

Novel Perspectives on the Design and Development of a Long-Acting Subcutaneous Raltegravir Injection for Treatment of HIV—In Vitro and In Vivo Evaluation



Abstract
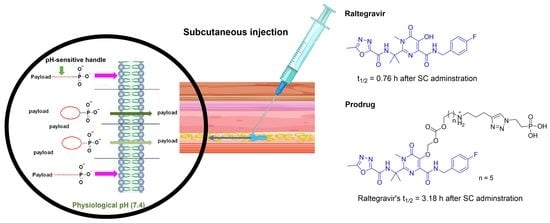
:
1. Introduction
2. Materials and Methods
2.1. Synthetic Procedures and Analytical Data
2.2. In Vitro Release Tests
2.3. Pharmacokinetic Study of Prodrug 25
2.3.1. Study Design
2.3.2. Blood and Sample Preparation
2.3.3. Data Analysis
2.3.4. Ethics
3. Results and Discussion
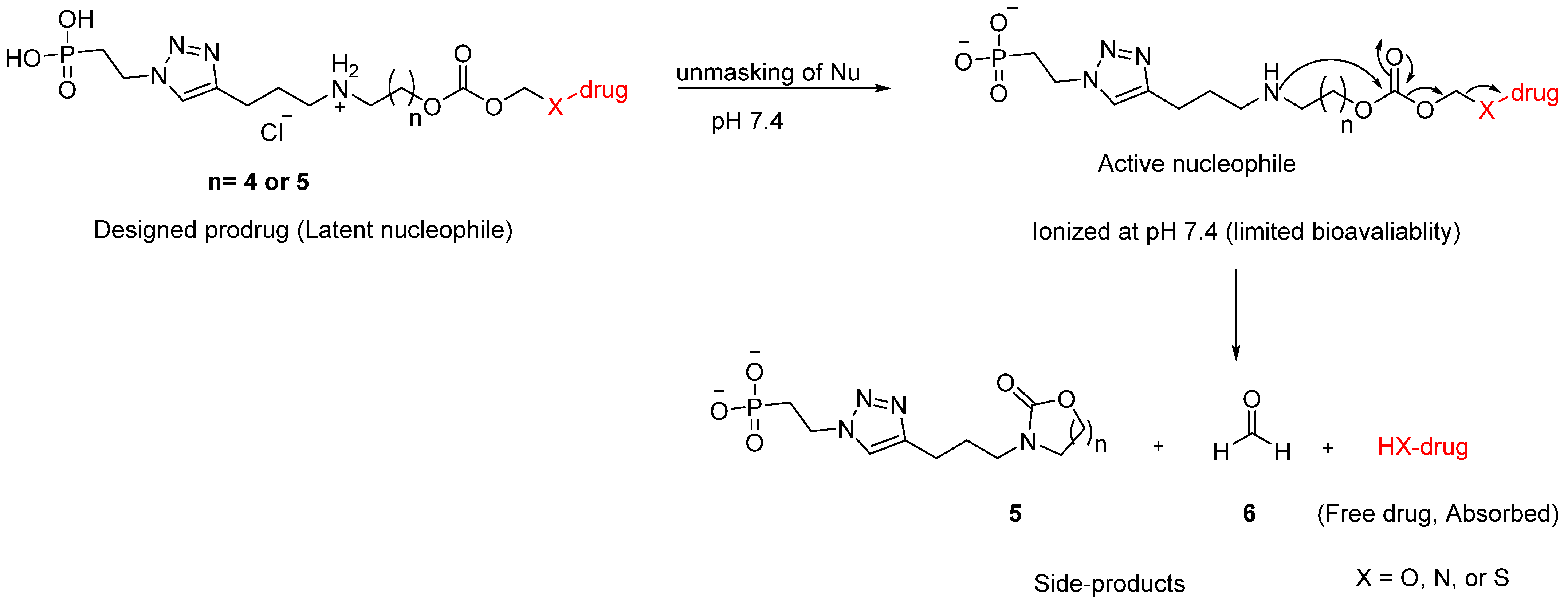
3.1. Proposed Release Mechanism
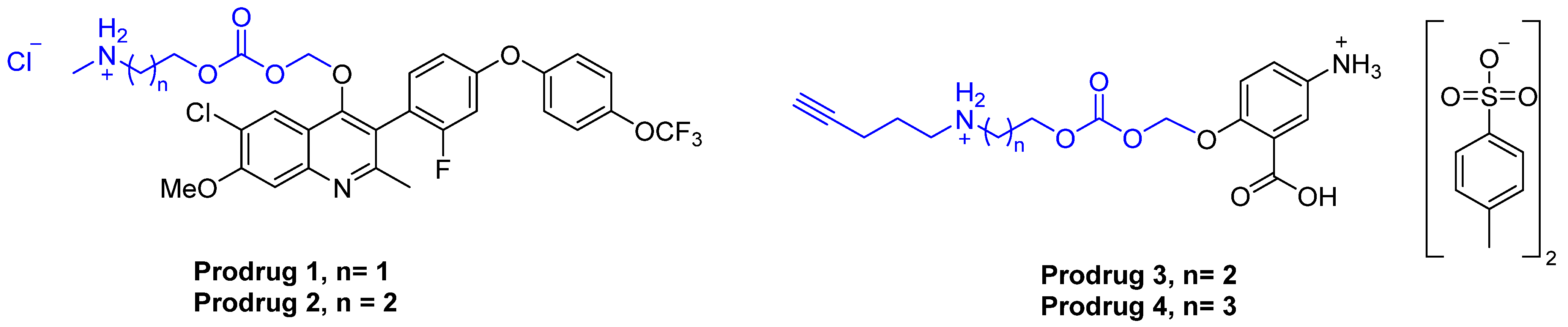
3.2. Synthesis
3.3. In Vitro Release Study of Probes 14–15 and 20–21
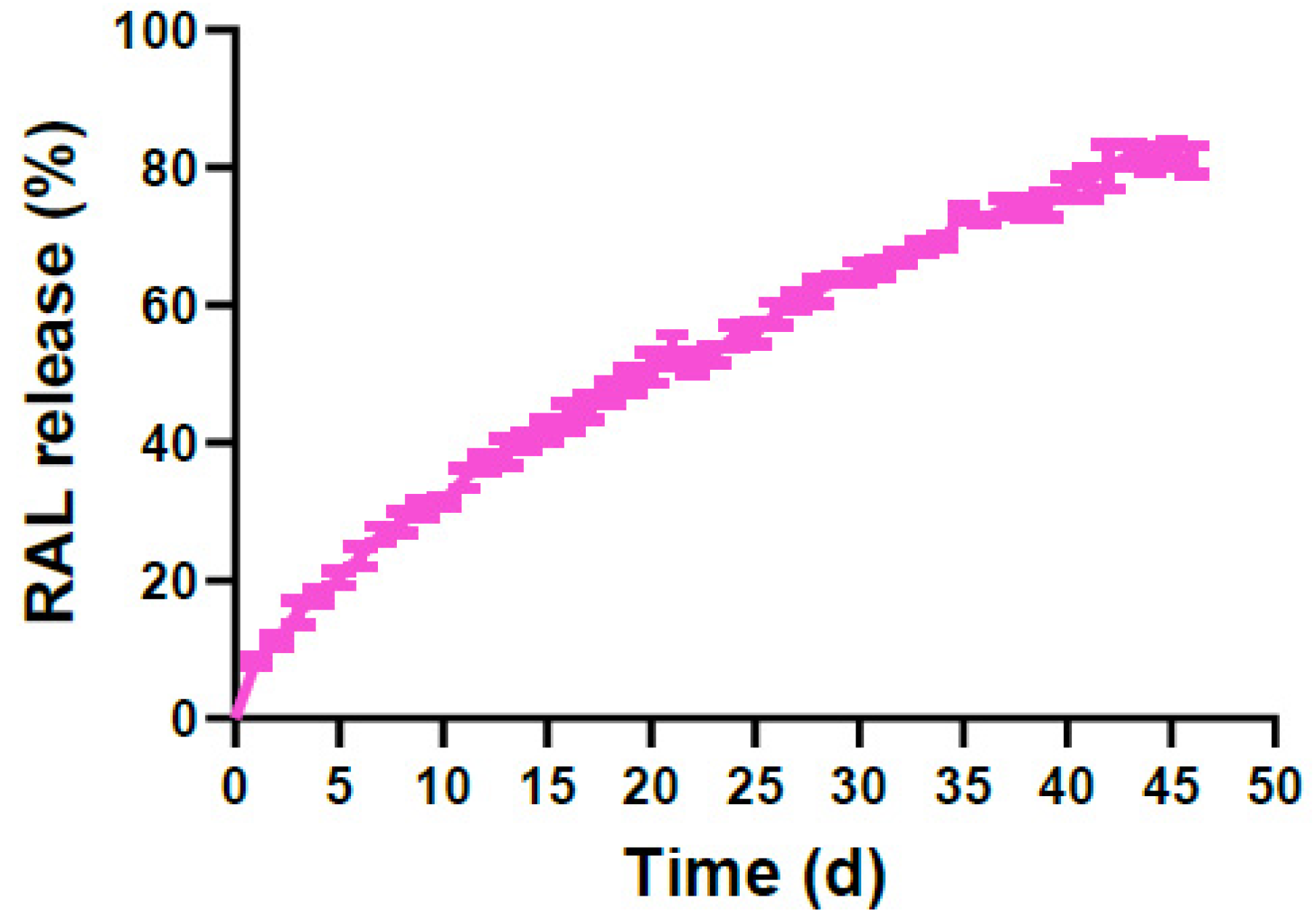
3.4. In Vitro Release Study of Prodrug 25
3.5. Pharmacokinetic Release Profile of 25 in BALB/c Mice
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ARVs | Antiretrovirals |
| Amino-AOCOM | Amino-alkoxycarbonyloxymethyl |
| CTG | 4-Carboxy-2-methyl Tokyo Green |
| SC | Subcutaneous |
| RAL | Raltegravir |
| t½ | Half-life |
| PrEP | Pre-exposure prophylaxis |
| ECM | Extracellular matrix |
| GAGs | Glycosaminoglycans |
| LLOQ | Lower limit of quantification |
Appendix A
Appendix A.1. General Procedure for the Synthesis of Compounds 8(a–b)
Appendix A.2. General Procedure for the Synthesis of Compounds 9(a–b)
Appendix A.3. General Procedure for the Synthesis of Compounds 10(a–b)
Appendix A.4. General Procedure for the Synthesis of Compounds 11(a–b)
Appendix A.5. General Procedure for the Synthesis of CTG-Based Conjugates 13(a–b)
Appendix A.6. General Procedures for the Synthesis of Probes 14–15
Appendix A.7. Procedure for the Synthesis of Diethyl (2-Azidoethyl)Phosphonate (17)
Appendix A.8. Preparation of (2-Azidoethyl)Phosphonic Acid (18)
Appendix A.9. General Procedures to Synthesise Compounds 19(a–b)
Appendix A.10. Preparation of 5-(((((9-(4-Carboxy-2-Methylphenyl)-3-Oxo-3H-Xanthen-6-yl)Oxy)Methoxy)Carbonyl)Oxy)-N-(3-(1-(2-Phosphonoethyl)-1H-1,2,3-triazol-4-yl)Propyl)Pentan-1-Aminium Chloride (20)
Appendix A.11. Preparation of 6-(((((9-(4-(Tert-Butoxycarbonyl)-2-Methylphenyl)-3-Oxo-3H-Xanthen-6-yl)Oxy)Methoxy)Carbonyl)Oxy)-N-(3-(1-(2-Phosphonoethyl)-1H-1,2,3-Triazol-4-yl)Propyl)Hexan-1-Aminium Chloride (21)
Appendix A.12. N-(2-(3-(4-Fluorobenzyl)-7-Methyl-4,8-Dioxo-3,4,7,8-Tetrahydro-2H-Pyrimido[4,5-E][1,3]Oxazin-6-yl)Propan-2-yl)-5-Methyl-1,3,4-Oxadiazole-2-Carboxamide (22)
Appendix A.13. Preparation of Tert-Butyl(6-(((((4-((4-Fluorobenzyl)Carbamoyl)-1-Methyl-2-(2-(5-Methyl-1,3,4-Oxa-Diazole-2-Carboxamido)Propan-2-yl)-6-Oxo-1,6-Dihydropyrimidin-5yl)Oxy)Methoxy)Carbonyl)Oxy)Hexyl)(Pent-4-yn-1-yl)Carbamate (23)
Appendix A.14. Preparation of (2-(4-(3-((Tert-Butoxycarbonyl)(6-(((((4-((4-Fluorobenzyl)Carbamoyl)-1-Methyl-2-(2-(5-Methyl-1,3,4-Oxadiazole-2-Carboxamido)Propan-2-yl)-6-Oxo-1,6-Dihydropyrimidin-5-yl)Oxy)Methoxy)Carbonyl)Oxy)Hexyl)Amino)Propyl)-1H-1,2,3-Triazol-1-yl)Ethyl)Phosphonic Acid (24)
Appendix A.15. Preparation of 6-(((((4-((4-Fluorobenzyl)Carbamoyl)-1-Methyl-2-(2-(5-Methyl-1,3,4-Oxadiazole-2-Carboxa-Mido)Propan-2-Yl)-6-Oxo-1,6-Dihydropyrimidin-5yl)Oxy)Methoxy)Carbonyl)Oxy)-N-(3-(1-(2-Phosphonoethyl)-1H-1,2,3-Triazol-4-Yl)Propyl)Hexan-1-Aminium Chloride (25)
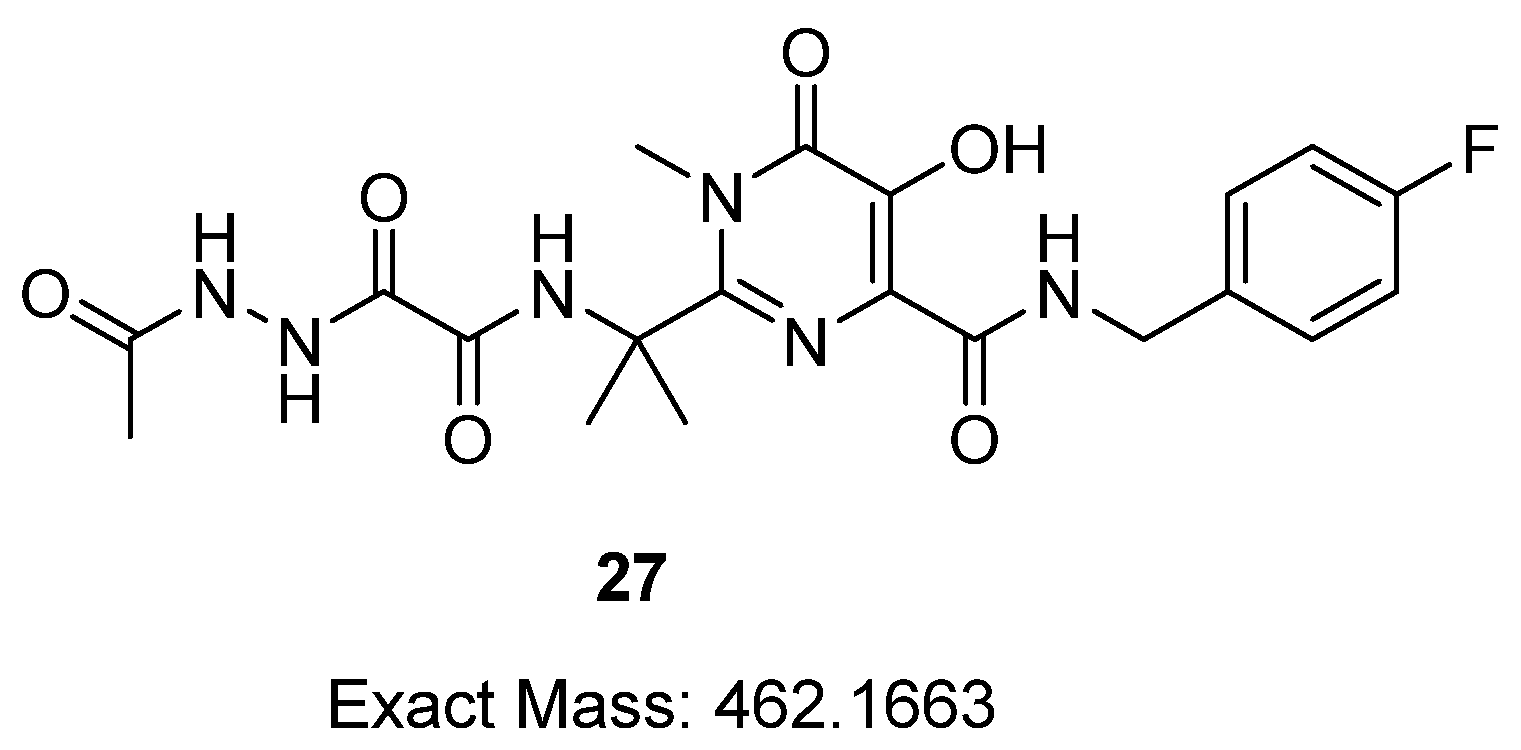
Appendix A.16. (E)-6-(((((4-((4-Fluorobenzyl)Carbamoyl)-2-(2-(2-(2-(1-Hydroxyethylidene)Hydrazineyl)-2-Oxoacetamido)Propan-2-yl)-1-Methyl-6-Oxo-1,6-Dihydropyrimidin-5-yl)Oxy)Methoxy) Carbonyl)Oxy)-N-(3-(1-(2-Phosphonoethyl)-1H-1,2,3-Triazol-4-yl)Propyl)Hexan-1-Aminium Chloride (26)
References
- Barnhart, M. Long-acting HIV treatment and prevention: Closer to the threshold. Glob. Health Sci. Pract. 2017, 5, 182–187. [Google Scholar] [CrossRef]
- Walensky, R.P.; Paltiel, A.D.; Losina, E.; Mercincavage, L.M.; Schackman, B.R.; Sax, P.E.; Weinstein, M.C.; Freedberg, K.A. The survival benefits of AIDS treatment in the united states. J. Infect. Dis. 2006, 194, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Blanc, F.X.; Sok, T.; Laureillard, D.; Borand, L.; Rekacewicz, C.; Nerrienet, E.; Madec, Y.; Marcy, O.; Chan, S.; Prak, N.; et al. Earlier versus later start of antiretroviral therapy in HIV-infected adults with tuberculosis. N. Engl. J. Med. 2011, 365, 1471–1481. [Google Scholar] [CrossRef] [PubMed]
- Wada, N.; Jacobson, L.P.; Cohen, M.; French, A.; Phair, J.; Muñoz, A. Cause-specific life expectancies after 35 years of age for human immunodeficiency syndrome-infected and human immunodeficiency syndrome-negative individuals followed simultaneously in long-term cohort studies, 1984–2008. Am. J. Epidemiololgy 2013, 177, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Chesney, M.A. Factors affecting adherence to antiretroviral therapy. Clin. Infect. Dis. 2000, 30 (Suppl. S2), S171–S176. [Google Scholar] [CrossRef]
- Gonzalez, J.S.; Batchelder, A.W.; Psaros, C.; Safren, S.A. Depression and HIV/AIDS treatment nonadherence: A review and meta-analysis. J. Acquiedr Immune Defic. Syndr. 2011, 58, 181–187. [Google Scholar] [CrossRef]
- Barnabas, R.V.; Celum, C. Closing the gaps in the HIV care continuum. PLoS Med. 2017, 14, 1002443. [Google Scholar] [CrossRef]
- Ortego, C.; Huedo-Medina, T.B.; Llorca, J.; Sevilla, L.; Santos, P.; Rodríguez, E.; Warren, M.R.; Vejo, J. Adherence to highly active antiretroviral therapy (HAART): A meta-analysis. AIDS Behavoir 2011, 15, 1381–1396. [Google Scholar] [CrossRef]
- Amico, K.R.; Stirratt, M.J. Adherence to preexposure prophylaxis: Current, emerging, and anticipated bases of evidence. Clin. Infect. Dis. 2014, 59 (Suppl. S1), S55–S60. [Google Scholar] [CrossRef]
- Williams, J.; Sayles, H.R.; Meza, J.L.; Sayre, P.; Sandkovsky, U.; Gendelman, H.E.; Flexner, C.; Swindells, S. Long-acting parenteral nanoformulated antiretroviral therapy: Interest and attitudes of HIV-infected patients. Nanomedicine 2013, 8, 1807–1813. [Google Scholar] [CrossRef]
- Margolis, D.A.; Gonzalez-Garcia, J.; Stellbrink, H.-J.; Eron, J.J.; Yazdanpanah, Y.; Podzamczer, D.; Lutz, T.; Angel, J.B.; Richmond, G.J.; Clotet, B.; et al. Long-acting intramuscular cabotegravir and rilpivirine in adults with HIV-1 infection (LATTE-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. Lancet 2017, 390, 1499–1510. [Google Scholar] [CrossRef]
- Swindells, S.; Andrade-Villanueva, J.-F.; Richmond, G.J.; Rizzardini, G.; Baumgarten, A.; Masiá, M.; Latiff, G.; Pokrovsky, V.; Bredeek, F.; Smith, G.; et al. Long-Acting Cabotegravir and Rilpivirine for Maintenance of HIV-1 Suppression. N. Engl. J. Med. 2020, 382, 1112–1123. [Google Scholar] [CrossRef]
- Orkin, C.; Arasteh, K.; Górgolas Hernández-Mora, M.; Pokrovsky, V.; Overton, E.T.; Girard, P.-M.; Oka, S.; Walmsley, S.; Bettacchi, C.; Brinson, C.; et al. Long-Acting cabotegravir and rilpivirine after oral induction for HIV-1 infection. N. Engl. J. Med. 2020, 382, 1124–1135. [Google Scholar] [CrossRef]
- Markham, A. Cabotegravir plus rilpivirine: First approval. Drugs 2020, 80, 915–922. [Google Scholar] [CrossRef]
- FDA Approves First Extended-Release, Injectable Drug Regimen for Adults Living with HIV. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-extended-release-injectable-drug-regimen-adults-living-hiv (accessed on 29 November 2022).
- Harrison, T.S.; Goa, K.L. Long-acting risperidone: A review of its use in schizophrenia. CNS Drugs 2004, 18, 113–132. [Google Scholar] [CrossRef]
- Draper, B.H.; Morroni, C.; Hoffman, M.; Smit, J.; Beksinska, M.; Hapgood, J.; Van der Merwe, L. Depot medroxyprogesterone versus norethisterone oenanthate for long-acting progestogenic contraception. Cochrane Database Syst. Rev. 2006, 19, Cd005214. [Google Scholar] [CrossRef]
- Rabinow, B.E. Nanosuspensions in drug delivery. Nat. Rev. Drug Discov. 2004, 3, 785–796. [Google Scholar] [CrossRef]
- Müller, R.H.; Peters, K. Nanosuspensions for the formulation of poorly soluble drugs: I. Preparation by a size-reduction technique. Int. J. Pharm. 1998, 160, 229–237. [Google Scholar] [CrossRef]
- Paik, J.; Duggan, S.T.; Keam, S.J. Triamcinolone acetonide extended-release: A review in osteoarthritis pain of the knee. Drugs 2019, 79, 455–462. [Google Scholar] [CrossRef]
- Kanwar, N.; Sinha, V.R. In Situ Forming Depot as Sustained-Release Drug Delivery Systems. Crit. Rev. Ther. Drug Carr. Syst. 2019, 36, 93–136. [Google Scholar] [CrossRef]
- Shi, Y.; Lu, A.; Wang, X.; Belhadj, Z.; Wang, J.; Zhang, Q. A review of existing strategies for designing long-acting parenteral formulations: Focus on underlying mechanisms, and future perspectives. Acta Pharm. Sin. B 2021, 11, 2396–2415. [Google Scholar] [CrossRef] [PubMed]
- Benhabbour, S.R.; Kovarova, M.; Jones, C.; Copeland, D.J.; Shrivastava, R.; Swanson, M.D.; Sykes, C.; Ho, P.T.; Cottrell, M.L.; Sridharan, A.; et al. Ultra-long-acting tunable biodegradable and removable controlled release implants for drug delivery. Nat. Commun. 2019, 10, 4324. [Google Scholar] [CrossRef] [PubMed]
- Monastyrskyi, A.; Brockmeyer, F.; LaCrue, A.N.; Zhao, Y.; Maher, S.P.; Maignan, J.R.; Padin-Irizarry, V.; Sakhno, Y.I.; Parvatkar, P.T.; Asakawa, A.H. Aminoalkoxycarbonyloxymethyl ether prodrugs with a pH-triggered release mechanism: A case study improving the solubility, bioavailability, and efficacy of antimalarial 4 (1H)-quinolones with single dose cures. J. Med. Chem. 2021, 64, 6581–6595. [Google Scholar] [CrossRef] [PubMed]
- Abd-Ellah, H.S.; Mudududdla, R.; Carter, G.P.; Baell, J.B. Design, development, and optimisation of smart linker chemistry for targeted colonic delivery— in vitro evaluation. Pharmaceutics 2023, 15, 303. [Google Scholar] [CrossRef] [PubMed]
- Croxtall, J.D.; Keam, S.J. Raltegravir: A review of its use in the management of HIV infection in treatment-experienced patients. Drugs 2009, 69, 1059–1075. [Google Scholar] [CrossRef]
- Evering, T.H.; Markowitz, M. Raltegravir: An integrase inhibitor for HIV-1. Expert Opin. Investig. Drugs 2008, 17, 413–422. [Google Scholar] [CrossRef]
- Kovarova, M.; Swanson, M.D.; Sanchez, R.I.; Baker, C.E.; Steve, J.; Spagnuolo, R.A.; Howell, B.J.; Hazuda, D.J.; Garcia, J.V. A long-acting formulation of the integrase inhibitor raltegravir protects humanized BLT mice from repeated high-dose vaginal HIV challenges. J. Antimicrob. Chemother. 2016, 71, 1586–1596. [Google Scholar] [CrossRef]
- Cottrell, M.L.; Patterson, K.B.; Prince, H.M.; Jones, A.; White, N.; Wang, R.; Kashuba, A.D. Effect of HIV infection and menopause status on raltegravir pharmacokinetics in the blood and genital tract. Antivir. Ther. 2015, 20, 795–803. [Google Scholar] [CrossRef]
- Clavel, C.; Peytavin, G.; Tubiana, R.; Soulié, C.; Crenn-Hebert, C.; Heard, I.; Bissuel, F.; Ichou, H.; Ferreira, C.; Katlama, C.; et al. Raltegravir concentrations in the genital tract of HIV-1-infected women treated with a raltegravir-containing regimen (DIVA 01 study). Antimicrob. Agents Chemother. 2011, 55, 3018–3021. [Google Scholar] [CrossRef]
- Heck, H.D.; Casanova-schm1tz, M.; Dodd, P.B.; Schachter, E.N.; Witek, T.J.; Tosun, T. Formaldehyde (CH2O) concentrations in the blood of humans and Fischer-344 rats exposed to CH2O under controlled conditions. Am. Ind. Hyg. Assoc. J. 1985, 46, 1–3. [Google Scholar] [CrossRef]
- Arda, O.; Göksügür, N.; Tüzün, Y. Basic histological structure and functions of facial skin. Clin. Dermatol. 2014, 32, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Viola, M.; Sequeira, J.; Seiça, R.; Veiga, F.; Serra, J.; Santos, A.C.; Ribeiro, A.J. Subcutaneous delivery of monoclonal antibodies: How do we get there? J. Control. Release 2018, 286, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.; Scioli Montoto, S. Routes of Drug Administration: Dosage, Design, and Pharmacotherapy Success; Springer: Berlin/Heidelberg, Germany, 2018; pp. 97–133. [Google Scholar] [CrossRef]
- Supersaxo, A.; Hein, W.R.; Steffen, H. Effect of molecular weight on the lymphatic absorption of water-soluble compounds following subcutaneous administration. Pharm. Res. Off. J. Am. Assoc. Pharm. Sci. 1990, 7, 167–169. [Google Scholar] [CrossRef]
- Usach, I.; Martinez, R.; Festini, T.; Peris, J.E. Subcutaneous injection of drugs: Literature review of factors influencing pain sensation at the injection site. Adv. Ther. 2019, 36, 2986–2996. [Google Scholar] [CrossRef] [PubMed]
- Hecker, S.J.; Erion, M.D. Prodrugs of phosphates and phosphonates. J. Med. Chem. 2008, 51, 2328–2345. [Google Scholar] [CrossRef]
- Schull, T.L.; Fettinger, J.C.; Knight, D.A. Synthesis and characterization of palladium(ii) and platinum(ii) complexes containing water-soluble hybrid phosphine−phosphonate ligands. Inorg. Chem. 1996, 35, 6717–6723. [Google Scholar] [CrossRef]
- Wiemer, A.J.; Wiemer, D.F. Prodrugs of phosphonates and phosphates: Crossing the membrane barrier. Top. Curr. Chem. 2015, 360, 115–160. [Google Scholar] [CrossRef]
- Jiang, J.; Yang, E.; Reddy, K.R.; Niedzwiedzki, D.M.; Kirmaier, C.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Synthetic bacteriochlorins bearing polar motifs (carboxylate, phosphonate, ammonium and a short PEG). Water-solubilization, bioconjugation, and photophysical properties. New J. Chem. 2015, 39, 5694–5714. [Google Scholar] [CrossRef]
- Bittner, B.; Richter, W.; Schmidt, J. Subcutaneous administration of biotherapeutics: An overview of current challenges and opportunities. BioDrugs 2018, 32, 425–440. [Google Scholar] [CrossRef]
- Mineno, T.; Ueno, T.; Urano, Y.; Kojima, H.; Nagano, T. Creation of superior carboxyfluorescein dyes by blocking donor-excited photoinduced electron transfer. Org. Lett. 2006, 8, 5963–5966. [Google Scholar] [CrossRef]
- Yamagishi, K.; Sawaki, K.; Murata, A.; Takeoka, S. A Cu-free clickable fluorescent probe for intracellular targeting of small biomolecules. Chem. Commun. 2015, 51, 7879–7882. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Lee, D.J.; Brimble, M.A. Synthesis of an NDPK phosphocarrier domain peptide containing a novel triazolylalanine analogue of phosphohistidine using click chemistry. Org. Lett. 2011, 13, 5604–5607. [Google Scholar] [CrossRef] [PubMed]
- Patil, G.D.; Kshirsagar, S.W.; Shinde, S.B.; Patil, P.S.; Deshpande, M.S.; Chaudhari, A.T.; Sonawane, S.P.; Maikap, G.C.; Gurjar, M.K. Identification, synthesis, and strategy for minimization of potential impurities observed in raltegravir potassium drug substance. Org. Process Res. Dev. 2012, 16, 1422–1429. [Google Scholar] [CrossRef]
- Webb, P. Temperatures of skin, subcutaneous tissue, muscle and core in resting men in cold, comfortable and hot conditions. Eur. J. Appl. Physiol. Occup. Physiol. 1992, 64, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P. Photobleaching of organic fluorophores: Quantitative characterization, mechanisms, protection. Methods Appl. Fluoresc. 2020, 8, 022001. [Google Scholar] [CrossRef]
- Kabadi, P.G.; Sankaran, P.K.; Palanivelu, D.V.; Adhikary, L.; Khedkar, A.; Chatterjee, A. Mass spectrometry based mechanistic insights into formation of tris conjugates: Implications on protein biopharmaceutics. J. Am. Soc. Mass Spectrom. 2016, 27, 1677–1685. [Google Scholar] [CrossRef]
- Saari, W.S.; Schwering, J.E.; Lyle, P.A.; Smith, S.J.; Engelhardt, E.L. Cyclization-activated prodrugs. Basic esters of 5-bromo-2’-deoxyuridine. J. Med. Chem. 1990, 33, 2590–2595. [Google Scholar] [CrossRef]
- Thomsen, K.F.; Bundgaard, H. Cyclization-activated phenyl carbamate prodrug forms for protecting phenols against first-pass metabolism. Int. J. Pharm. 1993, 91, 39–49. [Google Scholar] [CrossRef]
- Matsumoto, H.; Sohma, Y.; Kimura, T.; Hayashi, Y.; Kiso, Y. Controlled drug release: New water-soluble prodrugs of an HIV protease inhibitor. Bioorganic Med. Chem. Lett. 2001, 11, 605–609. [Google Scholar] [CrossRef]
- Sharma, I.; Kaminski, G.A. Calculating pKa values for substituted phenols and hydration energies for other compounds with the first-order Fuzzy-Border continuum solvation model. J. Comput. Chem. 2012, 33, 2388–2399. [Google Scholar] [CrossRef]
- Moss, D.M.; Siccardi, M.; Back, D.J.; Owen, A. Predicting intestinal absorption of raltegravir using a population-based ADME simulation. J. Antimicroibial Chemother. 2013, 68, 1627–1634. [Google Scholar] [CrossRef]
- Fredholt, K.; Mørk, N.; Begtrup, M. Hemiesters of aliphatic dicarboxylic acids as cyclization-activated prodrug forms for protecting phenols against first-pass metabolism. Int. J. Pharm. 1995, 123, 209–216. [Google Scholar] [CrossRef]
- Sánchez-Félix, M.; Burke, M.; Chen, H.H.; Patterson, C.; Mittal, S. Predicting bioavailability of monoclonal antibodies after subcutaneous administration: Open innovation challenge. Adv. Drug Deliv. Rev. 2020, 167, 66–77. [Google Scholar] [CrossRef]
- Pitiot, A.; Heuzé-Vourc’h, N.; Sécher, T. Alternative routes of administration for therapeutic antibodies-state of the art. Antibodies 2022, 11, 56. [Google Scholar] [CrossRef]
- Bumbaca Yadav, D.; Sharma, V.K.; Boswell, C.A.; Hotzel, I.; Tesar, D.; Shang, Y.; Ying, Y.; Fischer, S.K.; Grogan, J.L.; Chiang, E.Y.; et al. Evaluating the use of antibody variable region (Fv) charge as a risk assessment tool for predicting typical cynomolgus monkey pharmacokinetics. J. Biol. Chem. 2015, 290, 29732–29741. [Google Scholar] [CrossRef]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K.M. Polymeric systems for controlled drug release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef]
- Luginbuhl, K.M.; Schaal, J.L.; Umstead, B.; Mastria, E.M.; Li, X.; Banskota, S.; Arnold, S.; Feinglos, M.; D’Alessio, D.; Chilkoti, A. One-week glucose control via zero-order release kinetics from an injectable depot of glucagon-like peptide-1 fused to a thermosensitive biopolymer. Nat. Biomed. Eng. 2017, 1, 0078. [Google Scholar] [CrossRef]
- Lau, J.; Bloch, P.; Schäffer, L.; Pettersson, I.; Spetzler, J.; Kofoed, J.; Madsen, K.; Knudsen, L.B.; McGuire, J.; Steensgaard, D.B.; et al. Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) Analogue semaglutide. J. Med. Chem. 2015, 58, 7370–7380. [Google Scholar] [CrossRef]
- Lee, P.Y.; Cobain, E.; Huard, J.; Huang, L. Thermosensitive hydrogel PEG-PLGA-PEG enhances engraftment of muscle-derived stem cells and promotes healing in diabetic wound. Mol. Ther. 2007, 15, 1189–1194. [Google Scholar] [CrossRef]
- Kuzma, P.; Moo-Young, A.J.; Mora, D.; Quandt, H.; Bardin, C.W.; Schlegel, P.H. Subcutaneous hydrogel reservoir system for controlled drug delivery. Macromol. Symp. 1996, 109, 15–26. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, S.; Kang, Y.; Huang, K.; Gu, Z.; Wu, J. Advances in long-circulating drug delivery strategy. Curr. Drug Metab. 2018, 19, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Van Witteloostuijn, S.B.; Pedersen, S.L.; Jensen, K.J. Half-life extension of biopharmaceuticals using chemical methods: Alternatives to PEGylation. ChemMedChem 2016, 11, 2474–2495. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Cheng, S.; Zhang, Y.; Ding, Y.; Chong, H.; Xing, H.; Jiang, S.; Li, X.; Ma, L. Long-Acting HIV-1 fusion inhibitory peptides and their mechanisms of action. Viruses 2019, 11, 811. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Wang, Y.; Zhang, Z.; Lv, X.; Gao, G.F.; Shao, Y.; Ma, L.; Li, X. Enfuvirtide-PEG conjugate: A potent HIV fusion inhibitor with improved pharmacokinetic properties. Eur. J. Med. Chem. 2016, 121, 232–237. [Google Scholar] [CrossRef]
- Sherman, M.R.; Williams, L.D.; Sobczyk, M.A.; Michaels, S.J.; Saifer, M.G. Role of the methoxy group in immune responses to mPEG-protein conjugates. Bioconjugate Chem. 2012, 23, 485–499. [Google Scholar] [CrossRef]
- Ma, C.; Bian, T.; Yang, S.; Liu, C.; Zhang, T.; Yang, J.; Li, Y.; Li, J.; Yang, R.; Tan, W. Fabrication of versatile cyclodextrin-functionalized upconversion luminescence nanoplatform for biomedical imaging. Anal. Chem. 2014, 86, 6508–6515. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | kobs [d−1] b | t½ [d] c |
|---|---|---|
| 14 | 4.1 × 10−1 | 1.69 |
| 15 | 3.5 × 10−1 | 1.98 |
| 20 | 2.4 × 10−1 | 2.88 |
| 21 | 2.2 × 10−1 | 3.15 |
| 25 | 0.36 × 10−1 | 19.3 |
| Compound | Dose (mg/kg) | Cmax (µg/mL) b | tmax (h) b | AUC 0–96 h (µg h/mL) c | t½ (h) b |
|---|---|---|---|---|---|
| 25 a | 30 | 4.55 | 0.25 | 3.27 | 0.85 |
| Metabolite (released RAL) | – | 6.59 | 0.42 | 5.59 | 3.18 |
| Control (RAL) a | 30 | 6.23 | 0.33 | 9.89 | 0.76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abd-Ellah, H.S.; Mudududdla, R.; Carter, G.P.; Baell, J.B. Novel Perspectives on the Design and Development of a Long-Acting Subcutaneous Raltegravir Injection for Treatment of HIV—In Vitro and In Vivo Evaluation. Pharmaceutics 2023, 15, 1530. https://doi.org/10.3390/pharmaceutics15051530
Abd-Ellah HS, Mudududdla R, Carter GP, Baell JB. Novel Perspectives on the Design and Development of a Long-Acting Subcutaneous Raltegravir Injection for Treatment of HIV—In Vitro and In Vivo Evaluation. Pharmaceutics. 2023; 15(5):1530. https://doi.org/10.3390/pharmaceutics15051530
Chicago/Turabian StyleAbd-Ellah, Heba S., Ramesh Mudududdla, Glen P. Carter, and Jonathan B. Baell. 2023. "Novel Perspectives on the Design and Development of a Long-Acting Subcutaneous Raltegravir Injection for Treatment of HIV—In Vitro and In Vivo Evaluation" Pharmaceutics 15, no. 5: 1530. https://doi.org/10.3390/pharmaceutics15051530
APA StyleAbd-Ellah, H. S., Mudududdla, R., Carter, G. P., & Baell, J. B. (2023). Novel Perspectives on the Design and Development of a Long-Acting Subcutaneous Raltegravir Injection for Treatment of HIV—In Vitro and In Vivo Evaluation. Pharmaceutics, 15(5), 1530. https://doi.org/10.3390/pharmaceutics15051530