
Adoptive Transfer of Photosensitizer-Loaded Cytotoxic T Cells for Combinational Photodynamic Therapy and Cancer Immuno-Therapy
 , ,
, , 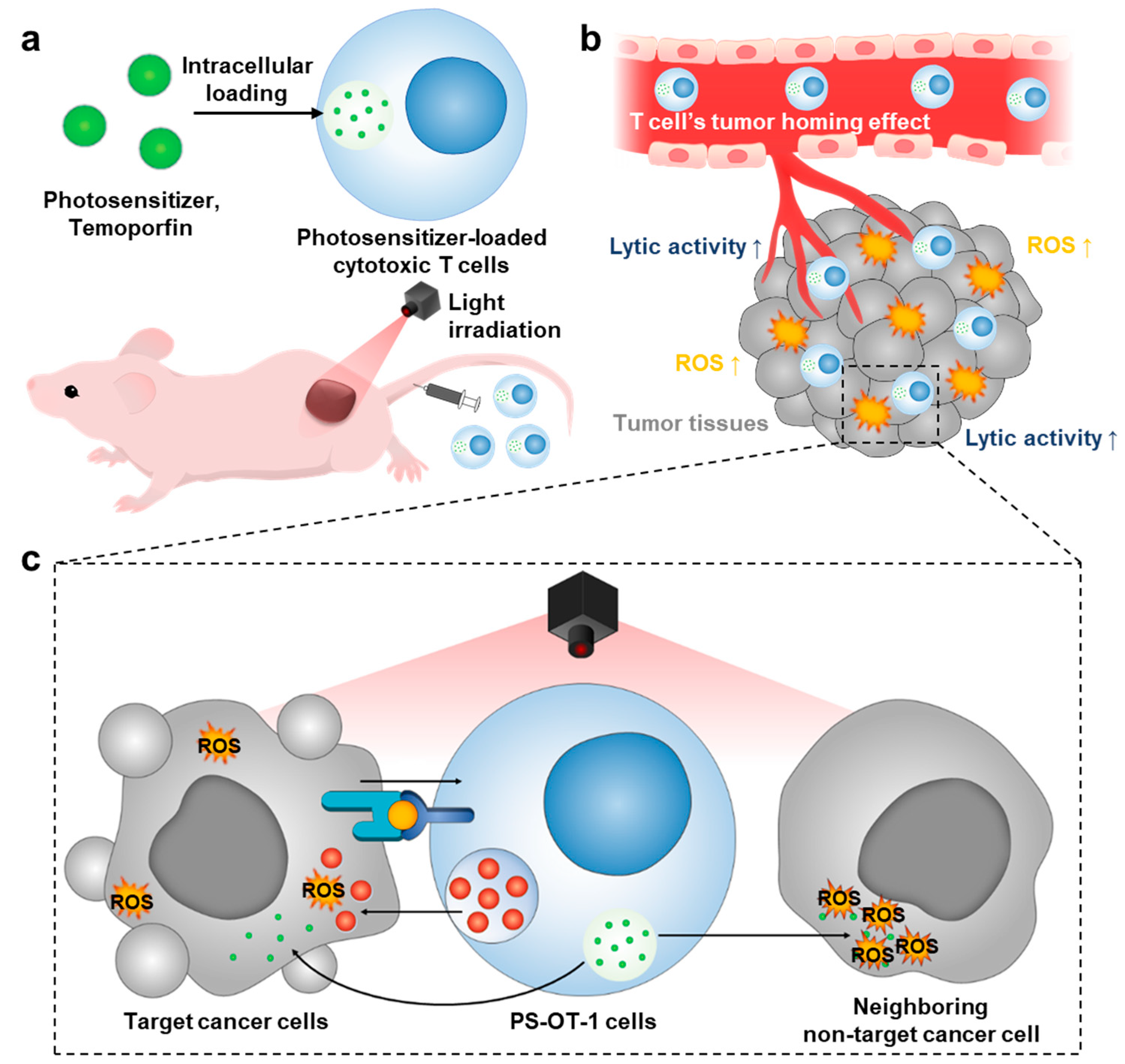{kind=link}
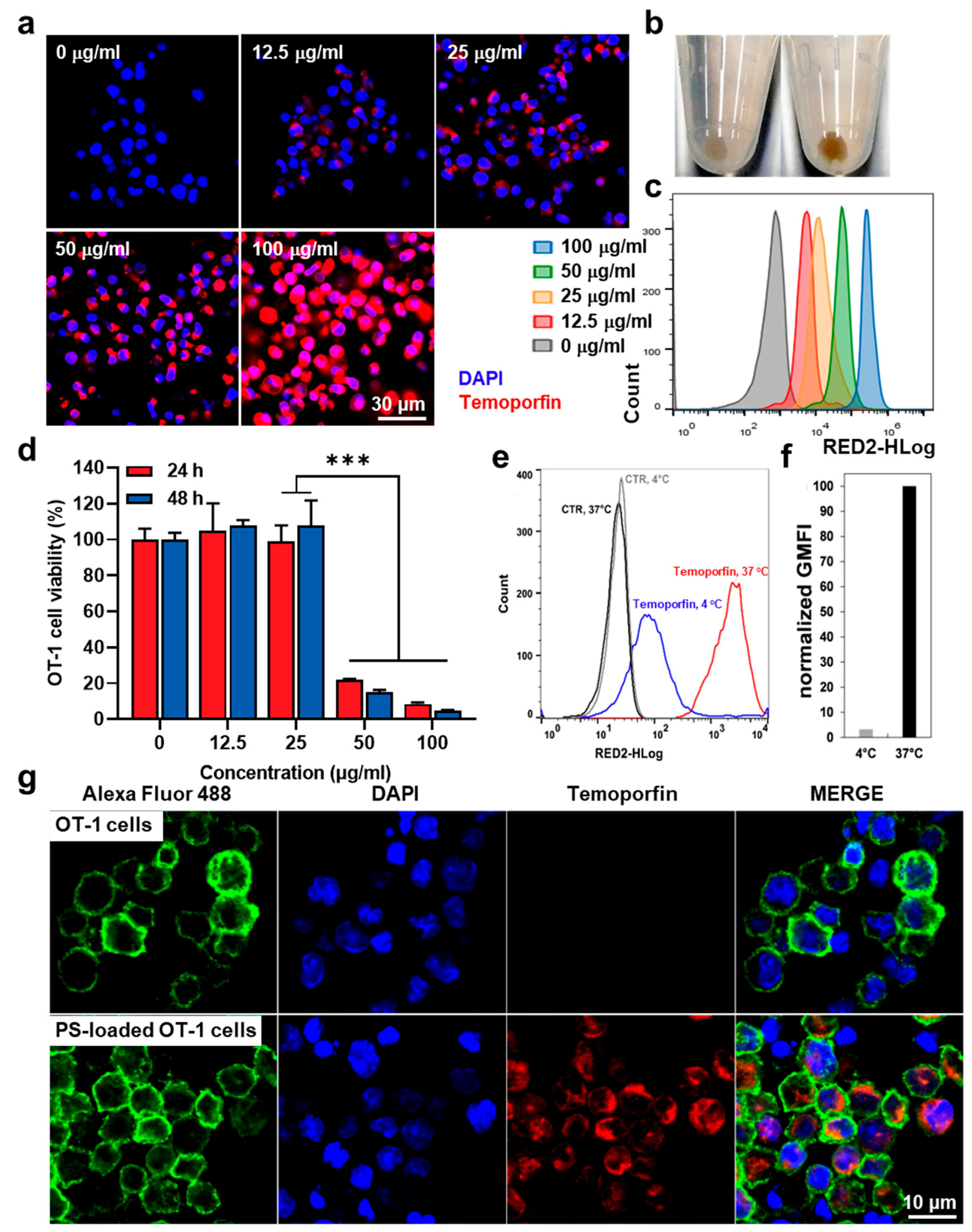{kind=link}
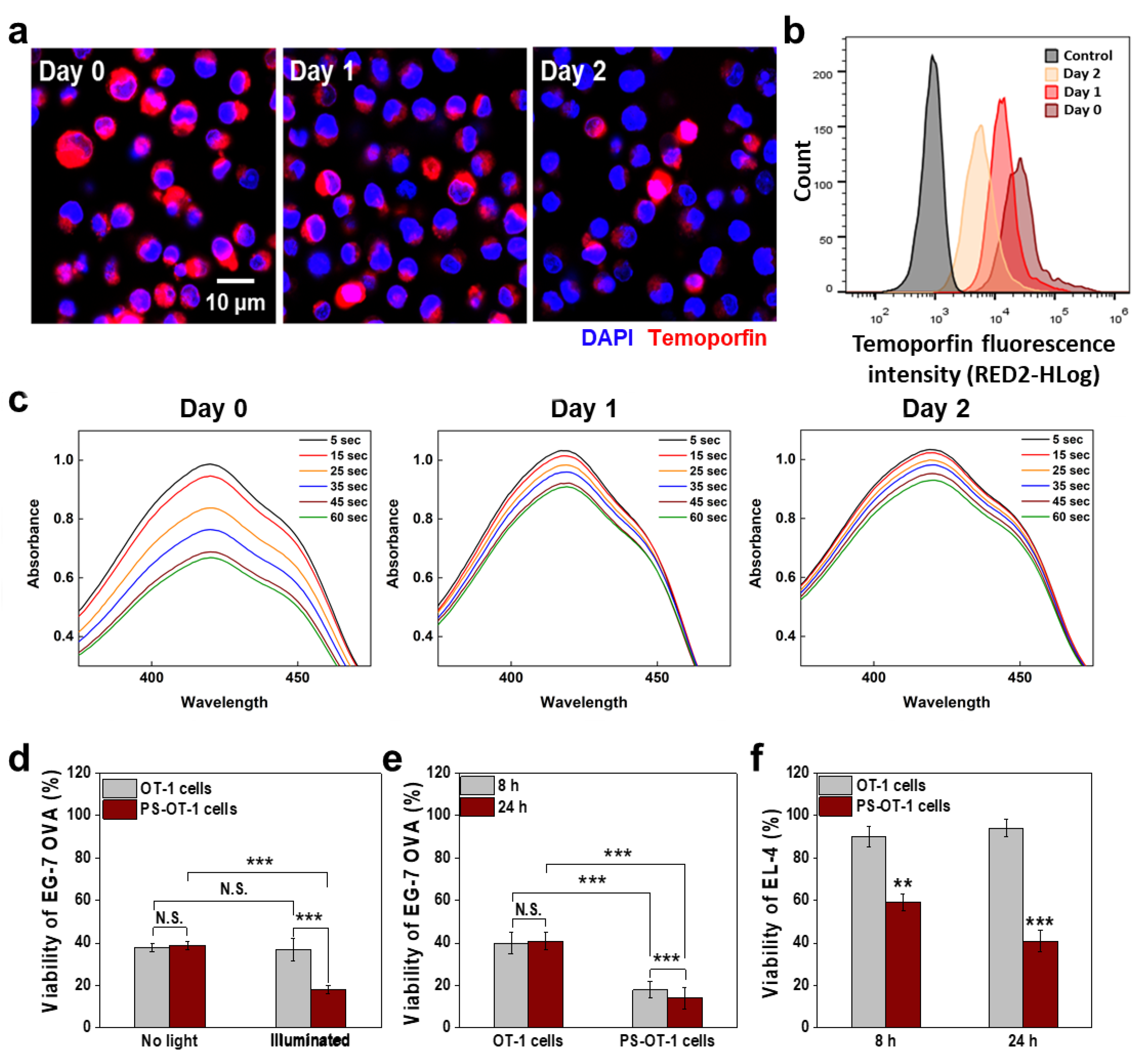{kind=link}
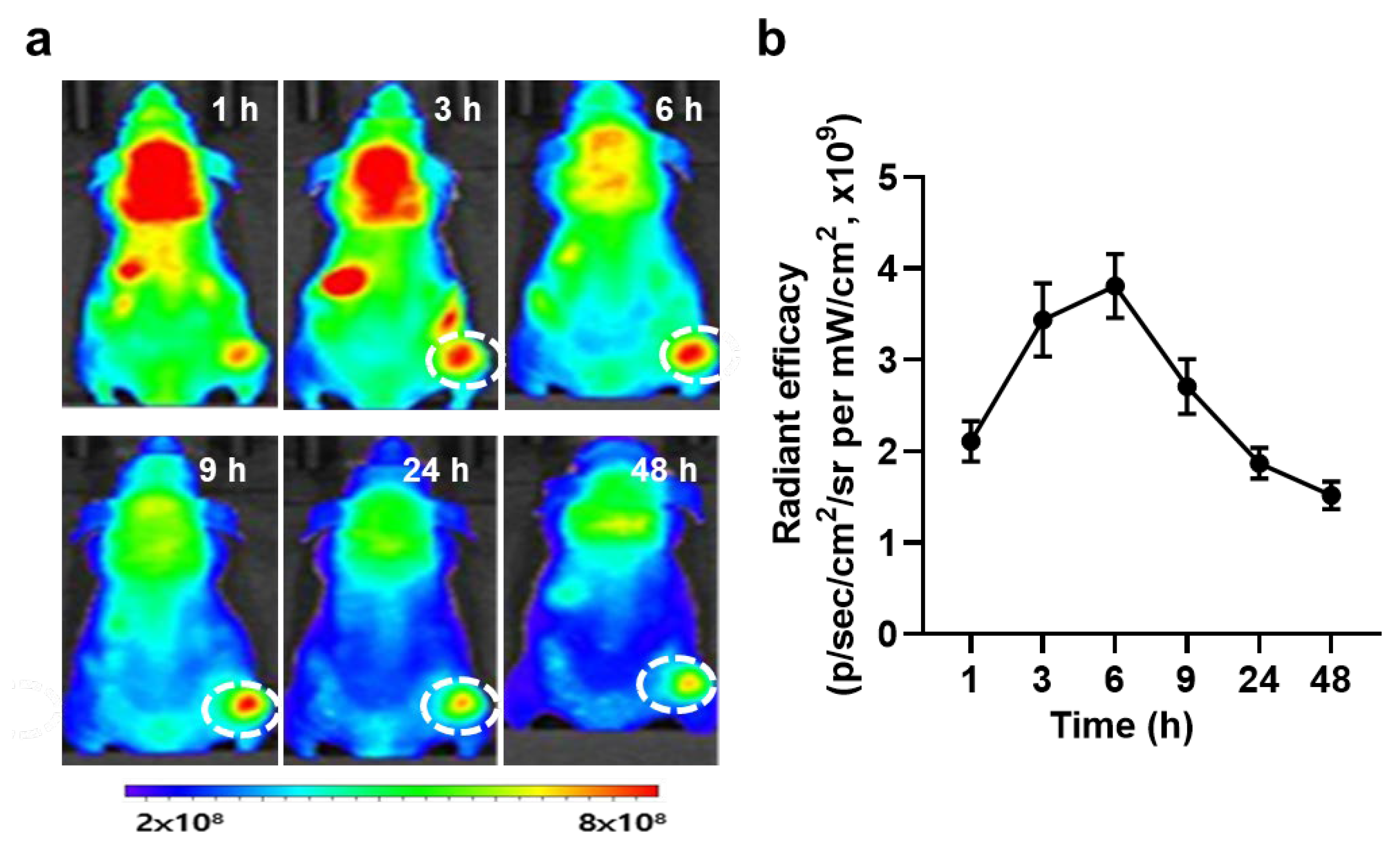{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Preparation of OT-I Cells
2.3. Preparation of Temoporfin (PS)-Loaded OT-1 Cells (PS-OT-1 Cells)
2.4. Antitumor Efficacy of PS-OT-1 Cells In Vitro
2.5. Antitumor Efficacy of PS-OT-1 Cells in a Murinelymphoma Model
2.6. Statistical Analysis
2.7. Data Availability
3. Results and Discussion
3.1. Preparation of Temoporfin-Loaded OT-1 Cells (PS-OT-1 Cells)
3.2. In Vitro Combinational Effect of PDT and ACT by PS-OT-1 Cells
3.3. In Vivo Biodistribution and Antitumor Efficacy of PS-OT-1 Cells in EG.7-OVA Tumor-bearing Mice
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shafer, P.; Kelly, L.M.; Hoyos, V. Cancer therapy with TCR-engineered T cells: Current strategies, challenges, and prospects. Front. Immunol. 2022, 13, 835762. [Google Scholar] [CrossRef] [PubMed]
- Legut, M.; Sewell, A.K. Designer T-cells and T-cell receptors for customized cancer immunotherapies. Curr. Opin. Pharmacol. 2018, 41, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Cappell, K.M.; Kochenderfer, J.N. Long-term outcomes following CAR T cell therapy: What we know so far. Nat. Rev. Clin. Oncol. 2023, 1–13. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Hiltensperger, M.; Krackhardt, A.M. Current and future concepts for the generation and application of genetically engineered CAR-T and TCR-T cells. Front. Immunol. 2023, 14, 1121030. [Google Scholar] [CrossRef]
- Blache, U.; Popp, G.; Dünkel, A.; Koehl, U.; Fricke, S. Potential solutions for manufacture of CAR T cells in cancer immunotherapy. Nat. Commun. 2022, 13, 5225. [Google Scholar] [CrossRef]
- McKenzie, B.; Valitutti, S. Resisting T cell attack: Tumor-cell-intrinsic defense and reparation mechanisms. Trends Cancer 2023, 9, 198–211. [Google Scholar]
- Cassioli, C.; Baldari, C.T. The expanding arsenal of cytotoxic T cells. Front. Immunol. 2022, 13, 883010. [Google Scholar] [CrossRef]
- Westin, J.R.; Kersten, M.J.; Salles, G.; Abramson, J.S.; Schuster, S.J.; Locke, F.L.; Andreadis, C. Efficacy and safety of CD19-directed CAR-T cell therapies in patients with relapsed/refractory aggressive B-cell lymphomas: Observations from the JULIET, ZUMA-1, and TRANSCEND trials. Am. J. Hematol. 2021, 96, 1295–1312. [Google Scholar] [CrossRef]
- Filley, A.C.; Henriquez, M.; Dey, M. CART Immunotherapy: Development, Success, and Translation to Malignant Gliomas and Other Solid Tumors. Front. Oncol. 2018, 8, 453. [Google Scholar] [CrossRef] [PubMed]
- Yeku, O.; Li, X.; Brentjens, R.J. Adoptive T-Cell Therapy for Solid Tumors. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Stephan, M.T.; Moon, J.J.; Um, S.H.; Bershteyn, A.; Irvine, D.J. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat. Med. 2010, 16, 1035–1041. [Google Scholar] [CrossRef]
- Cerundolo, V.; Zanovello, P.; McIntosh, D.; Fabbris, R.; Davies, A.J.; Collavo, D. Temporary inhibition of Moloney-murine sarcoma virus (M-MSV) induced-tumours by adoptive transfer of ricin-treated T-lymphocytes. Br. J. Cancer 1987, 55, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Zanovello, P.; Rosato, A.; Bronte, V.; Mandruzzato, S.; Cerundolo, V.; Collavo, D. Antitumour efficacy of lymphokine-activated killer cells loaded with ricin against experimentally induced lung metastases. Cancer Immunol. Immunother. 1992, 35, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Mandruzzato, S.; Rosato, A.; Bronte, V.; Zanovello, P.; Amboldi, N.; Ballinari, D.; Collavo, D. Adoptive transfer of lymphokine-activated killer cells loaded with 4′-deoxy-4′-iododoxorubicin: Therapeutic effect in mice bearing lung metastases. Cancer Res. 1994, 54, 1016–1020. [Google Scholar]
- Blaudszun, A.R.; Lian, Q.; Schnabel, M.; Loretz, B.; Steinfeld, U.; Lee, H.H.; Wenz, G.; Lehr, C.M.; Schneider, M.; Philippi, A. Polyester-idarubicin nanoparticles and a polymer-photosensitizer complex as potential drug formulations for cell-mediated drug delivery. Int. J. Pharm. 2014, 474, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Blaudszun, A.-R.; Moldenhauer, G.; Schneider, M.; Philippi, A. A photosensitizer delivered by bispecific antibody redirected T lymphocytes enhances cytotoxicity against EpCAM-expressing carcinoma cells upon light irradiation. J. Control. Release 2015, 197, 58–68. [Google Scholar] [CrossRef]
- Wiehe, A.; Senge, M.O. The Photosensitizer Temoporfin (m THPC)–Chemical, Pre-clinical and Clinical Developments in the Last Decade. Photochem. Photobiol. 2022, 99, 356–419. [Google Scholar] [CrossRef]
- Ma, L.; Moan, J.; Berg, K. Evaluation of a new photosensitizer, meso-tetra-hydroxyphenyl-chlorin, for use in photodynamic therapy: A comparison of its photobiological properties with those of two other photosensitizers. Int. J. Cancer 1994, 57, 883–888. [Google Scholar] [CrossRef]
- Silverstein, S.C.; Steinman, R.M.; Cohn, Z.A. Endocytosis. Annu. Rev. Biochem. 1977, 46, 669–722. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blaudszun, A.-R.; Kim, W.J.; Um, W.; Yoon, H.Y.; Shim, M.K.; Kim, K. Adoptive Transfer of Photosensitizer-Loaded Cytotoxic T Cells for Combinational Photodynamic Therapy and Cancer Immuno-Therapy. Pharmaceutics 2023, 15, 1295. https://doi.org/10.3390/pharmaceutics15041295
Blaudszun A-R, Kim WJ, Um W, Yoon HY, Shim MK, Kim K. Adoptive Transfer of Photosensitizer-Loaded Cytotoxic T Cells for Combinational Photodynamic Therapy and Cancer Immuno-Therapy. Pharmaceutics. 2023; 15(4):1295. https://doi.org/10.3390/pharmaceutics15041295
Chicago/Turabian StyleBlaudszun, André-René, Woo Jun Kim, Wooram Um, Hong Yeol Yoon, Man Kyu Shim, and Kwangmeyung Kim. 2023. "Adoptive Transfer of Photosensitizer-Loaded Cytotoxic T Cells for Combinational Photodynamic Therapy and Cancer Immuno-Therapy" Pharmaceutics 15, no. 4: 1295. https://doi.org/10.3390/pharmaceutics15041295
APA StyleBlaudszun, A. -R., Kim, W. J., Um, W., Yoon, H. Y., Shim, M. K., & Kim, K. (2023). Adoptive Transfer of Photosensitizer-Loaded Cytotoxic T Cells for Combinational Photodynamic Therapy and Cancer Immuno-Therapy. Pharmaceutics, 15(4), 1295. https://doi.org/10.3390/pharmaceutics15041295