.1 | 34.9 ± 4.8 | 8.2 ± 1.3 | |
| MRT (h) | 13.4 ± 6.3 | 7.3 ± 5.4 | 22.0 ± 2.7 | 26.0 ± 4.9 | 23.2 ± 5.3 | |
| PLGA-PEG/P80 | Tmax (h) | 4 | 8 | 24 | 8 | 24 |
| Cmax (mg/mL) | 0.12 ± 0.04 | 0.025 ± 0.01 | 0.89 ± 0.1 | 0.53 ± 0.1 | 0.065 ± 0.02 | |
| Vd (mL) | 15.7 ± 12.5 | 114.8 ± 107.0 | 2.1 ± 0.6 | 7.4 ± 2.9 | 48.1 ± 22.0 | |
| Vss (mL) | 28.2 ± 41.2 | 96.8 ± 152.0 | 2.5 ± 2.3 | 4.1 ± 4.3 | 27.3 ± 30.8 | |
| Clearance (mL/h) | 2.2 ± 0.8 | 9.9 ± 3.8 | 0.11 ± 0.01 | 0.24 ± 0.05 | 1.4 ± 0.4 | |
| T1/2 (h) | 4.9 ± 2.3 | 8.0 ± 1.8 | 13.5 ± 2.4 | 21.2 ± 4.1 | 24.0 ± 2.2 | |
| AUC (h-mg/mL) | 1.7 ± 0.6 | 0.39 ± 0.2 | 36.3 ± 4.9 | 15.8 ± 3.2 | 2.8 ± 0.7 | |
| MRT (h) | 12.6 ± 9.0 | 9.7 ± 8.3 | 24.1 ± 6.3 | 17.0 ± 5.7 | 19.6 ± 7.3 | |
| Formulation | PK Parameter | |
|---|---|---|
| PLGA-PEG/F127 | Tmax (h) | 4 |
| Cmax (mg/mL) | 0.013 ± 0.01 | |
| Vd (mL) | 261.5 ± 281.7 | |
| Vss (mL) | 208.0 ± 256.4 | |
| Clearance (mL/h) | 13.6 ± 7.2 | |
| T1/2 (h) | 13.3 ± 4.4 | |
| AUC (h-mg/mL) | 0.28 ± 0.2 | |
| MRT (h) | 15.3 ± 14.4 | |
| PLGA-PEG/P80 | Tmax (h) | 4 |
| Cmax (mg/mL) | 0.020 ± 0.01 | |
| Vd (mL) | 38.7 ± 38.5 | |
| Vss (mL) | 379.2 ± 628.2 | |
| Clearance (mL/h) | 16.7 ± 8.8 | |
| T1/2 (h) | 1.6 ± 0.9 | |
| AUC (h-mg/mL) | 0.23 ± 0.1 | |
| MRT (h) | 22.7 ± 19.1 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, N.; Wong, M.; Balistreri, G.; Nance, E. Neonatal Pharmacokinetics and Biodistribution of Polymeric Nanoparticles and Effect of Surfactant. Pharmaceutics 2023, 15, 1176. https://doi.org/10.3390/pharmaceutics15041176
Xu N, Wong M, Balistreri G, Nance E. Neonatal Pharmacokinetics and Biodistribution of Polymeric Nanoparticles and Effect of Surfactant. Pharmaceutics. 2023; 15(4):1176. https://doi.org/10.3390/pharmaceutics15041176
Chicago/Turabian StyleXu, Nuo, Megan Wong, Gabrielle Balistreri, and Elizabeth Nance. 2023. "Neonatal Pharmacokinetics and Biodistribution of Polymeric Nanoparticles and Effect of Surfactant" Pharmaceutics 15, no. 4: 1176. https://doi.org/10.3390/pharmaceutics15041176
APA StyleXu, N., Wong, M., Balistreri, G., & Nance, E. (2023). Neonatal Pharmacokinetics and Biodistribution of Polymeric Nanoparticles and Effect of Surfactant. Pharmaceutics, 15(4), 1176. https://doi.org/10.3390/pharmaceutics15041176