
Emerging Preclinical Applications of Humanized Mouse Models in the Discovery and Validation of Novel Immunotherapeutics and Their Mechanisms of Action for Improved Cancer Treatment



Abstract
:1. Introduction
2. Applications of Humanized Mice in Oncology
2.1. Humanized Mouse Models for Testing ICIs and Antibody-Based Drugs

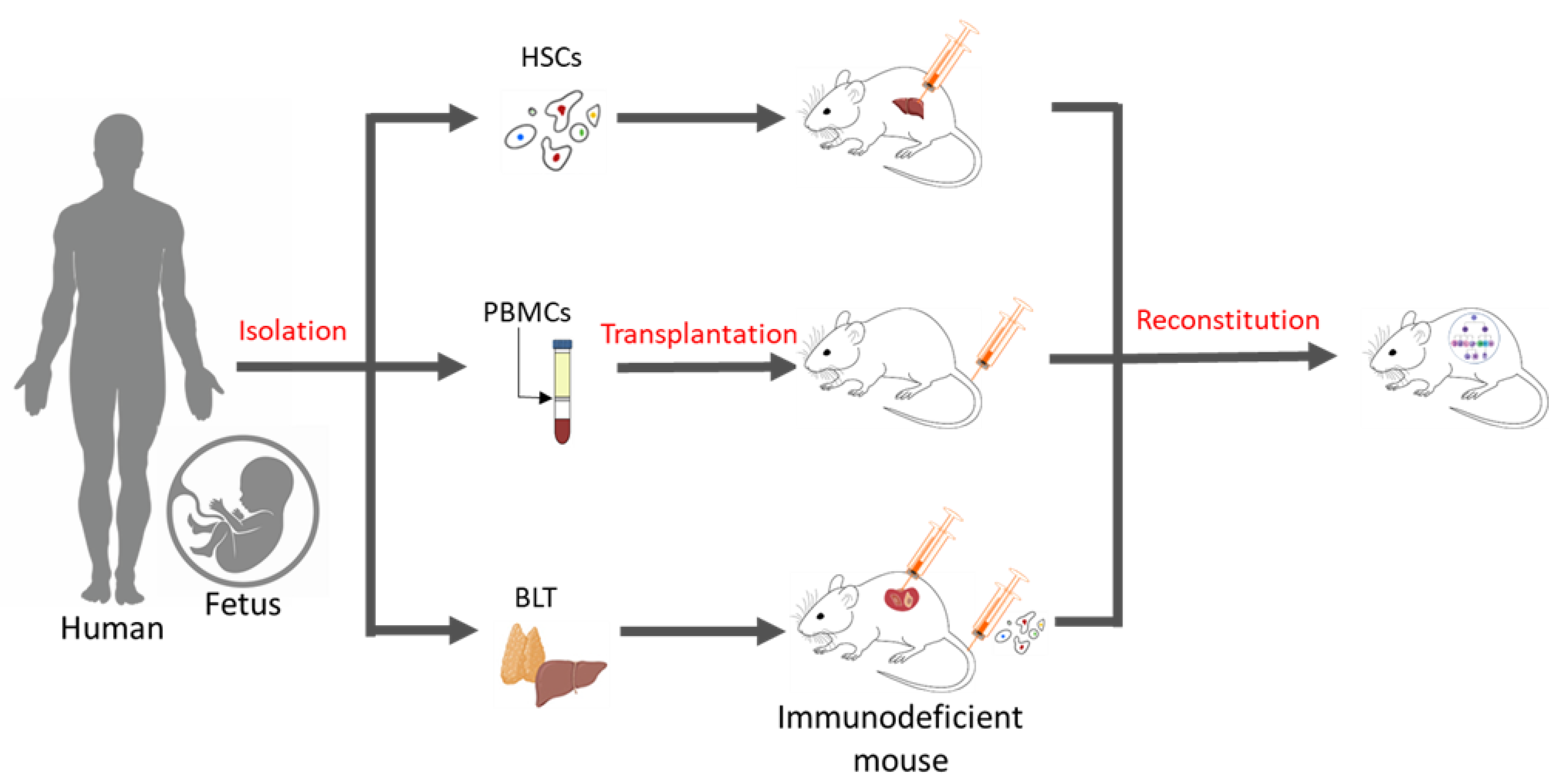{kind=link}
{kind=link}
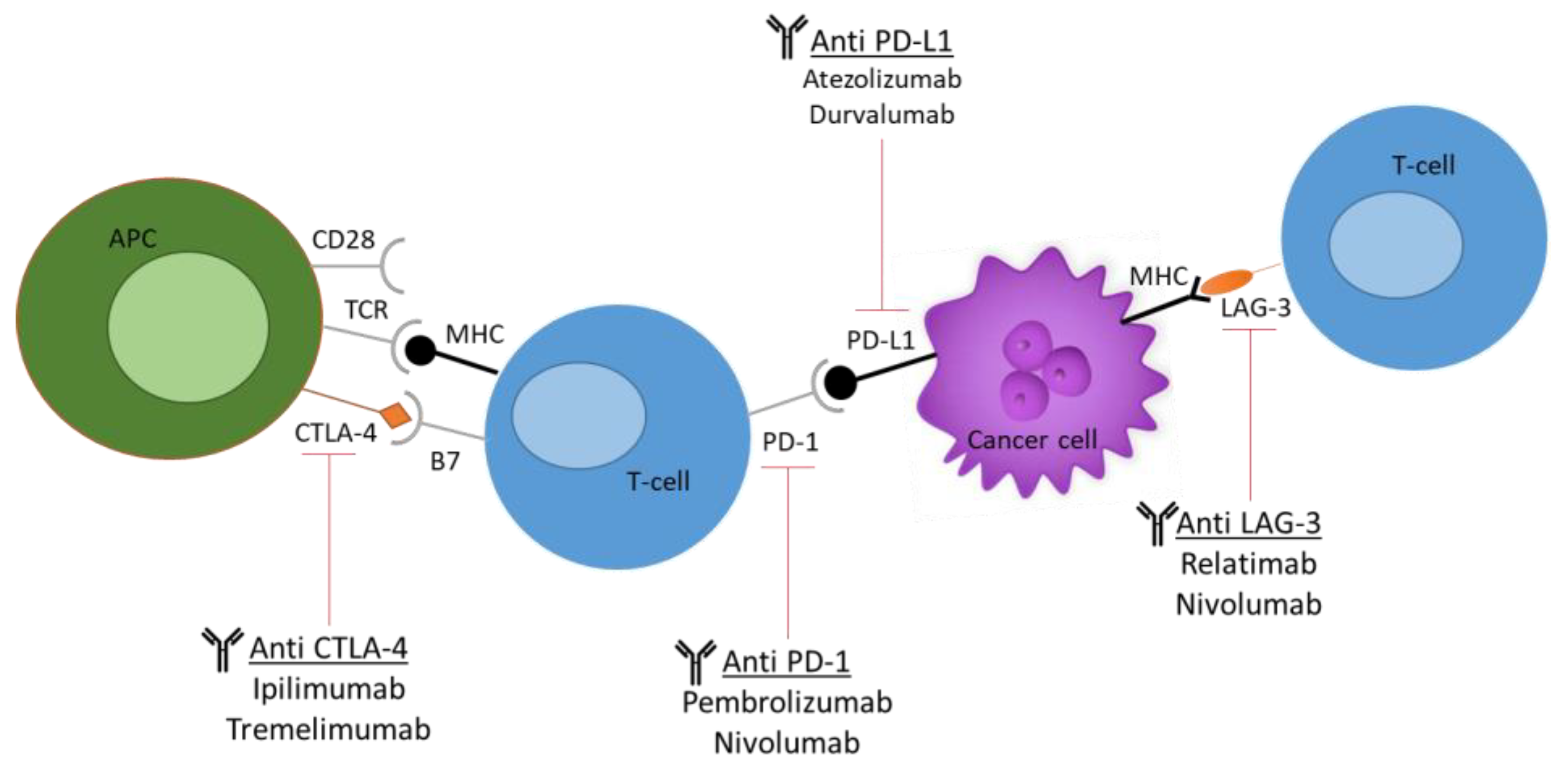{kind=link}
| Mouse model | Target | Safety | Efficacy | Model Description | Refs. |
|---|---|---|---|---|---|
| Colon | CD137 and PD-1 | √ | √ | CDX-model with intraperitoneal injection of PBMCs in Rag2−/−IL2Rγnull strain | [41] |
| Gastric | CD137 and PD-1 | √ | √ | PDX-model with intraperitoneal injection of PBMCs in Rag2−/−IL2Rγnull strain | [41] |
| HCC 1 | PD-1 and CTLA-4 | √ | √ | PDX-model with intrahepatic injection of human CD34+ HSCs in NSG mice | [40,49] |
| NPC 2 | PD-1 and CTLA-4 | √ | √ | PDX-model with intrahepatic injection of human CD34+ HSCs in NSG mice | [39] |
| Lymphoma | PD-1 and CTLA-4 | √ | √ | CDX-model with subcutaneous engraftment of human PBMCs in NOG mice | [53,54] |
| Sarcoma | PD-1 | √ | √ | PDX-model with intravenous injection of human CD34+ CB cells in NSG mice | [52] |
| NSCLC 3 | PD-1 | √ | √ | CDX and PDX-models with intravenous injection of human CD34+ HPSCs in NSG mice | [51,55] |
| PD-L1 | √ | √ | CDX and PDX-models with intravenous injection of either PBMCs or human CD34+ HSCs in NSG mice | [56] | |
| Bladder | PD-1 | √ | √ | PDX-model with intravenous injection of human CD34+ HPSCs in NSG mice | [51] |
| TNBC 4 | PD-1 | √ | √ | CDX and PDX-models with intravenous injection of human CD34+ HPSCs in NSG mice | [51] |
| Lung adenocarcinoma | PD-1 | √ | √ | CDX-model with intravenous injection of CD34+ human HSCs in NOG and NOG-FcγR−/− mice | [57] |
| PD-L1 | √ | √ | CDX-model with intravenous injection of cord-blood derived CD34+ human HSCs in NSG mice | [58] | |
| HNSCC 5 | PD-1 | √ | √ | CDX-model with intravenous injection of CD34+ human HSCs in NOG and NOG-FcγR−/− mice | [57] |
| Ovarian carcinoma | PD-L1 | √ | √ | CDX-model with intravenous injection of fetal liver-derived CD34+ human HSCs in NOG mice | [58] |
2.2. Safety and Efficacy Profiling of ACT in Humanized Mouse Models
2.2.1. CAR-T Cells
2.2.2. CAR-NK Cells
2.3. Improving Cytokine-Based Immunotherapy Using Humanized Mouse Models
2.4. Immune Response of Cancer Vaccines in Humanized Mouse Models
2.5. Targeted Tumour Lysis by Oncolytic Viruses (OVs) Using Humanized Mice
2.6. Combination Therapy with ICIs
3. Emerging Applications of Humanized Mouse Models
3.1. Drug Discovery Models
3.2. Drug Repurposing Using Humanized Mice
3.3. Uncovering New Disease and Resistance Mechanisms Using Humanized Mouse Models
3.4. Identifying Biomarkers of ICIs to Improve Predictability of Drug Responses
4. Next-Generation Humanized Mice Models
4.1. Hydrodynamic Injection Method for Improved Cytokine Model
4.2. Transgenic Mice Using Pronuclear Injection
4.3. Knock-in Models of Improved Humanized Mouse Models
5. Limitations
6. Future Directions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 121, 3335–3337. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 361, 711–723, Erratum in N. Engl. J. Med. 2010, 361, 1290. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, J.; Liu, W.N.; Zhao, Y. Cancer Immunotherapies and Humanized Mouse Drug Testing Platforms. Transl. Oncol. 2019, 11, 987–995. [Google Scholar] [CrossRef]
- Mobasheri, A. Comparative Medicine in the Twenty-First Century: Where are We Now and Where Do We Go from Here? Front. Vet. Sci. 2015, 1, 2. [Google Scholar] [CrossRef]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. 2017, 11, 187–215. [Google Scholar] [CrossRef]
- Ogilvie, L.A.; Kovachev, A.; Wierling, C.; Lange, B.M.H.; Lehrach, H. Models of Models: A Translational Route for Cancer Treatment and Drug Development. Front. Oncol. 2017, 1, 219. [Google Scholar] [CrossRef]
- Vandamme, T.F. Use of rodents as models of human diseases. J. Pharm. Bioallied Sci. 2014, 1, 2–9. [Google Scholar] [CrossRef]
- Lampreht Tratar, U.; Horvat, S.; Cemazar, M. Transgenic Mouse Models in Cancer Research. Front. Oncol. 2018, 1, 268. [Google Scholar] [CrossRef]
- Sakurai, T.; Kamiyoshi, A.; Kawate, H.; Watanabe, S.; Sato, M.; Shindo, T. Production of genetically engineered mice with higher efficiency, lower mosaicism, and multiplexing capability using maternally expressed Cas9. Sci. Rep. 2020, 11, 1091. [Google Scholar] [CrossRef]
- Kersten, K.; de Visser, K.E.; van Miltenburg, M.H.; Jonkers, J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol. Med. 2017, 1, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Zschaler, J.; Schlorke, D.; Arnhold, J. Differences in innate immune response between man and mouse. Crit. Rev. Immunol. 2014, 31, 433–454. [Google Scholar] [CrossRef]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 351, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Bannerji, R.; Allan, J.N.; Arnason, J.E.; Brown, J.R.; Advani, R.; Ansell, S.M.; O’Brien, S.M.; Duell, J.; Martin, P.; Joyce, R.M.; et al. Odronextamab (REGN1979), a Human CD20 x CD3 Bispecific Antibody, Induces Durable, Complete Responses in Patients with Highly Refractory B-Cell Non-Hodgkin Lymphoma, Including Patients Refractory to CAR T Therapy. Blood 2020, 136 (Suppl. S1), 42–43. [Google Scholar] [CrossRef]
- Regeneron’s Star Bispecific Is Linked to 2 Deaths in a Small Study—Which Was No Help for Its Q1 Call. John Carroll. Available online: https://endpts.com/regeneons-star-bispecific-is-linked-to-2-deaths-in-a-small-study-which-was-no-help-for-its-q1-call/ (accessed on 15 February 2023).
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 371, 449–459. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 11, 197–218. [Google Scholar] [CrossRef]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 1, 11. [Google Scholar] [CrossRef]
- Flora, A. Evidence-Based Selection of the Starting Dose in First-in-Human Clinical Trials Using Humanized Mouse Models. The Jackson Laboratory. Available online: https://www.jax.org/news-and-insights/jax-blog/2022/april/evidence-based-selection-starting-dose-in-human-clinical-trials (accessed on 15 February 2023).
- Mosier, D.E.; Gulizia, R.J.; Baird, S.M.; Wilson, D.B. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature 1988, 331, 256–259. [Google Scholar] [CrossRef]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005, 171, 6477–6489. [Google Scholar] [CrossRef]
- Ishikawa, F.; Yasukawa, M.; Lyons, B.; Yoshida, S.; Miyamoto, T.; Yoshimoto, G.; Watanabe, T.; Akashi, K.; Shultz, L.D.; Harada, M. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood 2005, 101, 1565–1573. [Google Scholar] [CrossRef]
- Bosma, G.C.; Custer, R.P.; Bosma, M.J. A severe combined immunodeficiency mutation in the mouse. Nature 1983, 301, 527–530. [Google Scholar] [CrossRef]
- Makino, S.; Kunimoto, K.; Muraoka, Y.; Mizushima, Y.; Katagiri, K.; Tochino, Y. Breeding of a non-obese, diabetic strain of mice. Exp. Anim. 1980, 21, 1–13. [Google Scholar] [CrossRef]
- Mian, S.A.; Anjos-Afonso, F.; Bonnet, D. Advances in Human Immune System Mouse Models for Studying Human Hematopoiesis and Cancer Immunotherapy. Front. Immunol. 2021, 11, 619236. [Google Scholar] [CrossRef] [PubMed]
- Mullen, Y. Development of the Nonobese Diabetic Mouse and Contribution of Animal Models for Understanding Type 1 Diabetes. Pancreas 2017, 41, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, K.; Prasolava, T.K.; Wang, J.C.; Mortin-Toth, S.M.; Khalouei, S.; Gan, O.I.; Dick, J.E.; Danska, J.S. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat. Immunol. 2007, 1, 1313–1323. [Google Scholar] [CrossRef]
- Shultz, L.D.; Schweitzer, P.A.; Christianson, S.W.; Gott, B.; Schweitzer, I.B.; Tennent, B.; McKenna, S.; Mobraaten, L.; Rajan, T.V.; Greiner, D.L.; et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J. Immunol. 1995, 151, 180–191. [Google Scholar] [CrossRef]
- Serreze, D.V.; Gaedeke, J.W.; Leiter, E.H. Hematopoietic stem-cell defects underlying abnormal macrophage development and maturation in NOD/Lt mice: Defective regulation of cytokine receptors and protein kinase C. Proc. Natl. Acad. Sci. USA 1993, 91, 9625–9629. [Google Scholar] [CrossRef]
- Ménoret, S.; Fontanière, S.; Jantz, D.; Tesson, L.; Thinard, R.; Rémy, S.; Usal, C.; Ouisse, L.H.; Fraichard, A.; Anegon, I. Generation of Rag1-knockout immunodeficient rats and mice using engineered meganucleases. FASEB J. 2013, 21, 703–711. [Google Scholar] [CrossRef]
- Lee, J.Y.; Han, A.R.; Lee, D.R. T Lymphocyte Development and Activation in Humanized Mouse Model. Dev. Reprod. 2019, 21, 79–92. [Google Scholar] [CrossRef]
- Shultz, L.D.; Banuelos, S.; Lyons, B.; Samuels, R.; Burzenski, L.; Gott, B.; Lang, P.; Leif, J.; Appel, M.; Rossini, A.; et al. NOD/LtSz-Rag1nullPfpnull mice: A new model system with increased levels of human peripheral leukocyte and hematopoietic stem-cell engraftment. Transplantation 2003, 71, 1036–1042. [Google Scholar] [CrossRef]
- Azuma, H.; Paulk, N.; Ranade, A.; Dorrell, C.; Al-Dhalimy, M.; Ellis, E.; Strom, S.; Kay, M.A.; Finegold, M.; Grompe, M. Robust expansion of human hepatocytes in Fah−/−/Rag2−/−/Il2rg−/− mice. Nat. Biotechnol. 2007, 21, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Lai, F.; Wang, Y.; Sutter, K.; Dittmer, U.; Ye, J.; Zai, W.; Liu, M.; Shen, F.; et al. Functional Comparison of Interferon-α Subtypes Reveals Potent Hepatitis B Virus Suppression by a Concerted Action of Interferon-α and Interferon-γ Signaling. Hepatology 2021, 71, 486–502. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Fajerman, Y.; Kollet, O. Immune-deficient SCID and NOD/SCID mice models as functional assays for studying normal and malignant human hematopoiesis. J. Mol. Med. 1997, 71, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Bosma, G.C.; Fried, M.; Custer, R.P.; Carroll, A.; Gibson, D.M.; Bosma, M.J. Evidence of functional lymphocytes in some (leaky) scid mice. J. Exp. Med. 1988, 161, 1016–1033. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Wang, X.J.; Chen, D.X.; Liu, X.N.; Wang, X.J. Humanized mouse model: A review on preclinical applications for cancer immunotherapy. Am. J. Cancer Res. 2020, 11, 4568–4584. [Google Scholar]
- Cogels, M.M.; Rouas, R.; Ghanem, G.E.; Martinive, P.; Awada, A.; Van Gestel, D.; Krayem, M. Humanized Mice as a Valuable Pre-Clinical Model for Cancer Immunotherapy Research. Front. Oncol. 2021, 11, 784947. [Google Scholar] [CrossRef]
- Liu, W.N.; Fong, S.Y.; Tan, W.W.S.; Tan, S.Y.; Liu, M.; Cheng, J.Y.; Lim, S.; Suteja, L.; Huang, E.K.; Chan, J.K.Y.; et al. Establishment and Characterization of Humanized Mouse NPC-PDX Model for Testing Immunotherapy. Cancers 2020, 11, 1025. [Google Scholar] [CrossRef]
- Zhao, Y.; Shuen, T.W.H.; Toh, T.B.; Chan, X.Y.; Liu, M.; Tan, S.Y.; Fan, Y.; Yang, H.; Lyer, S.G.; Bonney, G.K.; et al. Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut 2018, 61, 1845–1854. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Rodriguez, I.; Schalper, K.A.; Oñate, C.; Azpilikueta, A.; Rodriguez-Ruiz, M.E.; Morales-Kastresana, A.; Labiano, S.; Pérez-Gracia, J.L.; Martín-Algarra, S.; et al. Nivolumab and Urelumab Enhance Antitumor Activity of Human T Lymphocytes Engrafted in Rag2−/−IL2Rγnull Immunodeficient Mice. Cancer Res. 2015, 71, 3466–3478. [Google Scholar] [CrossRef]
- Roth, M.D.; Harui, A. Human tumor infiltrating lymphocytes cooperatively regulate prostate tumor growth in a humanized mouse model. J. Immunother. Cancer 2015, 1, 12. [Google Scholar] [CrossRef]
- Pan, C.X.; Shi, W.; Ma, A.H.; Zhang, H.; Lara, P.; Keck, J.G.; Palucka, K.; Airhart, S.D.; White, R.D. Humanized mice (humice) carrying patient-derived xenograft (PDX) as a platform to develop immunotherapy in bladder cancer (BCa). Clin. Oncol. 2017, 31 (Suppl. S6), 381. [Google Scholar] [CrossRef]
- Morton, J.J.; Bird, G.; Keysar, S.B.; Astling, D.P.; Lyons, T.R.; Anderson, R.T.; Glogowska, M.J.; Estes, P.; Eagles, J.R.; Le, P.N.; et al. XactMice: Humanizing mouse bone marrow enables microenvironment reconstitution in a patient-derived xenograft model of head and neck cancer. Oncogene 2016, 31, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Ishida, T.; Yano, H.; Inagaki, A.; Suzuki, S.; Sato, F.; Takino, H.; Mori, F.; Ri, M.; Kusumoto, S.; et al. Defucosylated anti-CCR4 monoclonal antibody exercises potent ADCC-mediated antitumor effect in the novel tumor-bearing humanized NOD/Shi-scid, IL-2Rgamma(null) mouse model. Cancer Immunol. Immunother. 2009, 51, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Tsoneva, D.; Minev, B.; Frentzen, A.; Zhang, Q.; Wege, A.K.; Szalay, A.A. Humanized Mice with Subcutaneous Human Solid Tumors for Immune Response Analysis of Vaccinia Virus-Mediated Oncolysis. Mol. Ther. Oncolytics 2017, 1, 41–61. [Google Scholar] [CrossRef]
- Schupp, J.; Christians, A.; Zimmer, N.; Gleue, L.; Jonuleit, H.; Helm, M.; Tuettenberg, A. In-Depth Immune-Oncology Studies of the Tumor Microenvironment in a Humanized Melanoma Mouse Model. Int. J. Mol. Sci. 2021, 21, 1011. [Google Scholar] [CrossRef]
- Franzin, R.; Netti, G.S.; Spadaccino, F.; Porta, C.; Gesualdo, L.; Stallone, G.; Castellano, G.; Ranieri, E. The Use of Immune Checkpoint Inhibitors in Oncology and the Occurrence of AKI: Where Do We Stand? Front. Immunol. 2020, 11, 574271. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, J.; Liu, W.N.; Fong, S.Y.; Shuen, T.W.H.; Liu, M.; Harden, S.; Tan, S.Y.; Cheng, J.Y.; Tan, W.W.S.; et al. Analysis and Validation of Human Targets and Treatments Using a Hepatocellular Carcinoma-Immune Humanized Mouse Model. Hepatology 2021, 71, 1395–1410. [Google Scholar] [CrossRef]
- He, Q.F.; Xu, Y.; Li, J.; Huang, Z.M.; Li, X.H.; Wang, X. CD8+ T-cell exhaustion in cancer: Mechanisms and new area for cancer immunotherapy. Brief Funct. Genom. 2019, 11, 99–106. [Google Scholar] [CrossRef]
- Wang, M.; Yao, L.C.; Cheng, M.; Cai, D.; Martinek, J.; Pan, C.X.; Shi, W.; Ma, A.H.; De Vere White, R.W.; Airhart, S.; et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB J. 2018, 31, 1537–1549. [Google Scholar] [CrossRef]
- Choi, B.; Lee, J.S.; Kim, S.J.; Hong, D.; Park, J.B.; Lee, K.Y. Anti-tumor effects of anti-PD-1 antibody, pembrolizumab, in humanized NSG PDX mice xenografted with dedifferentiated liposarcoma. Cancer Lett. 2020, 471, 56–69. [Google Scholar] [CrossRef]
- Donnou, S.; Galand, C.; Touitou, V.; Sautès-Fridman, C.; Fabry, Z.; Fisson, S. Murine models of B-cell lymphomas: Promising tools for designing cancer therapies. Adv. Hematol. 2012, 2011, 701704. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Xu, X.; Jones, R.; Delecluse, H.J.; Zumwalde, N.A.; Sharma, A.; Gumperz, J.E.; Kenney, S.C. PD-1/CTLA-4 Blockade Inhibits Epstein-Barr Virus-Induced Lymphoma Growth in a Cord Blood Humanized-Mouse Model. PLoS Pathog. 2016, 12, e1005642. [Google Scholar] [CrossRef] [PubMed]
- Qiao, T.; Xiong, Y.; Feng, Y.; Guo, W.; Zhou, Y.; Zhao, J.; Jiang, T.; Shi, C.; Han, Y. Inhibition of LDH-A by Oxamate Enhances the Efficacy of Anti-PD-1 Treatment in an NSCLC Humanized Mouse Model. Front. Oncol. 2021, 11, 632364. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Huang, G.; Cheng, L.; Li, Z.; Xiao, Y.; Deng, Q.; Jiang, Y.; Li, B.; Lin, S.; Wang, S.; et al. Establishment of peripheral blood mononuclear cell-derived humanized lung cancer mouse models for studying efficacy of PD-L1/PD-1 targeted immunotherapy. MAbs 2018, 11, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Katano, I.; Hanazawa, A.; Otsuka, I.; Yamaguchi, T.; Mochizuki, M.; Kawai, K.; Ito, R.; Goto, M.; Kagawa, T.; Takahashi, T. Development of a novel humanized mouse model for improved evaluation of in vivo anti-cancer effects of anti-PD-1 antibody. Sci. Rep. 2021, 11, 21087. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Carpenito, C.; Wang, G.; Surguladze, D.; Forest, A.; Malabunga, M.; Murphy, M.; Zhang, Y.; Sonyi, A.; Chin, D.; et al. Discovery and preclinical characterization of the antagonist anti-PD-L1 monoclonal antibody LY3300054. J. Immunother. Cancer 2018, 1, 31, Erratum in J. Immunother. Cancer 2018, 1, 45. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Dudley, M.E. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr. Opin. Immunol. 2009, 21, 233–240. [Google Scholar] [CrossRef]
- Vanegas, Y.M.; Mohty, R.; Gadd, M.E.; Luo, Y.; Aljurf, M.; Qin, H.; Kharfan-Dabaja, M.A. CAR-T cell Therapies for B-cell Lymphoid Malignancies: Identifying Targets Beyond CD19. Hematol. Oncol. Stem Cell Ther. 2022, 11, 81–93. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 91, 720–724. [Google Scholar] [CrossRef]
- Marofi, F.; Achmad, H.; Bokov, D.; Abdelbasset, W.K.; Alsadoon, Z.; Chupradit, S.; Suksatan, W.; Shariatzadeh, S.; Hasanpoor, Z.; Yazdanifar, M.; et al. Hurdles to breakthrough in CAR T cell therapy of solid tumors. Stem Cell Res. Ther. 2022, 11, 140. [Google Scholar] [CrossRef]
- Havard, R.; Stephens, D.M. Anti-CD19 chimeric antigen receptor T cell therapies: Harnessing the power of the immune system to fight diffuse large b cell lymphoma. Curr. Hematol. Malig. Rep. 2018, 11, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 121, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 21, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Diorio, C.; Murray, R.; Naniong, M.; Barrera, L.; Camblin, A.; Chukinas, J.; Coholan, L.; Edwards, A.; Fuller, T.; Gonzales, C.; et al. Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood 2022, 141, 619–629. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfo, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 1, 304. [Google Scholar] [CrossRef]
- Jin, C.H.; Xia, J.; Rafiq, S.; Huang, X.; Hu, Z.; Zhou, X.; Brentjens, R.J.; Yang, Y.G. Modeling anti-CD19 CAR T cell therapy in humanized mice with human immunity and autologous leukemia. EBioMedicine 2019, 31, 173–181. [Google Scholar] [CrossRef]
- Li, H.; Song, W.; Li, Z.; Zhang, M. Preclinical and clinical studies of CAR-NK-cell therapies for malignancies. Front. Immunol. 2022, 11, 992232. [Google Scholar] [CrossRef]
- Liu, W.N.; So, W.Y.; Harden, S.L.; Fong, S.Y.; Wong, M.X.Y.; Tan, W.W.S.; Tan, S.Y.; Ong, J.K.L.; Rajarethinam, R.; Liu, M.; et al. Successful targeting of PD-1/PD-L1 with chimeric antigen receptor-natural killer cells and nivolumab in a humanized mouse cancer model. Sci. Adv. 2022, 8, eadd1187. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 31, 520–531. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; Green, A.R.; Rizvanov, A.A.; Solovyeva, V.V. Molecular Aspects and Future Perspectives of Cytokine-Based Anti-cancer Immunotherapy. Front. Cell Dev. Biol. 2020, 1, 402. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in cancer immunotherapy. Oncoimmunology 2016, 5, e1163462. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Frana, L.W.; Sharrow, S.O.; Robb, R.J.; Rosenberg, S.A. In vivo administration of purified human interleukin 2. I. Half-life and immunologic effects of the Jurkat cell line-derived interleukin 2. J. Immunol. 1985, 131, 157–166. [Google Scholar] [CrossRef]
- Lotze, M.T.; Matory, Y.L.; Ettinghausen, S.E.; Rayner, A.A.; Sharrow, S.O.; Seipp, C.A.; Custer, M.C.; Rosenberg, S.A. In vivo administration of purified human interleukin 2. II. Half life, immunologic effects, and expansion of peripheral lymphoid cells in vivo with recombinant IL 2. J. Immunol. 1985, 131, 2865–2875. [Google Scholar] [CrossRef]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 191, 5451–5458. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Chang, A.E.; Avis, F.P.; Leitman, S.; Linehan, W.M.; Robertson, C.N.; Lee, R.E.; Rubin, J.T.; et al. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N. Engl. J. Med. 1987, 311, 889–897. [Google Scholar] [CrossRef]
- Bankert, R.B.; Balu-Iyer, S.V.; Odunsi, K.; Shultz, L.D.; Kelleher RJJr Barnas, J.L.; Simpson-Abelson, M.; Parsons, R.; Yokota, S.J. Humanized mouse model of ovarian cancer recapitulates patient solid tumor progression, ascites formation, and metastasis. PLoS ONE 2011, 6, e24420. [Google Scholar] [CrossRef]
- Stanley, M. Tumour virus vaccines: Hepatitis B virus and human papillomavirus. Philos. Trans. R Soc. Lond. B Biol. Sci. 2017, 371, 20160268. [Google Scholar] [CrossRef]
- Roudko, V.; Greenbaum, B.; Bhardwaj, N. Computational Prediction and Validation of Tumor-Associated Neoantigens. Front. Immunol. 2020, 11, 27. [Google Scholar] [CrossRef]
- Spranger, S.; Frankenberger, B.; Schendel, D.J. NOD/scid IL-2Rg(null) mice: A preclinical model system to evaluate human dendritic cell-based vaccine strategies in vivo. J. Transl. Med. 2012, 11, 30. [Google Scholar] [CrossRef]
- Jhajharia, S.; Lai, F.; Low, H.B.; Purushotorman, K.; Shunmuganathan, B.D.; Chan, C.E.Z.; Hammond, R.; Netter, H.J.; Chen, Q.; Lim, S.G.; et al. Defining the specificity and function of a human neutralizing antibody for Hepatitis B virus. NPJ Vaccines 2022, 1, 121. [Google Scholar] [CrossRef]
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 1, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.A.; Shabahang, S.; Timiryasova, T.M.; Zhang, Q.; Beltz, R.; Gentschev, I.; Goebel, W.; Szalay, A.A. Visualization of tumors and metastases in live animals with bacteria and vaccinia virus encoding light-emitting proteins. Nat. Biotechnol. 2004, 21, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, Y.A.; Wang, E.; Chen, N.; Danner, R.L.; Munson, P.J.; Marincola, F.M.; Szalay, A.A. Eradication of solid human breast tumors in nude mice with an intravenously injected light-emitting oncolytic vaccinia virus. Cancer Res. 2007, 61, 10038–10046. [Google Scholar] [CrossRef] [PubMed]
- Dey, M.; Yu, D.; Kanojia, D.; Li, G.; Sukhanova, M.; Spencer, D.A.; Pituch, K.C.; Zhang, L.; Han, Y.; Ahmed, A.U.; et al. Intranasal Oncolytic Virotherapy with CXCR4-Enhanced Stem Cells Extends Survival in Mouse Model of Glioma. Stem Cell Rep. 2016, 1, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Minev, B.; Kohrt, H.; Kilinc, M.; Chen, N.; Feng, A.; Pessian, M.; Geissinger, U.; Haefner, E.; Tsoneva, D.; Bozhilov, K.; et al. Combination immunotherapy with oncolytic vaccinia virus and checkpoint inhibitor following local tumor irradiation. J. Immunother. Cancer 2014, 2 (Suppl. S3), P112. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 1, 86. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 361, 122–133, Erratum in N. Engl. J. Med. 2018, 371, 2185. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. RELATIVITY-047 Investigators. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 381, 24–34. [Google Scholar] [CrossRef]
- Sinha, D.; Srihari, S.; Beckett, K.; Le Texier, L.; Solomon, M.; Panikkar, A.; Ambalathingal, G.R.; Lekieffre, L.; Crooks, P.; Rehan, S.; et al. ‘Off-the-shelf’ allogeneic antigen-specific adoptive T-cell therapy for the treatment of multiple EBV-associated malignancies. J. Immunother. Cancer 2021, 9, e001608. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, X.; Chen, S.; Lai, Y.; Wei, X.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; Liang, Q.; et al. Anti-GPC3-CAR T Cells Suppress the Growth of Tumor Cells in Patient-Derived Xenografts of Hepatocellular Carcinoma. Front. Immunol. 2017, 1, 690. [Google Scholar] [CrossRef]
- Huang, R.Y.; Francois, A.; McGray, A.R.; Miliotto, A.; Odunsi, K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology 2016, 6, e1249561. [Google Scholar] [CrossRef] [PubMed]
- Odunsi, A.; McGray, A.J.R.; Miliotto, A.; Zhang, Y.; Wang, J.; Abiola, A.; Eppolito, C.; Huang, R.Y. Fidelity of human ovarian cancer patient-derived xenografts in a partially humanized mouse model for preclinical testing of immunotherapies. J. Immunother. Cancer 2020, 8, e001237. [Google Scholar] [CrossRef] [PubMed]
- Capasso, A.; Lang, J.; Pitts, T.M.; Jordan, K.R.; Lieu, C.H.; Davis, S.L.; Diamond, J.R.; Kopetz, S.; Barbee, J.; Peterson, J.; et al. Characterization of immune responses to anti-PD-1 mono and combination immunotherapy in hematopoietic humanized mice implanted with tumor xenografts. J. Immunother. Cancer 2019, 1, 37. [Google Scholar] [CrossRef] [PubMed]
- Ha, W.; Sevim-Nalkiran, H.; Zaman, A.M.; Matsuda, K.; Khasraw, M.; Nowak, A.K.; Chung, L.; Baxter, R.C.; McDonald, K.L. Ibudilast sensitizes glioblastoma to temozolomide by targeting Macrophage Migration Inhibitory Factor (MIF). Sci. Rep. 2019, 1, 2905. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 11, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Dang, S. Anti-Cancer Potential of Some Commonly Used Drugs. Curr. Pharm. Des. 2021, 21, 4530–4538. [Google Scholar] [CrossRef]
- Moullan, N.; Mouchiroud, L.; Wang, X.; Ryu, D.; Williams, E.G.; Mottis, A.; Jovaisaite, V.; Frochaux, M.V.; Quiros, P.M.; Deplancke, B.; et al. Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: A Call for Caution in Biomedical Research. Cell Rep. 2015, 11, 1681–1691. [Google Scholar] [CrossRef]
- Vendramin, R.; Katopodi, V.; Cinque, S.; Konnova, A.; Knezevic, Z.; Adnane, S.; Verheyden, Y.; Karras, P.; Demesmaeker, E.; Bosisio, F.M.; et al. Activation of the integrated stress response confers vulnerability to mitoribosome-targeting antibiotics in melanoma. J. Exp. Med. 2021, 218, e20210571. [Google Scholar] [CrossRef]
- Ravà, M.; D’Andrea, A.; Nicoli, P.; Gritti, I.; Donati, G.; Doni, M.; Giorgio, M.; Olivero, D.; Amati, B. Therapeutic synergy between tigecycline and venetoclax in a preclinical model of MYC/BCL2 double-hit B cell lymphoma. Sci. Transl. Med. 2018, 10, eaan8723. [Google Scholar] [CrossRef]
- Alamino, V.A.; Mascanfroni, I.D.; Montesinos, M.M.; Gigena, N.; Donadio, A.C.; Blidner, A.G.; Milotich, S.I.; Cheng, S.Y.; Masini-Repiso, A.M.; Rabinovich, G.A.; et al. Antitumor Responses Stimulated by Dendritic Cells Are Improved by Triiodothyronine Binding to the Thyroid Hormone Receptor β. Cancer Res. 2015, 71, 1265–1274. [Google Scholar] [CrossRef]
- Thuru, X.; Magnez, R.; El-Bouazzati, H.; Vergoten, G.; Quesnel, B.; Bailly, C. Drug Repurposing to Enhance Antitumor Response to PD-1/PD-L1 Immune Checkpoint Inhibitors. Cancers 2022, 11, 3368. [Google Scholar] [CrossRef] [PubMed]
- Pourzardosht, N.; Hashemi, Z.S.; Mard-Soltani, M.; Jahangiri, A.; Rahbar, M.R.; Zakeri, A.; Mirzajani, E.; Khalili, S. Liothyronine could block the programmed death-ligand 1 (PDL1) activity: An e-Pharmacophore modeling and virtual screening study. J. Recept. Signal Transduct. Res. 2022, 41, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.S.M.; Her, Z.; Tan, S.Y.; Tan, W.W.S.; Liu, M.; Lai, F.; Heng, S.M.; Fan, Y.; Chang, K.T.E.; Wang, C.I.; et al. Humanized Mouse as a Tool to Predict Immunotoxicity of Human Biologics. Front. Immunol. 2020, 11, 553362. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, M.; Chan, X.Y.; Tan, S.Y.; Subramaniam, S.; Fan, Y.; Loh, E.; Chang, K.T.E.; Tan, T.C.; Chen, Q. Uncovering the mystery of opposite circadian rhythms between mouse and human leukocytes in humanized mice. Blood 2017, 131, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Ladinsky, M.S.; Khamaikawin, W.; Jung, Y.; Lin, S.; Lam, J.; An, D.S.; Bjorkman, P.J.; Kieffer, C. Mechanisms of virus dissemination in bone marrow of HIV-1-infected humanized BLT mice. Elife 2019, 8, e46916. [Google Scholar] [CrossRef] [PubMed]
- Tan-Garcia, A.; Lai, F.; Sheng Yeong, J.P.; Irac, S.E.; Ng, P.Y.; Msallam, R.; Tatt Lim, J.C.; Wai, L.E.; Tham, C.Y.L.; Choo, S.P.; et al. Liver fibrosis and CD206+ macrophage accumulation are suppressed by anti-GM-CSF therapy. JHEP Rep. 2019, 1, 100062. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 1, 13. [Google Scholar] [CrossRef]
- Tan-Garcia, A.; Wai, L.E.; Zheng, D.; Ceccarello, E.; Jo, J.; Banu, N.; Khakpoor, A.; Chia, A.; Tham, C.Y.L.; Tan, A.T.; et al. Intrahepatic CD206+ macrophages contribute to inflammation in advanced viral-related liver disease. J. Hepatol. 2017, 61, 490–500. [Google Scholar] [CrossRef]
- Kaur, M.; Drake, A.C.; Hu, G.; Rudnick, S.; Chen, Q.; Phennicie, R.; Attar, R.; Nemeth, J.; Gaudet, F.; Chen, J. Induction and Therapeutic Targeting of Human NPM1c+ Myeloid Leukemia in the Presence of Autologous Immune System in Mice. J. Immunol. 2019, 201, 1885–1894. [Google Scholar] [CrossRef]
- Schmidt, B. Proof of Principle studies. Epilepsy Res. 2006, 61, 48–52. [Google Scholar] [CrossRef]
- Somasundaram, R.; Connelly, T.; Choi, R.; Choi, H.; Samarkina, A.; Li, L.; Gregorio, E.; Chen, Y.; Thakur, R.; Abdel-Mohsen, M.; et al. Tumor-infiltrating mast cells are associated with resistance to anti-PD-1 therapy. Nat. Commun. 2021, 11, 346. [Google Scholar] [CrossRef]
- Dobson, S.M.; García-Prat, L.; Vanner, R.J.; Wintersinger, J.; Waanders, E.; Gu, Z.; McLeod, J.; Gan, O.I.; Grandal, I.; Payne-Turner, D.; et al. Relapse-Fated Latent Diagnosis Subclones in Acute B Lineage Leukemia Are Drug Tolerant and Possess Distinct Metabolic Programs. Cancer Discov. 2020, 11, 568–587. [Google Scholar] [CrossRef] [PubMed]
- Sebestyén, A.; Kopper, L.; Dankó, T.; Tímár, J. Hypoxia Signaling in Cancer: From Basics to Clinical Practice. Pathol. Oncol. Res. 2021, 21, 1609802. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zhao, Y.; Lee, W.C.; Bae, S.; Diéguez-Martínez, N.; Khoury, A.; Lai, X.; Li, J.; Li, W.; Liu, J.; et al. Hypoxia induces HIF1α-dependent epigenetic vulnerability in triple negative breast cancer to confer immune effector dysfunction and resistance to anti-PD-1 immunotherapy. Nat. Commun. 2022, 11, 4118. [Google Scholar] [CrossRef] [PubMed]
- Kuick, R.; Misek, D.E.; Monsma, D.J.; Webb, C.P.; Wang, H.; Peterson, K.J.; Pisano, M.; Omenn, G.S.; Hanash, S.M. Discovery of cancer biomarkers through the use of mouse models. Cancer Lett. 2007, 241, 40–48. [Google Scholar] [CrossRef]
- Pillai, S.G.; Li, S.; Siddappa, C.M.; Ellis, M.J.; Watson, M.A.; Aft, R. Identifying biomarkers of breast cancer micrometastatic disease in bone marrow using a patient-derived xenograft mouse model. Breast Cancer Res. 2018, 21, 2. [Google Scholar] [CrossRef]
- Sundar, R.; Huang, K.K.; Kumar, V.; Ramnarayanan, K.; Demircioglu, D.; Her, Z.; Ong, X.; Bin Adam Isa, Z.F.; Xing, M.; Tan, A.L.; et al. Epigenetic promoter alterations in GI tumour immune-editing and resistance to immune checkpoint inhibition. Gut 2022, 71, 1277–1288. [Google Scholar] [CrossRef]
- Huin, V.; Buée, L.; Behal, H.; Labreuche, J.; Sablonnière, B.; Dhaenens, C.M. Alternative promoter usage generates novel shorter MAPT mRNA transcripts in Alzheimer’s disease and progressive supranuclear palsy brains. Sci. Rep. 2017, 1, 12589. [Google Scholar] [CrossRef]
- Eisenman, J.; Ahdieh, M.; Beers, C.; Brasel, K.; Kennedy, M.K.; Le, T.; Bonnert, T.P.; Paxton, R.J.; Park, L.S. Interleukin-15 interactions with interleukin-15 receptor complexes: Characterization and species specificity. Cytokine 2002, 21, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Khoury, M.; Chen, J. Expression of human cytokines dramatically improves reconstitution of specific human-blood lineage cells in humanized mice. Proc. Natl. Acad. Sci. USA 2009, 101, 21783–21788. [Google Scholar] [CrossRef]
- Mencarelli, A.; Gunawan, M.; Yong, K.S.M.; Bist, P.; Tan, W.W.S.; Tan, S.Y.; Liu, M.; Huang, E.K.; Fan, Y.; Chan, J.K.Y.; et al. A humanized mouse model to study mast cells mediated cutaneous adverse drug reactions. J. Leukoc. Biol. 2020, 101, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Q.; Zheng, D.; Yin, L.; Chionh, Y.H.; Wong, L.H.; Tan, S.Q.; Tan, T.C.; Chan, J.K.; Alonso, S.; et al. Induction of functional human macrophages from bone marrow promonocytes by M-CSF in humanized mice. J. Immunol. 2013, 191, 3192–3199. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; He, F.; Kwang, J.; Chan, J.K.; Chen, J. GM-CSF and IL-4 stimulate antibody responses in humanized mice by promoting T, B, and dendritic cell maturation. J. Immunol. 2012, 181, 5223–5229. [Google Scholar] [CrossRef] [PubMed]
- Iwabuchi, R.; Ikeno, S.; Kobayashi-Ishihara, M.; Takeyama, H.; Ato, M.; Tsunetsugu-Yokota, Y.; Terahara, K. Introduction of Human Flt3-L and GM-CSF into Humanized Mice Enhances the Reconstitution and Maturation of Myeloid Dendritic Cells and the Development of Foxp3+CD4+ T Cells. Front. Immunol. 2018, 1, 1042. [Google Scholar] [CrossRef]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 21, 739–748. [Google Scholar] [CrossRef]
- Nicolini, F.E.; Cashman, J.D.; Hogge, D.E.; Humphries, R.K.; Eaves, C.J. NOD/SCID mice engineered to express human IL-3, GM-CSF and Steel factor constitutively mobilize engrafted human progenitors and compromise human stem cell regeneration. Leukemia 2004, 11, 341–347. [Google Scholar] [CrossRef]
- Martinov, T.; McKenna, K.M.; Tan, W.H.; Collins, E.J.; Kehret, A.R.; Linton, J.D.; Olsen, T.M.; Shobaki, N.; Rongvaux, A. Building the Next Generation of Humanized Hemato-Lymphoid System Mice. Front. Immunol. 2021, 11, 643852. [Google Scholar] [CrossRef]
- Aryee, K.E.; Burzenski, L.M.; Yao, L.C.; Keck, J.G.; Greiner, D.L.; Shultz, L.D.; Brehm, M.A. Enhanced development of functional human NK cells in NOD-scid-IL2rgnull mice expressing human IL15. FASEB J. 2022, 36, e22476. [Google Scholar] [CrossRef]
- Rongvaux, A.; Willinger, T.; Martinek, J.; Strowig, T.; Gearty, S.V.; Teichmann, L.L.; Saito, Y.; Marches, F.; Halene, S.; Palucka, A.K.; et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014, 31, 364–372, Erratum in Nat. Biotechnol. 2017, 35, 1211. [Google Scholar] [CrossRef]
- Suda, T.; Liu, D. Hydrodynamic gene delivery: Its principles and applications. Mol. Ther. 2007, 11, 2063–2069. [Google Scholar] [CrossRef]
- Amaladoss, A.; Chen, Q.; Liu, M.; Dummler, S.K.; Dao, M.; Suresh, S.; Chen, J.; Preiser, P.R. De Novo Generated Human Red Blood Cells in Humanized Mice Support Plasmodium falciparum Infection. PLoS ONE 2015, 10, e0129825. [Google Scholar] [CrossRef]
- Liu, C.; Xie, W.; Gui, C.; Du, Y. Pronuclear microinjection and oviduct transfer procedures for transgenic mouse production. Methods Mol. Biol. 2013, 1021, 217–232. [Google Scholar] [CrossRef]
- Das, R.; Strowig, T.; Verma, R.; Koduru, S.; Hafemann, A.; Hopf, S.; Kocoglu, M.H.; Borsotti, C.; Zhang, L.; Branagan, A.; et al. Microenvironment-dependent growth of preneoplastic and malignant plasma cells in humanized mice. Nat. Med. 2016, 21, 1351–1357. [Google Scholar] [CrossRef] [PubMed]
- Macchiarini, F.; Manz, M.G.; Palucka, A.K.; Shultz, L.D. Humanized mice: Are we there yet? J. Exp. Med. 2005, 201, 1307–1311. [Google Scholar] [CrossRef] [PubMed]
- Taneja, V.; David, C.S. HLA transgenic mice as humanized mouse models of disease and immunity. J. Clin. Investig. 1998, 101, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Strom, S.C.; Davila, J.; Grompe, M. Chimeric mice with humanized liver: Tools for the study of drug metabolism, excretion, and toxicity. Methods Mol. Biol. 2010, 641, 491–509. [Google Scholar] [CrossRef]
- Washburn, M.L.; Bility, M.T.; Zhang, L.; Kovalev, G.I.; Buntzman, A.; Frelinger, J.A.; Barry, W.; Ploss, A.; Rice, C.M.; Su, L. A humanized mouse model to study hepatitis C virus infection, immune response, and liver disease. Gastroenterology 2011, 141, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Gutti, T.L.; Knibbe, J.S.; Makarov, E.; Zhang, J.; Yannam, G.R.; Gorantla, S.; Sun, Y.; Mercer, D.F.; Suemizu, H.; Wisecarver, J.L.; et al. Human hepatocytes and hematolymphoid dual reconstitution in treosulfan-conditioned uPA-NOG mice. Am. J. Pathol. 2014, 181, 101–109. [Google Scholar] [CrossRef]
- Lai, F.; Wee, C.Y.Y.; Chen, Q. Establishment of Humanized Mice for the Study of HBV. Front. Immunol. 2021, 11, 638447. [Google Scholar] [CrossRef] [PubMed]
- Foquet, L.; Schafer, C.; Minkah, N.K.; Alanine, D.G.W.; Flannery, E.L.; Steel, R.W.J.; Sack, B.K.; Camargo, N.; Fishbaugher, M.; Betz, W.; et al. Plasmodium falciparum Liver Stage Infection and Transition to Stable Blood Stage Infection in Liver-Humanized and Blood-Humanized FRGN KO Mice Enables Testing of Blood Stage Inhibitory Antibodies (Reticulocyte-Binding Protein Homolog 5) In Vivo. Front. Immunol. 2018, 1, 524. [Google Scholar] [CrossRef]
- Mailly, L.; Xiao, F.; Lupberger, J.; Wilson, G.K.; Aubert, P.; Duong, F.H.T.; Calabrese, D.; Leboeuf, C.; Fofana, I.; Thumann, C.; et al. Clearance of persistent hepatitis C virus infection in humanized mice using a claudin-1-targeting monoclonal antibody. Nat. Biotechnol. 2015, 31, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Khoury, M.; Limmon, G.; Choolani, M.; Chan, J.K.; Chen, J. Human fetal hepatic progenitor cells are distinct from, but closely related to, hematopoietic stem/progenitor cells. Stem Cells 2013, 31, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.M.; Bial, J.; Tarlow, B.; Bial, G.; Jensen, B.; Greiner, D.L.; Brehm, M.A.; Grompe, M. Extensive double humanization of both liver and hematopoiesis in FRGN mice. Stem Cell Res. 2014, 13, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Keng, C.T.; Sze, C.W.; Zheng, D.; Zheng, Z.; Yong, K.S.; Tan, S.Q.; Ong, J.J.; Tan, S.Y.; Loh, E.; Upadya, M.H.; et al. Characterisation of liver pathogenesis, human immune responses and drug testing in a humanised mouse model of HCV infection. Gut 2016, 61, 1744–1753. [Google Scholar] [CrossRef]
- Seyhan, A.A. Lost in translation: The valley of death across preclinical and clinical divide—Identification of problems and overcoming obstacles. Transl. Med. Commun. 2019, 1, 18. [Google Scholar] [CrossRef]



| Models | Mutations | Advantages | Disadvantages | Refs. |
|---|---|---|---|---|
| SCID | Protein kinase DNA-activated catalytic peptide gene (Prkdc) | Lack humoral and cell-mediated immunity Absence of B and T cells | Leakiness due to age Reduced lifespan Presence of murine NK cells Complementation of activity | [20,23] |
| NOD/SCID | Signal regulatory protein alpha (Sirpa) | High affinity to human CD47Tolerance of host macrophages to the human cellsImmunological multi-dysfunction, including defective NK cell activitySupport higher levels of human engraftments | Residual NK cell activity, and some other innate compounds of the immune system, fail to develop mature monocytes, which eventually become thymic lymphomas with age | [24,25,26,27,28,29] |
| BRG Rag1/2 | Recombination-activating gene 1 (Rag1) or 2 (Rag2) | No leakiness Absence of functional B and T cellsLonger lifespans | Limited lymphoid reconstitution Residual NK cell activity | [30] |
| NOG NSG | Interleukin-2 receptor gamma chain (IL2rγ) null phenotype and Prkdc mutation (NOD/SCID-IL2Rγ−/−) | Absence of functional receptors for cytokines like IL-2 and IL-7 Could hamper the development of host NK cells Highest rate of human cell engraftments Longer lifespans | Weak human myeloid reconstitution Lack of human thymus tissue Radiation sensitivity | [21,22,28,31] |
| NRG | RAG1/2 null mutation and Interleukin-2 receptor gamma chain (IL2rγ) null (NOD/RAG1/2−/−- IL2Rγ−/−) | Improved myeloid engraftmentCan tolerate chemotherapy at higher doses | Higher radiation dose for preconditioning Lack of human thymus tissue Possible background activity | [32] |
| FRG | Fah/Rag2/IL-2rγ | Triple knockout Expand human hepatocytes robustly Gene and cell therapy | Remnant mouse hepatocytes Background activity | [33,34] |
| Immunotherapy | Humanized Mouse Model Application | Safety & Efficacy | Further Modifications | Limitations |
|---|---|---|---|---|
| Antibody-based | Help in discovering new targets for ICIs | Improved compared to wild type mice | Immune-transgenic models | Undefined resistance mechanisms |
| Adoptive Cell Therapy | Hints towards possible mechanisms underlying CRS | Higher safety and efficacy standards | Improved NK cell model for cytotoxic activity | CRS and neurotoxicity, limited effect on solid tumours |
| Cytokine | Gaining popularity to prevent tumour growth | Potential to improve | Fully humanized models with improved cytokine transgenic models | Lack of specificity |
| Cancer vaccines | Prophylactic and therapeutic vaccines are being established using this model | Dose escalation studies provide improved safety and efficacy | Dual humanized mouse models for specific viral cancers | Scarcity of suitable neoantigens |
| Oncolytic viruses | Discovering novel delivery systems and targeting tumour lysis | Improved safety and efficacy when translated to clinic | Dual humanized mouse models | Need for improved delivery strategies, tumour heterogeneity |
 |  |  | |||
|---|---|---|---|---|---|
| Method of Delivery | Hydrodynamic Injection | Pronuclear Injection | Knock-In | ||
| Cytokines/ Modifications | IL-1B, IL-2, IL-7, GM-CSF, Flt-3L | NSG-SGM3: SCF, GM-CSF and IL-3 | Hu-IL15: Human IL-15 | SRG-15: Human IL-15 and SIRPα | MITRG/MISTRG: Human M-CSF, human IL-3 and GM-CSF, and human TPO |
| Key Features | Improved NK cells Proliferation of myeloid cells In vivo transfection of hepatocytes | Development of abundant and functional mast cells | Enhanced human NK cell development | Functional human NK cells | Multilineage differentiation of human B, T, and myeloid cells supports development of NK cells |
| Advantages | Easy to implement Time-saving Improved antigen-specific antibody responses | High level human haematopoietic engraftment initially | Improved capacity for cytotoxic activity | Simple use, no cytokine administration | No radiation preconditioning Long-term haematopoiesis Closely resembles diversity and cell populations seen in humans |
| Disadvantages | Possible damage to target tissue due to high and rapid pressure | Loss of stemness and functional properties over time Long-term effects of irradiation | Maturation differences between NK cell subset from humans and HSC-engrafted NSG-Tg (Hu-IL15) mice | High concentration of IL-15 could result in impaired functionality | Thrombocytopenia, hemophagocytosis, short lifespan, anaemia |
| Application | Liver-associated, Infectious diseases, immune responses, adverse drug reactions | Systemic anaphylaxis | Study of PDX tumours | Studying human tissue-resident immune cells, combination therapy protocols | Studying haematological malignancies |
| References | [122,123,124,125,126] | [127,128,129] | [129,130] | [129] | [129,131] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karnik, I.; Her, Z.; Neo, S.H.; Liu, W.N.; Chen, Q. Emerging Preclinical Applications of Humanized Mouse Models in the Discovery and Validation of Novel Immunotherapeutics and Their Mechanisms of Action for Improved Cancer Treatment. Pharmaceutics 2023, 15, 1600. https://doi.org/10.3390/pharmaceutics15061600
Karnik I, Her Z, Neo SH, Liu WN, Chen Q. Emerging Preclinical Applications of Humanized Mouse Models in the Discovery and Validation of Novel Immunotherapeutics and Their Mechanisms of Action for Improved Cancer Treatment. Pharmaceutics. 2023; 15(6):1600. https://doi.org/10.3390/pharmaceutics15061600
Chicago/Turabian StyleKarnik, Isha, Zhisheng Her, Shu Hui Neo, Wai Nam Liu, and Qingfeng Chen. 2023. "Emerging Preclinical Applications of Humanized Mouse Models in the Discovery and Validation of Novel Immunotherapeutics and Their Mechanisms of Action for Improved Cancer Treatment" Pharmaceutics 15, no. 6: 1600. https://doi.org/10.3390/pharmaceutics15061600
APA StyleKarnik, I., Her, Z., Neo, S. H., Liu, W. N., & Chen, Q. (2023). Emerging Preclinical Applications of Humanized Mouse Models in the Discovery and Validation of Novel Immunotherapeutics and Their Mechanisms of Action for Improved Cancer Treatment. Pharmaceutics, 15(6), 1600. https://doi.org/10.3390/pharmaceutics15061600