
Modulation of Transcription Profile Induced by Antiproliferative Thiosemicarbazone Metal Complexes in U937 Cancer Cells

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
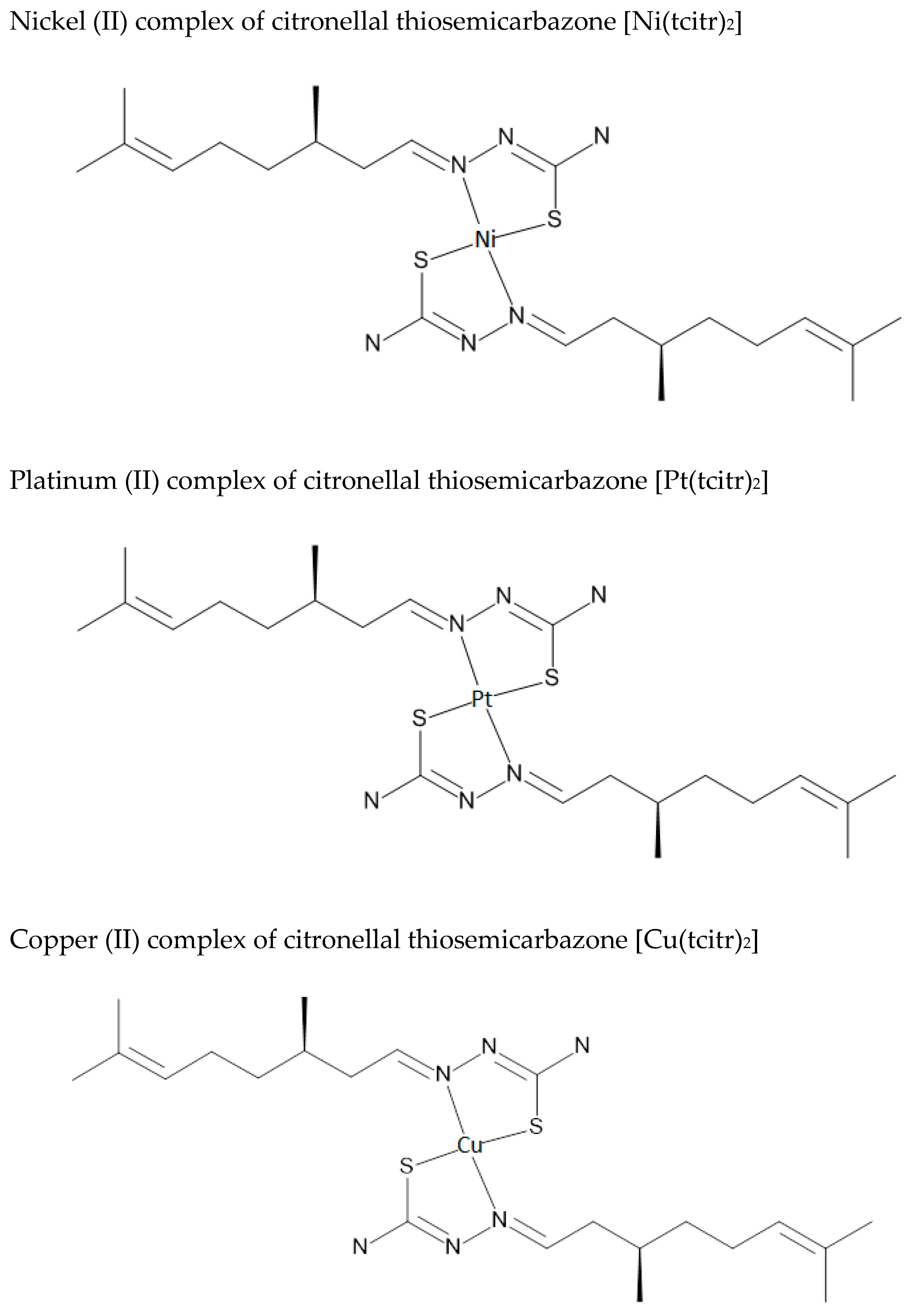
2.1. Synthesis and Chemical Characterization of Nickel [Ni(tcitr)2], Platinum [Pt(tcitr)2] and Copper [Cu(tcitr)2] Complexes
2.2. Cell Line and Culture Condition
2.3. Cell Cycle Analysis on U937 Cell Line by Flow Cytometry
2.4. mRNA Expression Studies on U937 Cell Line by Real Time qPCR
3. Results
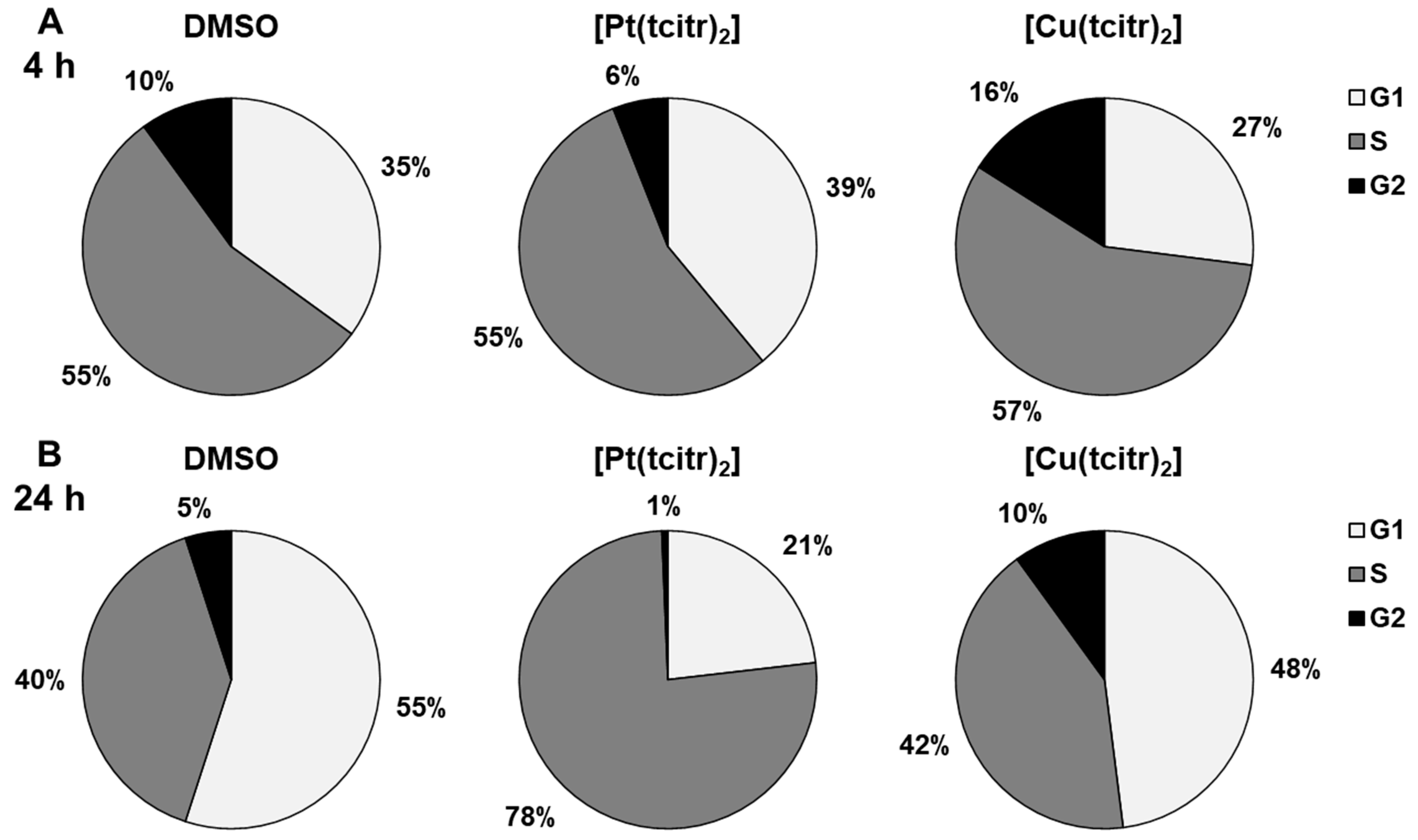
3.1. Cell Cycle Analysis
3.2. Analysis of mRNA Expression Upon Metal Complexes Treatment by qRT-PCR
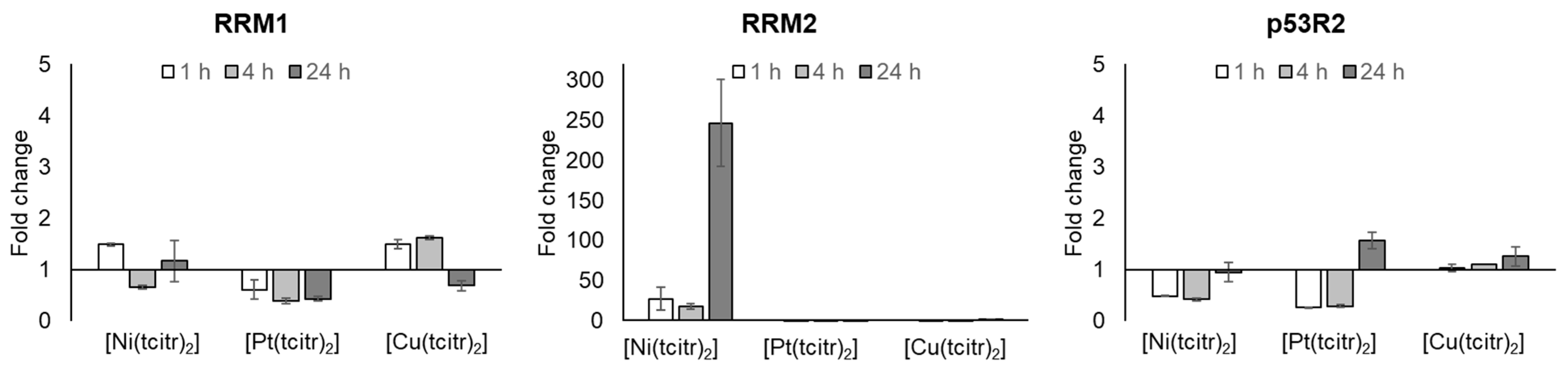
3.2.1. mRNA Expression Profile of Ribonucleotide Reductase Subunits
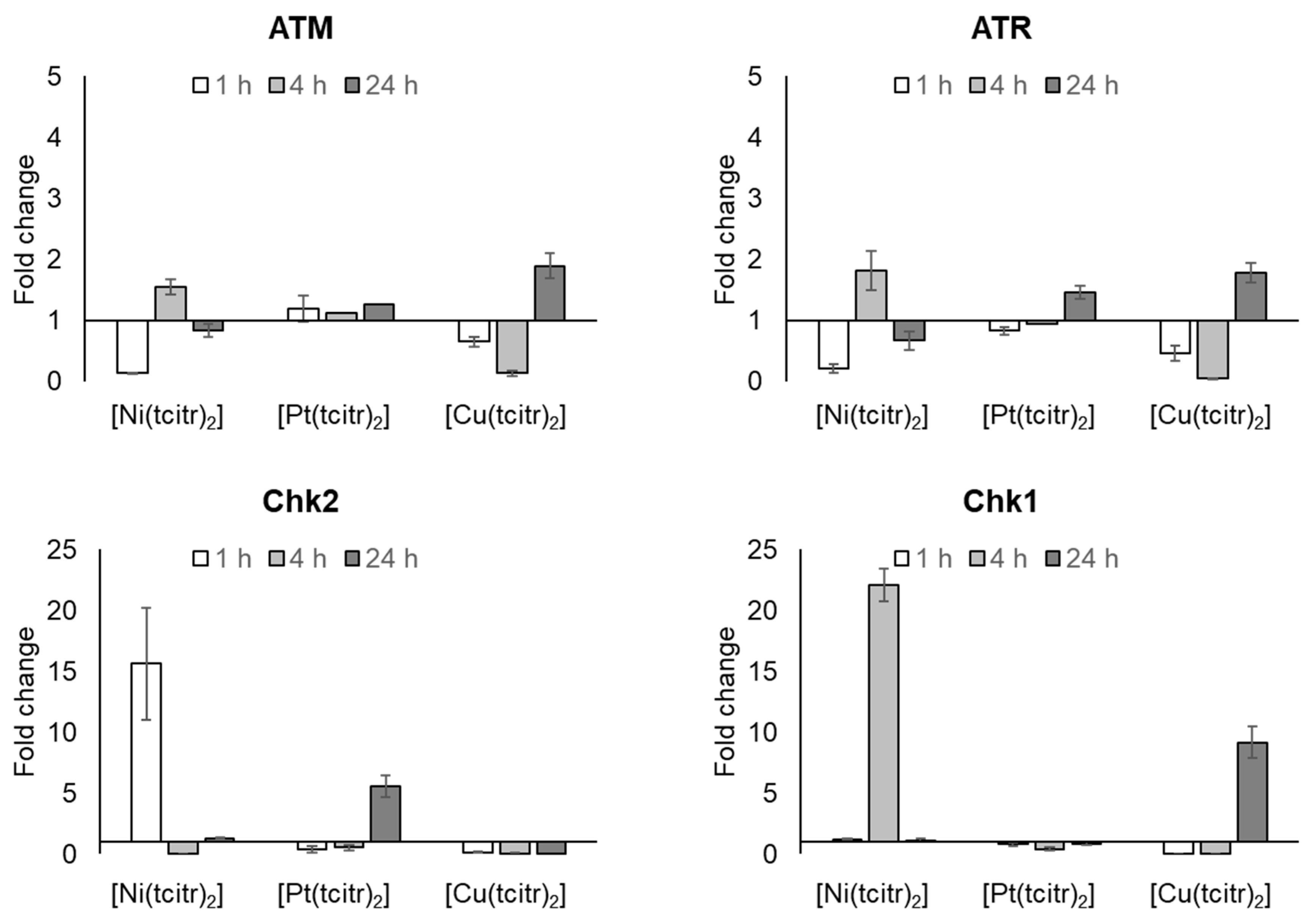
3.2.2. mRNA Expression Profile of DNA Damage Response Pathway
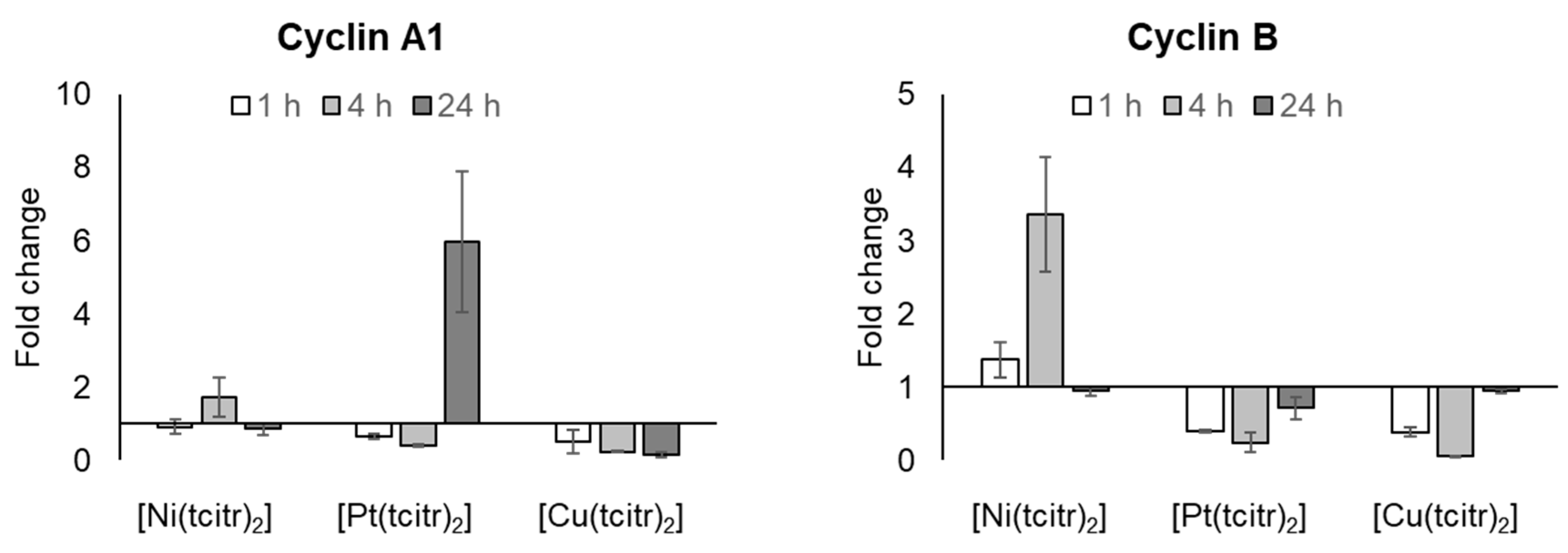
3.2.3. mRNA Expression Profile of Cyclins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Hannon, M.J. Metal-based anticancer drugs: From a past anchored in platinum chemistry to a post-genomic future of diverse chemistry and biology. Pure Appl. Chem. 2007, 79, 2243–2261. [Google Scholar] [CrossRef]
- Chovanec, M.; Cheng, L. Advances in diagnosis and treatment of testicular cancer. BMJ 2022, 379, e070499. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Prim. 2016, 2, 16061. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Zarrabi, A.; Hushmandi, K.; Hashemi, F.; Moghadam, E.R.; Owrang, M.; Hashemi, F.; Makvandi, P.; Goharrizi, M.A.S.B.; Najafi, M.; et al. Lung cancer cells and their sensitivity/resistance to cisplatin chemotherapy: Role of microRNAs and upstream mediators. Cell. Signal. 2021, 78, 109871. [Google Scholar] [CrossRef]
- Chan, W.L.; Choi, C.W.; Wong, I.Y.; Tsang, T.H.; Lam, A.T.; Tse, R.P.; Chan, K.K.; Wong, C.; Law, B.T.; Cheung, E.E.; et al. Docetaxel, Cisplatin, and 5-FU Triplet Therapy as Conversion Therapy for Locoregionally Advanced Unresectable Esophageal Squamous Cell Carcinoma. Ann. Surg. Oncol. 2023, 30, 861–870. [Google Scholar] [CrossRef]
- Ilson, D.H. Advances in the treatment of gastric cancer: 2019. Curr. Opin. Gastroenterol. 2019, 35, 551–554. [Google Scholar] [CrossRef]
- Hager, S.; Ackermann, C.J.; Joerger, M.; Gillessen, S.; Omlin, A. Anti-tumour activity of platinum compounds in advanced prostate cancer-a systematic literature review. Ann. Oncol. 2016, 27, 975–984. [Google Scholar] [CrossRef]
- Xiang, M.; Kidd, E.A. Benefit of Cisplatin With Definitive Radiotherapy in Older Women With Cervical Cancer. J. Natl. Compr. Cancer Netw. 2019, 17, 969–975. [Google Scholar] [CrossRef]
- Köberle, B.; Schoch, S. Platinum Complexes in Colorectal Cancer and Other Solid Tumors. Cancers 2021, 13, 2073. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, C.; Gan, Y.; Lu, F.; Qin, Y. Radiotherapy plus cetuximab or cisplatin in head and neck squamous cell carcinoma: An updated systematic review and meta-analysis of randomized controlled trials. Eur. Arch. Otorhinolaryngol. 2023, 280, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Batgi, H.; Merdin, A.; Dal, M.S.; Kızıl Çakar, M.; Yıldız, J.; Başçı, S.; Uncu Ulu, B.; Yiğenoğlu, T.N.; Darçın, T.; Şahin, D.; et al. The effect of gemcitabine, dexamethasone, and cisplatin chemotherapy in relapsed/refractory NHL and HL patients: A single center experience. J. Oncol. Pharm. Pract. 2020, 26, 1857–1863. [Google Scholar] [CrossRef] [PubMed]
- Durusu, İ.Z.; Hüsnügil, H.H.; Ataş, H.; Biber, A.; Gerekçi, S.; Güleç, E.A.; Özen, C. Anti-cancer effect of clofazimine as a single agent and in combination with cisplatin on U266 multiple myeloma cell line. Leuk. Res. 2017, 55, 33–40. [Google Scholar] [CrossRef]
- Karwaciak, I.; Sałkowska, A.; Karaś, K.; Dastych, J.; Ratajewski, M. Targeting SIRT2 Sensitizes Melanoma Cells to Cisplatin via an EGFR-Dependent Mechanism. Int. J. Mol. Sci. 2021, 22, 5034. [Google Scholar] [CrossRef]
- Sun, B.; Dong, Y.; Xu, J.; Wang, Z. Current status and progress in immunotherapy for malignant pleural mesothelioma. Chronic Dis. Transl. Med. 2022, 8, 91–99. [Google Scholar] [CrossRef]
- Kohno, K.; Wang, K.-Y.; Takahashi, M.; Kurita, T.; Yoshida, Y.; Hirakawa, M.; Harada, Y.; Kuma, A.; Izumi, H.; Matsumoto, S. Mitochondrial Transcription Factor A and Mitochondrial Genome as Molecular Targets for Cisplatin-Based Cancer Chemotherapy. Int. J. Mol. Sci. 2015, 16, 19836–19850. [Google Scholar] [CrossRef]
- Aminuddin, A.; Ng, P.Y.; Leong, C.O.; Chua, E.W. Mitochondrial DNA alterations may influence the cisplatin responsiveness of oral squamous cell carcinoma. Sci. Rep. 2020, 10, 7885. [Google Scholar] [CrossRef]
- Romani, A.M.P. Cisplatin in cancer treatment. Biochem. Pharmacol. 2022, 206, 115323. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Global Health Estimates 2020: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2019. 2020. Available online: who.int/data/gho/data/themes/mortality-and-global-health-estimates/ghe-leading-causes-of-death (accessed on 11 December 2020).
- Pelosi, G. Thiosemicarbazone Metal Complexes: From Structure to Activity. Open Crystallogr. J. 2010, 3, 16–28. [Google Scholar] [CrossRef]
- Matesanz, A.I.; Herrero, J.M.; Quiroga, A.G. Chemical and Biological Evaluation of Thiosemicarbazone-Bearing Heterocyclic Metal Complexes. Curr. Top. Med. Chem. 2021, 21, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.G.; Zheng, Y.; Qi, J. Advances in thiosemicarbazone metal complexes as anti-lung cancer agents. Front. Pharmacol. 2022, 13, 1018951. [Google Scholar] [CrossRef] [PubMed]
- Scarim, C.B.; Chin, C.M. Recent Trends in Drug Development for the Treatment of Adenocarcinoma Breast Cancer: Thiazole, Triazole, and Thiosemicarbazone Analogues as Efficient Scaffolds. Anticancer Agents Med. Chem. 2022, 22, 2204–2240. [Google Scholar] [CrossRef] [PubMed]
- Popović-Bijelić, A.; Kowol, C.R.; Lind, M.E.; Luo, J.; Himo, F.; Enyedy, E.A.; Arion, V.B.; Gräslund, A. Ribonucleotide reductase inhibition by metal complexes of Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone): A combined experimental and theoretical study. J. Inorg. Biochem. 2011, 105, 1422–1431. [Google Scholar] [CrossRef]
- Besleaga, I.; Stepanenko, I.; Petrasheuskaya, T.V.; Darvasiova, D.; Breza, M.; Hammerstad, M.; Marć, M.A.; Prado-Roller, A.; Spengler, G.; Popović-Bijelić, A.; et al. Triapine Analogues and Their Copper(II) Complexes: Synthesis, Characterization, Solution Speciation, Redox Activity, Cytotoxicity, and mR2 RNR Inhibition. Inorg. Chem. 2021, 60, 11297–11319. [Google Scholar] [CrossRef]
- Stepanenko, I.; Babak, M.V.; Spengler, G.; Hammerstad, M.; Popovic-Bijelic, A.; Shova, S.; Büchel, G.E.; Darvasiova, D.; Rapta, P.; Arion, V.B. Coumarin-Based Triapine Derivatives and Their Copper(II) Complexes: Synthesis, Cytotoxicity and mR2 RNR Inhibition Activity. Biomolecules 2021, 11, 862. [Google Scholar] [CrossRef]
- Da Silva Filho, F.A.; de Freitas Souza, T.; Ribeiro, A.G.; Alves, J.E.F.; de Oliveira, J.F.; de Lima Souza, T.R.C.; de Moura, R.O.; do Carmo Alves de Lima, M.; de Carvalho Junior, L.B.; de Almeida, S.M.V. Topoisomerase inhibition and albumin interaction studies of acridine-thiosemicarbazone derivatives. Int. J. Biol. Macromol. 2019, 138, 582–589. [Google Scholar] [CrossRef]
- Sousa, G.; de Almeida, M.C.F.; Lócio, L.L.; Dos Santos, V.L.; Bezerra, D.P.; Silva, V.R.; de Almeida, S.M.V.; Simon, A.; Honório, T.D.S.; Cabral, L.M.; et al. Synthesis and Evaluation of Antiproliferative Activity, Topoisomerase IIα Inhibition, DNA Binding and Non-Clinical Toxicity of New Acridine-Thiosemicarbazone Derivatives. Pharmaceuticals 2022, 15, 1098. [Google Scholar] [CrossRef]
- Ritacca, A.G.; Falcone, E.; Doumi, I.; Vileno, B.; Faller, P.; Sicilia, E. Dual Role of Glutathione as a Reducing Agent and Cu-Ligand Governs the ROS Production by Anticancer Cu-Thiosemicarbazone Complexes. Inorg. Chem. 2023, 62, 3957–3964. [Google Scholar] [CrossRef]
- Rouco, L.; Alvariño, R.; Alfonso, A.; Romero, M.J.; Pedrido, R.; Maneiro, M. Neuroprotective effects of fluorophore-labelled manganese complexes: Determination of ROS production, mitochondrial membrane potential and confocal fluorescence microscopy studies in neuroblastoma cells. J. Inorg. Biochem. 2022, 227, 111670. [Google Scholar] [CrossRef] [PubMed]
- Shyamsivappan, S.; Vivek, R.; Saravanan, A.; Arasakumar, T.; Suresh, T.; Athimoolam, S.; Mohan, P.S. A novel 8-nitro quinoline-thiosemicarbazone analogues induces G1/S & G2/M phase cell cycle arrest and apoptosis through ROS mediated mitochondrial pathway. Bioorg. Chem. 2020, 97, 103709. [Google Scholar] [CrossRef] [PubMed]
- Sinniah, S.K.; Tan, K.W.; Ng, S.W.; Sim, K.S. Thiosemicarbazone Derivative Induces in vitro Apoptosis in Metastatic PC-3 Cells via Activation of Mitochondrial Pathway. Anticancer Agents Med. Chem. 2017, 17, 741–753. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, J.; Guo, Y.; Hu, J.; Chen, X.; Ruan, H.; Cao, T.; Hou, H. Thiosemicarbazone N-Heterocyclic Cu(II) complexes inducing nuclei DNA and mitochondria damage in hepatocellular carcinoma cells. J. Inorg. Biochem. 2022, 236, 111964. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Z.; Kuang, H.; Zhang, Y.; Gu, W.; Zhu, Y.; Wang, S. Synthesis and antitumor activity of 2-isocamphanyl thiosemicarbazone derivatives via ROS-enhanced mitochondrial damage. Chem. Biol. Drug Des. 2019, 94, 1281–1291. [Google Scholar] [CrossRef]
- Balachandran, C.; Haribabu, J.; Jeyalakshmi, K.; Bhuvanesh, N.S.P.; Karvembu, R.; Emi, N.; Awale, S. Nickel(II) bis(isatin thiosemicarbazone) complexes induced apoptosis through mitochondrial signaling pathway and G0/G1 cell cycle arrest in IM-9 cells. J. Inorg. Biochem. 2018, 182, 208–221. [Google Scholar] [CrossRef]
- Emam, S.H.; Hassan, R.A.; Osman, E.O.; Hamed, M.I.A.; Abdou, A.M.; Kandil, M.M.; Elbaz, E.M.; Mikhail, D.S. Coumarin derivatives with potential anticancer and antibacterial activity: Design, synthesis, VEGFR-2 and DNA gyrase inhibition, and in silico studies. Drug Dev. Res. 2023. [Google Scholar] [CrossRef]
- Mehmood, H.; Musa, M.; Woodward, S.; Hossan, M.S.; Bradshaw, T.D.; Haroon, M.; Nortcliffe, A.; Akhtar, T. Design, and synthesis of selectively anticancer 4-cyanophenyl substituted thiazol-2-ylhydrazones. RSC Adv. 2022, 12, 34126–34141. [Google Scholar] [CrossRef]
- Khan, A.A.; Ahmad, R.; Alanazi, A.M.; Alsaif, N.; Abdullah, M.; Wani, T.A.; Bhat, M.A. Determination of anticancer potential of a novel pharmacologically active thiosemicarbazone derivative against colorectal cancer cell lines. Saudi Pharm. J. 2022, 30, 815–824. [Google Scholar] [CrossRef]
- Malarz, K.; Mrozek-Wilczkiewicz, A.; Serda, M.; Rejmund, M.; Polanski, J.; Musiol, R. The role of oxidative stress in activity of anticancer thiosemicarbazones. Oncotarget 2018, 9, 17689–17710. [Google Scholar] [CrossRef]
- Mrozek-Wilczkiewicz, A.; Kuczak, M.; Malarz, K.; Cieślik, W.; Spaczyńska, E.; Musiol, R. The synthesis and anticancer activity of 2-styrylquinoline derivatives. A p53 independent mechanism of action. Eur. J. Med. Chem. 2019, 177, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Mrozek-Wilczkiewicz, A.; Malarz, K.; Rejmund, M.; Polanski, J.; Musiol, R. Anticancer activity of the thiosemicarbazones that are based on di-2-pyridine ketone and quinoline moiety. Eur. J. Med. Chem. 2019, 171, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Moussa, R.S.; Kovacevic, Z.; Bae, D.H.; Lane, D.J.R.; Richardson, D.R. Transcriptional regulation of the cyclin-dependent kinase inhibitor, p21CIP1/WAF1, by the chelator, Dp44mT. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Suryo Rahmanto, Y.; Richardson, D.R. Bp44mT: An orally active iron chelator of the thiosemicarbazone class with potent anti-tumour efficacy. Br. J. Pharmacol. 2012, 165, 148–166. [Google Scholar] [CrossRef]
- Buschini, A.; Pinelli, S.; Pellacani, C.; Giordani, F.; Ferrari, M.B.; Bisceglie, F.; Giannetto, M.; Pelosi, G.; Tarasconi, P. Synthesis, characterization and deepening in the comprehension of the biological action mechanisms of a new nickel complex with antiproliferative activity. J. Inorg. Biochem. 2009, 103, 666–677. [Google Scholar] [CrossRef]
- Buschini, A.; Pinelli, S.; Alinovi, R.; Mussi, F.; Bisceglie, F.; Rivetti, C.; Doniselli, N.; Pelosi, G. Unravelling mechanisms behind the biological activity of bis(S-citronellalthiosemicarbazonato)nickel(II). Metallomics 2014, 6, 783–792. [Google Scholar] [CrossRef]
- Bisceglie, F.; Pinelli, S.; Alinovi, R.; Tarasconi, P.; Buschini, A.; Mussi, F.; Mutti, A.; Pelosi, G. Copper(II) thiosemicarbazonate molecular modifications modulate apoptotic and oxidative effects on U937 cell line. J. Inorg. Biochem. 2012, 116, 195–203. [Google Scholar] [CrossRef]
- Bisceglie, F.; Orsoni, N.; Pioli, M.; Bonati, B.; Tarasconi, P.; Rivetti, C.; Amidani, D.; Montalbano, S.; Buschini, A.; Pelosi, G. Cytotoxic activity of copper(ii), nickel(ii) and platinum(ii) thiosemicarbazone derivatives: Interaction with DNA and the H2A histone peptide. Metallomics 2019, 11, 1729–1742. [Google Scholar] [CrossRef]
- Belicchi Ferrari, M.; Bisceglie, F.; Pelosi, G.; Sassi, M.; Tarasconi, P.; Cornia, M.; Capacchi, S.; Albertini, R.; Pinelli, S. Synthesis, characterization and X-ray structures of new antiproliferative and proapoptotic natural aldehyde thiosemicarbazones and their nickel(II) and copper(II) complexes. J. Inorg. Biochem. 2002, 90, 113–126. [Google Scholar] [CrossRef]
- Bisceglie, F.; Alinovi, R.; Pinelli, S.; Goldoni, M.; Buschini, A.; Franzoni, S.; Mutti, A.; Tarasconi, P.; Pelosi, G. Ni(II) and Cu(II) N(4)-ethylmorpholine citronellalthiosemicarbazonate: A comparative analysis of cytotoxic effects in malignant human cancer cell lines. Metallomics 2013, 5, 1510–1518. [Google Scholar] [CrossRef]
- Bisceglie, F.; Pinelli, S.; Alinovi, R.; Goldoni, M.; Mutti, A.; Camerini, A.; Piola, L.; Tarasconi, P.; Pelosi, G. Cinnamaldehyde and cuminaldehyde thiosemicarbazones and their copper(II) and nickel(II) complexes: A study to understand their biological activity. J. Inorg. Biochem. 2014, 140, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Bisceglie, F.; Tavone, M.; Mussi, F.; Azzoni, S.; Montalbano, S.; Franzoni, S.; Tarasconi, P.; Buschini, A.; Pelosi, G. Effects of polar substituents on the biological activity of thiosemicarbazone metal complexes. J. Inorg. Biochem. 2018, 179, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, S.; Salari, S.; Kaveh, V.; Ghaffari, S.H.; Bashash, D. Alteration of PPAR-GAMMA (PPARG.; PPARγ) and PTEN gene expression in acute myeloid leukemia patients and the promising anticancer effects of PPARγ stimulation using pioglitazone on AML cells. Mol. Genet. Genomic Med. 2021, 9, e1818. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, M.; Gomez, N.; Remes Lenicov, F.; Echeverría, E.; Shayo, C.; Moglioni, A.; Fernández, N.; Davio, C. G2/M Cell Cycle Arrest and Tumor Selective Apoptosis of Acute Leukemia Cells by a Promising Benzophenone Thiosemicarbazone Compound. PLoS ONE 2015, 10, e0136878. [Google Scholar] [CrossRef] [PubMed]
- Raboni, S.; Montalbano, S.; Stransky, S.; Garcia, B.A.; Buschini, A.; Bettati, S.; Sidoli, S.; Mozzarelli, A. A Key Silencing Histon Mark on Chromatin Is Lost When Colorectal Adenocarcinoma Cells Are Depleted of Methionine by Methionine-Lyase. Front. Mol. Biosci. 2021, 8, 735303. [Google Scholar] [CrossRef] [PubMed]
- Baruffini, E.; Ruotolo, R.; Bisceglie, F.; Montalbano, S.; Ottonello, S.; Pelosi, G.; Buschini, A.; Lodi, T. Mechanistic insights on the mode of action of an antiproliferative thiosemicarbazone-nickel complex revealed by an integrated chemogenomic profiling study. Sci. Rep. 2020, 10, 10524. [Google Scholar] [CrossRef]
- Aye, Y.; Li, M.; Long, M.J.; Weiss, R.S. Ribonucleotide reductase and cancer: Biological mechanisms and targeted therapies. Oncogene 2015, 34, 2011–2021. [Google Scholar] [CrossRef]
- Graser-Loescher, G.; Schoenhuber, A.; Ciglenec, C.; Eberl, S.; Krupitza, G.; Mader, R.M.; Jadav, S.S.; Jayaprakash, V.; Fritzer-Szekeres, M.; Szekeres, T.; et al. Thiosemicarbazone derivatives, thiazolyl hydrazones, effectively inhibit leukemic tumor cell growth: Down-regulation of ribonucleotide reductase activity and synergism with arabinofuranosylcytosine. Food Chem. Toxicol. 2017, 108 Pt A, 53–62. [Google Scholar] [CrossRef]
- Tanaka, H.; Arakawa, H.; Yamaguchi, T.; Shiraishi, K.; Fukuda, S.; Matsui, K.; Takei, Y.; Nakamura, Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 2000, 404, 42–49. [Google Scholar] [CrossRef]
- Chabes, A.; Thelander, L. Controlled protein degradation regulates ribonucleotide reductase activity in proliferating mammalian cells during the normal cell cycle and in response to DNA damage and replication blocks. J. Biol. Chem. 2000, 275, 17747–17753. [Google Scholar] [CrossRef]
- Lindemann, A.; Patel, A.A.; Silver, N.L.; Tang, L.; Liu, Z.; Wang, L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, A Novel Thiosemicarbazone Derivative, Exhibits Antitumor Activity in HNSCC through p53-dependent and -independent Mechanisms. Clin. Cancer Res. 2019, 25, 5650–5662. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Bosotti, R.; Scacheri, E.; Belcastro, V.; Mithbaokar, P.; Ferriero, R.; Murino, L.; Tagliaferri, R.; Brunetti-Pierri, N.; Isacchi, A.; et al. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci. USA 2010, 107, 14621–14626. [Google Scholar] [CrossRef] [PubMed]
- Rahal, O.N.; Fatfat, M.; Hankache, C.; Osman, B.; Khalife, H.; Machaca, K.; Muhtasib, H.G. Chk1 and DNA-PK mediate TPEN-induced DNA damage in a ROS dependent manner in human colon cancer cells. Cancer Biol. Ther. 2016, 17, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Park, G.Y.; Lippard, S.J. Understanding and improving platinum anticancer drugs—Phenanthriplatin. Anticancer Res. 2014, 34, 471–476. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Forward (5′-3′) | Primer Reverse (5′-3′) | |
|---|---|---|
| GAPDH | ATGACATCAAGAAGGTGGTG | CATACCAGGAAATGAGCTTG |
| RRM1 | AAGAGCAGCGTGCCAGAGAT | ACACATCAAAGACCATCCTGATTAG |
| RRM2 | ACCAACTAGCCACACACCATGA | GGACTGTTTAATCCCGCTGT |
| p53R2 | CCTTGCGATGGATAGCAGATAGA | GCCAGAATATAGCAGCAAAAGATC |
| Cyclin A1 | GTCAGAGAGGGGATGGCAT | CCAGTCCACCAGAATCGTG |
| Cyclin B | CGGGAAGTCACTGGAAACAT | AAACATGGCAGTGACACCAA |
| Chk1 | GGTGCCTATGGAGAAGTTCAA | TCTACGGCACGCTTCATATC |
| Chk2 | CGGATGTTGAGGCTCACGA | TATGCCCTGGGACTGTGAGG |
| ATM | CAGCAGCTGTTACCTGTTTG | TAGATAGGCCAGCATTGGAT |
| ATR | TGTCTGTACTCTTCACGGCATGTT | AAGAGGTCCACATGTCCGTGTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montalbano, S.; Bisceglie, F.; Pelosi, G.; Lazzaretti, M.; Buschini, A. Modulation of Transcription Profile Induced by Antiproliferative Thiosemicarbazone Metal Complexes in U937 Cancer Cells. Pharmaceutics 2023, 15, 1325. https://doi.org/10.3390/pharmaceutics15051325
Montalbano S, Bisceglie F, Pelosi G, Lazzaretti M, Buschini A. Modulation of Transcription Profile Induced by Antiproliferative Thiosemicarbazone Metal Complexes in U937 Cancer Cells. Pharmaceutics. 2023; 15(5):1325. https://doi.org/10.3390/pharmaceutics15051325
Chicago/Turabian StyleMontalbano, Serena, Franco Bisceglie, Giorgio Pelosi, Mirca Lazzaretti, and Annamaria Buschini. 2023. "Modulation of Transcription Profile Induced by Antiproliferative Thiosemicarbazone Metal Complexes in U937 Cancer Cells" Pharmaceutics 15, no. 5: 1325. https://doi.org/10.3390/pharmaceutics15051325
APA StyleMontalbano, S., Bisceglie, F., Pelosi, G., Lazzaretti, M., & Buschini, A. (2023). Modulation of Transcription Profile Induced by Antiproliferative Thiosemicarbazone Metal Complexes in U937 Cancer Cells. Pharmaceutics, 15(5), 1325. https://doi.org/10.3390/pharmaceutics15051325