
Rapid In Vitro Assessment of Antimicrobial Drug Effect Bridging Clinically Relevant Pharmacokinetics: A Comprehensive Methodology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
- Automated collection of longitudinal optical density measurements of several bacterial cell suspensions by an optical density instrument, each suspension exposed to single or multiple drugs at a time-invariant concentration;
- Feeding the data collected in the previous step into a mathematical model to estimate the kill rate of the bacterial subpopulation least susceptible to the drug as a function of drug concentration;
- Use of the drug-concentration-dependent kill rate estimate from the previous step to design dosing regimens predicted to eradicate a bacterial population exposed to a drug following clinically relevant pharmacokinetics;
- Validation test of promising dosing regimens from the previous step in an in vitro hollow fiber infection model mimicking clinically relevant pharmacokinetics in humans;
- We elaborate on each of the above elements next.
2.1. Longitudinal Optical Density Measurements of Bacterial Cell Suspension under Drug Exposure
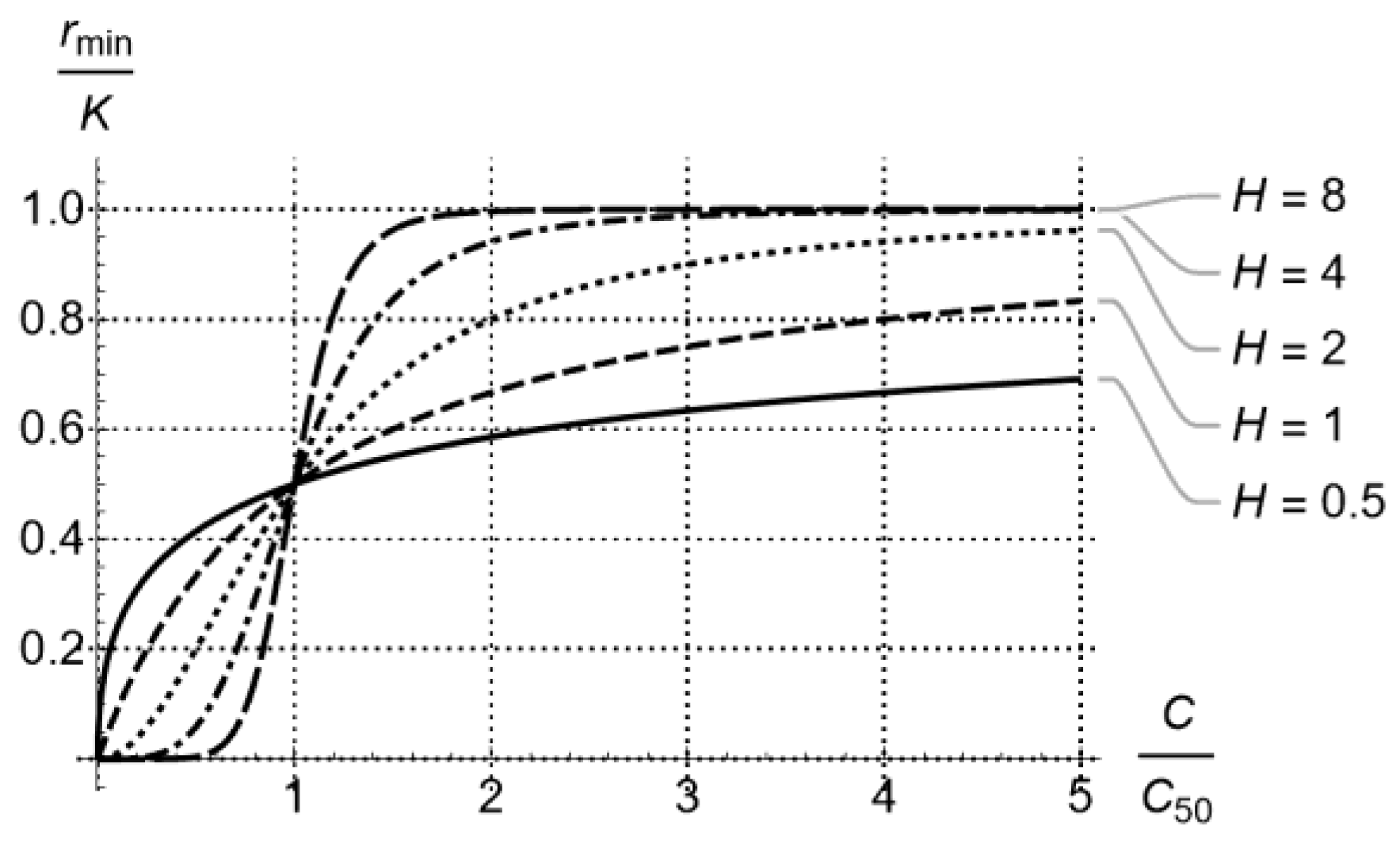
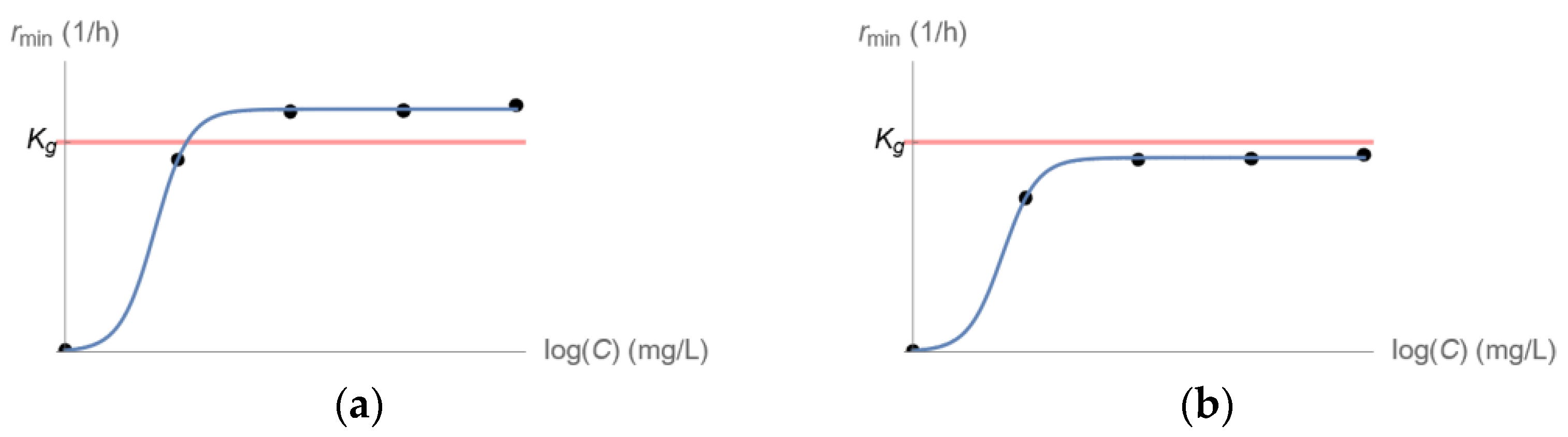
2.2. Kill Rate Estimation of Least Susceptible Bacteria as a Function of Drug Concentration
- is the live bacterial population size with an initial value of ;
- is the physiological net growth rate of the entire bacterial population, common for all subpopulations;
- is the kill rate induced by the antibiotic on the most resistant (least susceptible) subpopulation;
- is the maximum size of a bacterial population reaching saturation under growth conditions;
- is the kill rate average over the bacterial population at time ;
- is the kill rate variance over the bacterial population at time .
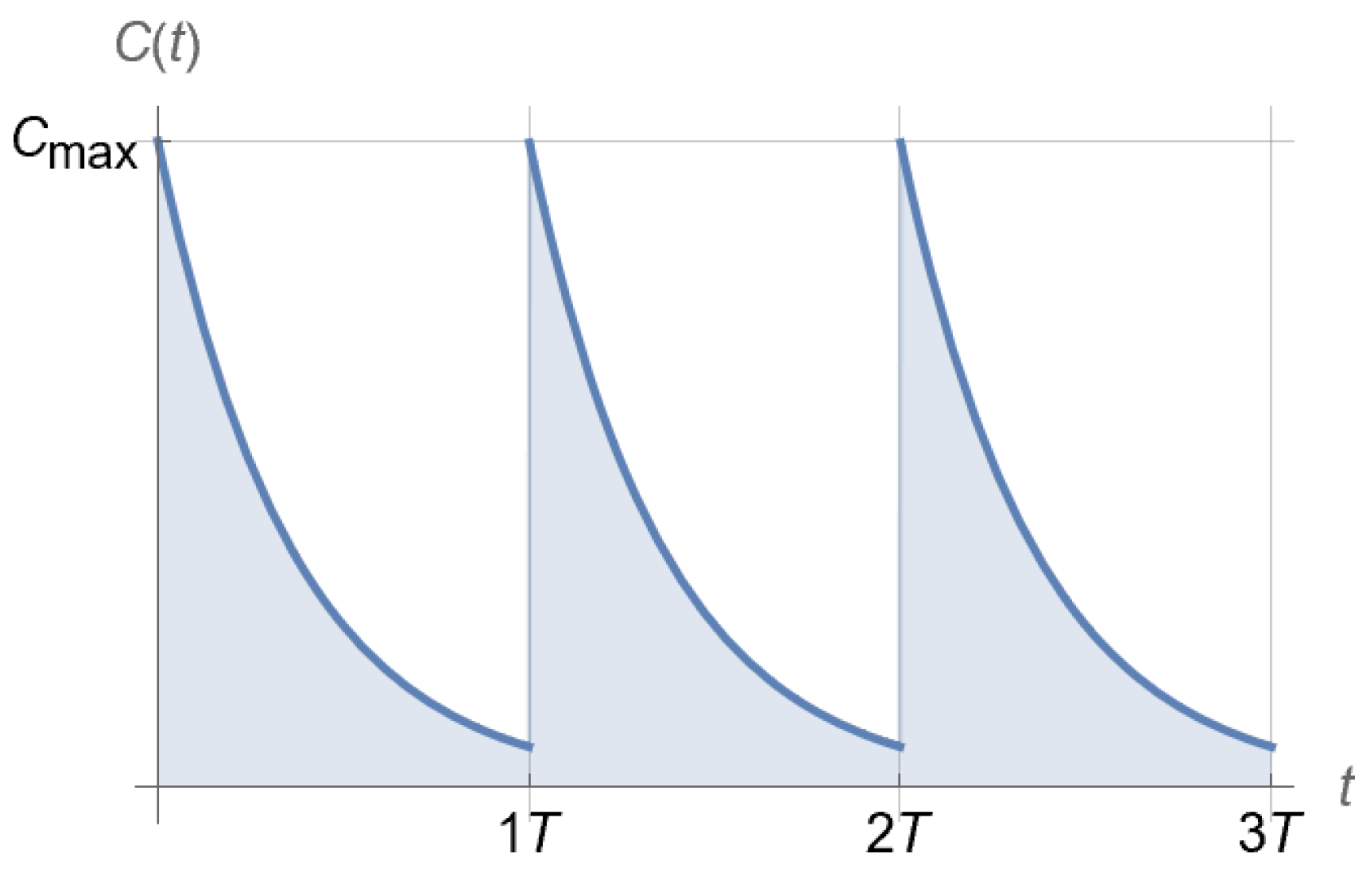
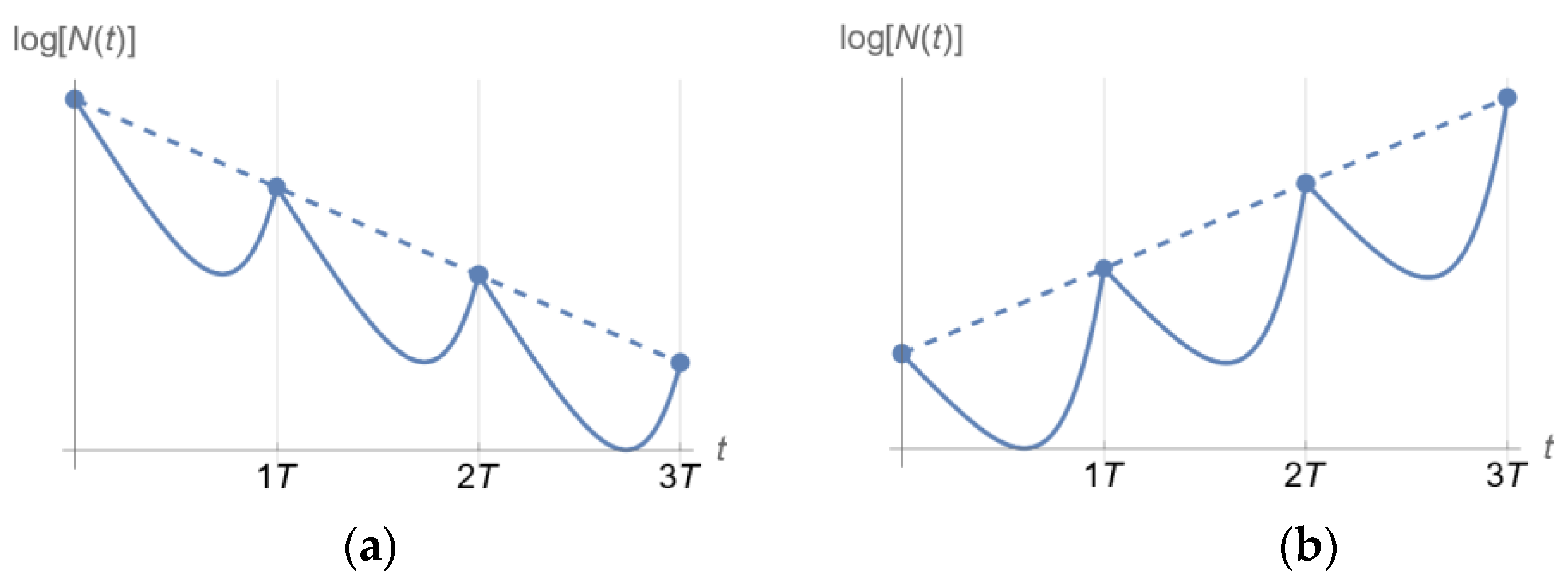
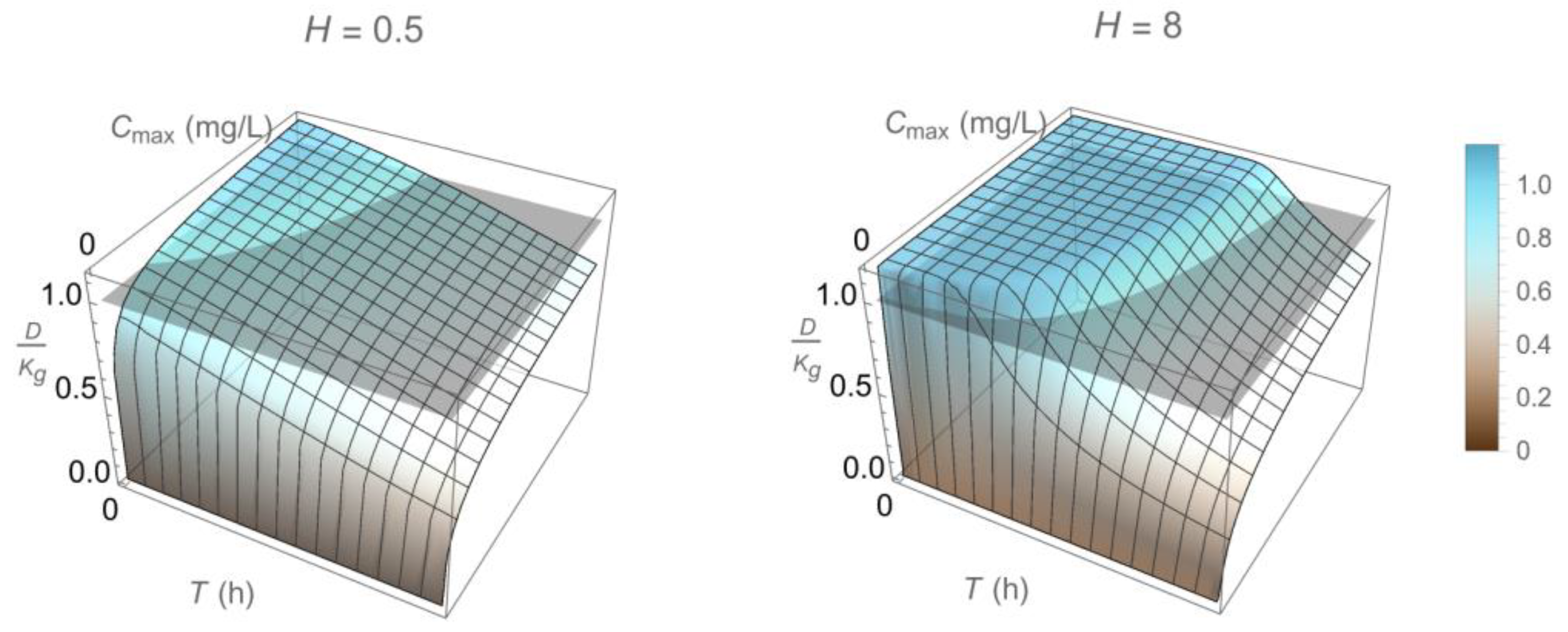
2.3. Dosing Regimen Design for Bacterial Eradication under Clinically Relevant Pharmacokinetics
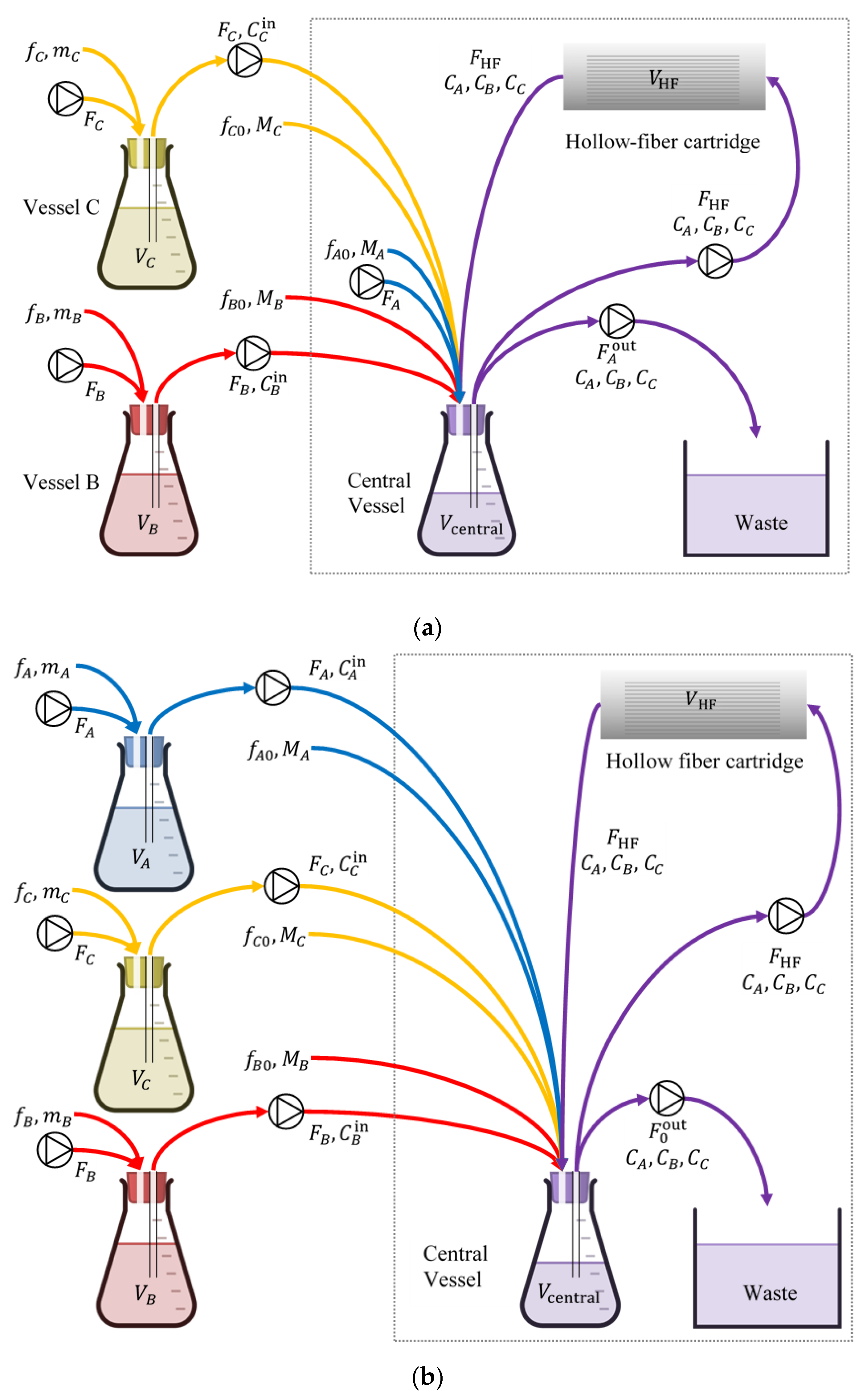
2.4. Validation Test of Promising Dosing Regimens in an In Vitro Hollow Fiber Infection Model
3. Results
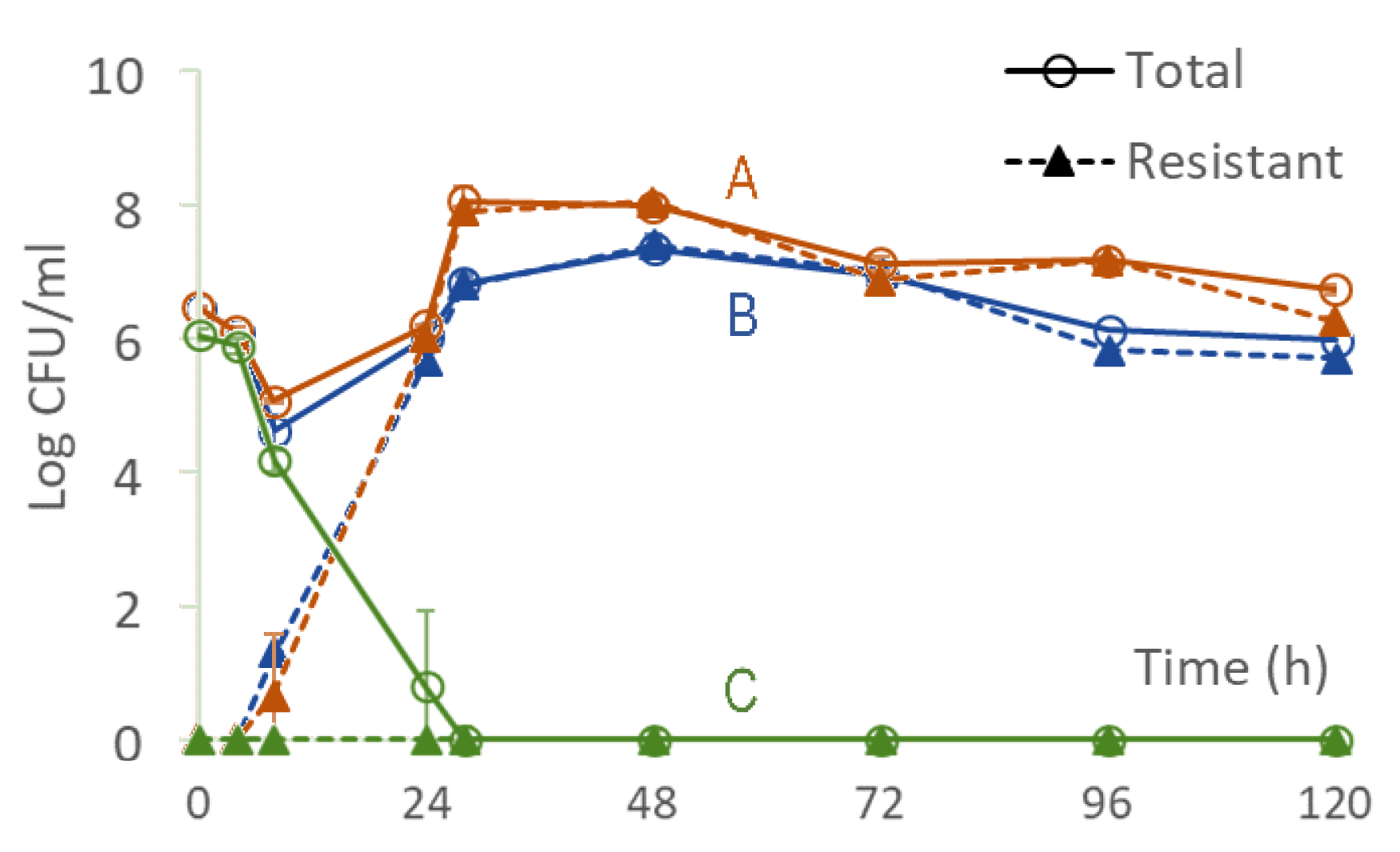
3.1. Longitudinal Optical Density Measurements of Bacterial Cell Suspension under Drug Exposure
3.2. From Optical Density Measurements to Dosing Regimen Design for Clinically Relevant Pharmacokinetics
4. Discussion
4.1. Optical Density Measurements and Alternatives
4.2. Clinically Relevant Pharmacokinetics and Design and In Vitro Testing of Dosing Regimens
5. Conclusions and Future Work
- Instrumentation improvements that may improve the quality of the data (reduction of systematic error);
- A wider range of clonally diverse bacteria;
- Bacteria with various resistance mechanisms;
- Different antibiotics, particularly with pharmacodynamics and pharmacokinetic differences;
- Combination therapy, particularly the interplay between pharmacodynamics and pharmacokinetics;
- Automation of computations through software development;
- Testing of in vivo relevance in animal infection models.
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Craig, W.A. Pharmacokinetic/Pharmacodynamic Parameters: Rationale for Antibacterial Dosing of Mice and Men. Clin. Infect. Dis. 1998, 26, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dadgostar, P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist. 2019, 12, 3903–3910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, E.R. Aseptic Laboratory Techniques: Plating Methods. J. Vis. Exp. 2012, 11, e3064. [Google Scholar] [CrossRef]
- Tam, V.H.; Nikolaou, M. A Novel Approach to Pharmacodynamic Assessment of Antimicrobial Agents: New Insights to Dosing Regimen Design. PLoS Comput. Biol. 2011, 7, e1001043. [Google Scholar] [CrossRef]
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001, 48, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouton, J.W.; Dudley, M.N.; Cars, O.; Derendorf, H.; Drusano, G.L. Standardization of pharmacokinetic/pharmacodynamic (PK/PD) terminology for anti-infective drugs: An update. J. Antimicrob. Chemother. 2005, 55, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef]
- Vogelman, B.; Craig, W.A. Kinetics of antimicrobial activity. J. Pediatr. 1986, 108, 835–840. [Google Scholar] [CrossRef]
- Giraldo, J.; Vivas, N.M.; Vila, E.; Badia, A. Assessing the (a)symmetry of concentration-effect curves: Empirical versus mechanistic models. Pharmacol. Ther. 2002, 95, 21–45. [Google Scholar] [CrossRef]
- Lipsitch, M.; Levin, B.R. The population dynamics of antimicrobial chemotherapy. Antimicrob. Agents Chemother. 1997, 41, 363–373. [Google Scholar] [CrossRef] [Green Version]
- Bhagunde, P.R.; Nikolaou, M.; Tam, V.H. Modeling heterogeneous bacterial populations exposed to antibiotics: The logistic-dynamics case. AIChE J. 2015, 61, 2385–2393. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, M.; Tam, V.H. A New Modeling Approach to the Effect of Antimicrobial Agents on Heterogeneous Microbial Populations. J. Math. Biol. 2005, 52, 154–182. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.; de la Pena, A.; Derendorf, H. Issues in Pharmacokinetics and Pharmacodynamics of Anti-Infective Agents: Kill Curves versus MIC. Antimicrob. Agents Chemother. 2004, 48, 369–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mytilinaios, I.; Salih, M.; Schofield, H.K.; Lambert, R.J.W. Growth curve prediction from optical density data. Int. J. Food Microbiol. 2012, 154, 169–176. [Google Scholar] [CrossRef] [Green Version]
- López, S.; Prieto, M.; Dijkstra, J.; Dhanoa, M.; France, J. Statistical evaluation of mathematical models for microbial growth. Int. J. Food Microbiol. 2004, 96, 289–300. [Google Scholar] [CrossRef]
- McMeekin, T.A.; Olley, J.N.; Ross, T.; Ratkowsky, D.A. Predictive Microbiology: Theory and Application; Wiley: New York, NY, USA, 1993. [Google Scholar]
- Davaro, E.; Tomaras, A.P.; Chamberland, R.R.; Isbell, T.S. Evaluation of a Novel Light Scattering Methodology for the Detection of Pathogenic Bacteria in Urine. J. Appl. Lab. Med. 2020, 5, 370–376. [Google Scholar] [CrossRef]
- Kesisoglou, I.; Tam, V.H.; Tomaras, A.P.; Nikolaou, M. Discerning in vitro pharmacodynamics from OD measurements: A model-based approach. Comput. Chem. Eng. 2022, 158. [Google Scholar] [CrossRef]
- Kesisoglou, I.; Eales, B.M.; Merlau, P.R.; Tam, V.H.; Nikolaou, M. Deciphering longitudinal optical-density measurements to guide clinical dosing regimen design: A model-based approach. Comput. Methods Programs Biomed. 2022, 227, 107212. [Google Scholar] [CrossRef]
- Schaechter, M. A brief history of bacterial growth physiology. Front. Microbiol. 2015, 6, 289. [Google Scholar] [CrossRef] [Green Version]
- Baranyi, J.; Roberts, T.A.; McClure, P. A non-autonomous different equation to model bacterial growth. Food Microbiol. 1993, 10, 43–59. [Google Scholar] [CrossRef]
- Verhulst, P.F. Resherches mathematiques sur la loi d’accroissement de la population. Nouv. Mem. L’academie R. Sci. 1845, 18, 1–41. [Google Scholar]
- Jusko, W.J. Pharmacodynamics of Chemotherapeutic Effects: Dose-Time-Response Relationships for Phase-Nonspecific Agents. J. Pharm. Sci. 1971, 60, 892–895. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J. Kinetics of pharmacologic response I. Proposed relationships between response and drug concentration in the intact animal and man. J. Theor. Biol. 1968, 20, 173–201. [Google Scholar] [CrossRef]
- Hill, A.V. The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J. Physiol. 1910, 40, iv–vii. [Google Scholar]
- Tamma, P.D.; Cosgrove, S.E.; Maragakis, L.L. Combination Therapy for Treatment of Infections with Gram-Negative Bacteria. Clin. Microbiol. Rev. 2012, 25, 450–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gloede, J.; Scheerans, C.; Derendorf, H.; Kloft, C. In vitro pharmacodynamic models to determine the effect of antibacterial drugs. J. Antimicrob. Chemother. 2010, 65, 186–201. [Google Scholar] [CrossRef] [Green Version]
- Doern, C.D. When Does 2 Plus 2 Equal 5? A Review of Antimicrobial Synergy Testing. J. Clin. Microbiol. 2014, 52, 4124–4128. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, S.E. (Ed.) Basic Pharmacokinetics and Pharmacodynamics: An Integrated Textbook and Computer Simulations, 2nd ed.; Wiley: Hoboken, NJ, USA, 2016. [Google Scholar]
- Nikolaou, M.; Schilling, A.N.; Vo, G.; Chang, K.-T.; Tam, V.H. Modeling of Microbial Population Responses to Time-Periodic Concentrations of Antimicrobial Agents. Ann. Biomed. Eng. 2007, 35, 1458–1470. [Google Scholar] [CrossRef]
- Blaser, J.; Stone, B.; Zinner, S. Two compartment kinetic model with multiple artificial capilary units. J. Antimicrob. Chemother. 1985, 15, 131–137. [Google Scholar] [CrossRef]
- Bulitta, J.B.; Hope, W.W.; Eakin, A.E.; Guina, T.; Tam, V.H.; Louie, A.; Drusano, G.L.; Hoover, J.L. Generating Robust and Informative Nonclinical In Vitro and In Vivo Bacterial Infection Model Efficacy Data to Support Translation to Humans. Antimicrob. Agents Chemother. 2019, 63, e02307-18. [Google Scholar] [CrossRef] [Green Version]
- Cadwell, J. The hollow fiber infection model: Principles and practice. Adv. Antibiot. Antibodies 2015, 1, 101. [Google Scholar]
- Bao, L.; Liu, B.; Lipscomb, G.G. Entry mass transfer in axial flows through randomly packed fiber bundles. AIChE J. 1999, 45, 2346–2356. [Google Scholar] [CrossRef]
- De Bartolo, L.; Salerno, S.; Curcio, E.; Piscioneri, A.; Rende, M.; Morelli, S.; Tasselli, F.; Bader, A.; Drioli, E. Human hepatocyte functions in a crossed hollow fiber membrane bioreactor. Biomaterials 2009, 30, 2531–2543. [Google Scholar] [CrossRef] [PubMed]
- Blaser, J. In-vitro model for simultaneous simulation of the serum kinetics of two drugs with different half-lives. J. Antimicrob. Chemother. 1985, 15, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Siwakoti, S.; Subedi, A.; Sharma, A.; Baral, R.; Bhattarai, N.R.; Khanal, B. Incidence and outcomes of multidrug-resistant gram-negative bacteria infections in intensive care unit from Nepal–a prospective cohort study. Antimicrob. Resist. Infect. Control 2018, 7, 114. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wei, H.; Zhao, Y.; Shang, L.; Di, L.; Lyu, C.; Liu, J. Analysis of multidrug-resistant bacteria in 3223 patients with hospital-acquired infections (HAI) from a tertiary general hospital in China. Bosn. J. Basic Med. Sci. 2019, 19, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Roy, I.; Gupta, M.K.; Kumar, M.; Agarwal, S.K. Prevalence of extended-spectrum β-lactamase-producing Gram-negative bacteria in septicaemic neonates in a tertiary care hospital. J. Med. Microbiol. 2003, 52, 421–425. [Google Scholar] [CrossRef] [Green Version]
- Pop-Vicas, A.; Mitchell, S.L.; Kandel, R.; Schreiber, R.; D’Agata, E.M.C. Multidrug-Resistant Gram-Negative Bacteria in a Long-Term Care Facility: Prevalence and Risk Factors. J. Am. Geriatr. Soc. 2008, 56, 1276–1280. [Google Scholar] [CrossRef]
- Saeed, S.; Naim, A.; Tariq, P. A study on prevalance of multi-drug-resistant gram negative bacteria. Int. J. Biol. Biotech. 2007, 4, 71–74. [Google Scholar]
- Tängdén, T. Combination antibiotic therapy for multidrug-resistant Gram-negative bacteria. Upsala J. Med. Sci. 2014, 119, 149–153. [Google Scholar] [CrossRef]
- Karakonstantis, S.; Kritsotakis, E.I.; Gikas, A. Pandrug-resistant Gram-negative bacteria: A systematic review of current epidemiology, prognosis and treatment options. J. Antimicrob. Chemother. 2020, 75, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Kesisoglou, I.; Eales, B.M.; Ledesma, K.R.; Merlau, P.R.; Tam, V.H.; Wang, W.; Nikolaou, M. Simultaneous in vitro simulation of multiple antimicrobial agents with different elimination half-lives in a pre-clinical infection model. Comput. Chem. Eng. 2021, 155, 107540. [Google Scholar] [CrossRef] [PubMed]
- Kralik, P.; Ricchi, M. A Basic Guide to Real Time PCR in Microbial Diagnostics: Definitions, Parameters, and Everything. Front. Microbiol. 2017, 8, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricchi, M.; Bertasio, C.; Boniotti, M.B.; Vicari, N.; Russo, S.; Tilola, M.; Bellotti, M.A.; Bertasi, B. Comparison among the Quantification of Bacterial Pathogens by qPCR, dPCR, and Cultural Methods. Front. Microbiol. 2017, 8, 1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Hammes, F.; De Roy, K.; Verstraete, W.; Boon, N. Past, present and future applications of flow cytometry in aquatic microbiology. Trends Biotechnol. 2010, 28, 416–424. [Google Scholar] [CrossRef]
- Hasan, M.; Alam, M.W.; Wahid, K.A.; Miah, S.; Lukong, K.E. A Low-Cost Digital Microscope with Real-Time Fluorescent Imaging Capability. PLoS ONE 2016, 11, e0167863. [Google Scholar] [CrossRef]
- Gracias, K.S.; McKillip, J.L. A review of conventional detection and enumeration methods for pathogenic bacteria in food. Can. J. Microbiol. 2004, 50, 883–890. [Google Scholar] [CrossRef] [Green Version]
- Pascaud, A.; Amellal, S.; Soulas, M.-L.; Soulas, G. A fluorescence-based assay for measuring the viable cell concentration of mixed microbial communities in soil. J. Microbiol. Methods 2009, 76, 81–87. [Google Scholar] [CrossRef]
- Feng, J.; Wang, T.; Zhang, S.; Shi, W.; Zhang, Y. An Optimized SYBR Green I/PI Assay for Rapid Viability Assessment and Antibiotic Susceptibility Testing for Borrelia burgdorferi. PLoS ONE 2014, 9, e111809. [Google Scholar] [CrossRef]
- Alakomi, H.-L.; Mättö, J.; Virkajärvi, I.; Saarela, M. Application of a microplate scale fluorochrome staining assay for the assessment of viability of probiotic preparations. J. Microbiol. Methods 2005, 62, 25–35. [Google Scholar] [CrossRef]
- Ou, F.; McGoverin, C.; Swift, S.; Vanholsbeeck, F. Absolute bacterial cell enumeration using flow cytometry. J. Appl. Microbiol. 2017, 123, 464–477. [Google Scholar] [CrossRef] [Green Version]
- Imade, G.E.; Badung, B.; Pam, S.; Agbaji, O.; Egah, D.; Sagay, A.S.; Sankale, J.-L.; Kapiga, S.; Idoko, J.; Kanki, P. Comparison of a New, Affordable Flow Cytometric Method and the Manual Magnetic Bead Technique for CD4 T-Lymphocyte Counting in a Northern Nigerian Setting. Clin. Vaccine Immunol. 2005, 12, 224–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.; McGoverin, C.; Swift, S.; Vanholsbeeck, F. A rapid and low-cost estimation of bacteria counts in solution using fluorescence spectroscopy. Anal. Bioanal. Chem. 2017, 409, 3959–3967. [Google Scholar] [CrossRef] [Green Version]
- Braissant, O.; Wirz, D.; GãPfert, B.; Daniels, A.U. Use of isothermal microcalorimetry to monitor microbial activities. FEMS Microbiol. Lett. 2010, 303, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Al-Madani, H.; Du, H.; Yao, J.; Peng, H.; Yao, C.; Jiang, B.; Wu, A.; Yang, F. Living Sample Viability Measurement Methods from Traditional Assays to Nanomotion. Biosensors 2022, 12, 453. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, M.; Tam, V.H.; Kesisoglou, I. Optical Based Methods for Determining Antimicrobial Dosing Regimens. U.S. Patent App. 17/636,079, 17 February 2022. [Google Scholar]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolaou, M.; Tam, V.H. Rapid In Vitro Assessment of Antimicrobial Drug Effect Bridging Clinically Relevant Pharmacokinetics: A Comprehensive Methodology. Pharmaceutics 2023, 15, 1671. https://doi.org/10.3390/pharmaceutics15061671
Nikolaou M, Tam VH. Rapid In Vitro Assessment of Antimicrobial Drug Effect Bridging Clinically Relevant Pharmacokinetics: A Comprehensive Methodology. Pharmaceutics. 2023; 15(6):1671. https://doi.org/10.3390/pharmaceutics15061671
Chicago/Turabian StyleNikolaou, Michael, and Vincent H. Tam. 2023. "Rapid In Vitro Assessment of Antimicrobial Drug Effect Bridging Clinically Relevant Pharmacokinetics: A Comprehensive Methodology" Pharmaceutics 15, no. 6: 1671. https://doi.org/10.3390/pharmaceutics15061671
APA StyleNikolaou, M., & Tam, V. H. (2023). Rapid In Vitro Assessment of Antimicrobial Drug Effect Bridging Clinically Relevant Pharmacokinetics: A Comprehensive Methodology. Pharmaceutics, 15(6), 1671. https://doi.org/10.3390/pharmaceutics15061671