1. Introduction
Attention deficit/hyperactivity disorder (ADHD) is characterized as a persistent pattern of inattention or hyperactivity and is considered to be the most common neurodevelopment disorder [
1]. Considering the latest official population figures in Brazil (2010) [
2], ADHD reaches between 5.3% of youths and 2.5% of adults [
3,
4]. If untreated, ADHD could negatively affect the general patient’s quality of life; social, family, and professional relationships; and increase accidental injury, substance abuse, or other psychiatric morbidity risks [
5]. Among the available treatments, central nervous system stimulants such as methylphenidate (MPH) and amphetamines are the most effective, in combination with psychological intervention. The indications include the treatment of children over six years of age, adolescents, and adults with significant impairment due to ADHD [
6].
Early ADHD treatment was initially quite limited by available formulations. The immediate-release formulations required the use of multiple daily doses, which contributed to treatment adherence problems for school-age children, as well as associated safety concerns and high variability individual response. Over the past 15 years, there has been a significant expansion in the number of formulations available to ADHD patients, in hopes of better adherence and better long-term outcomes. Some of the main advantages of ER formulations for school-age children are the better pharmacokinetic (PK) profile (lower peak concentrations) and the ability to provide coverage throughout the day, which avoids the need to administer doses during school hours [
7,
8].
Currently, there are MPH formulations commercialized as multiphasic drug release systems. They were designed to release an initial amount of the drug immediately after administration, followed by slower drug delivery throughout the day [
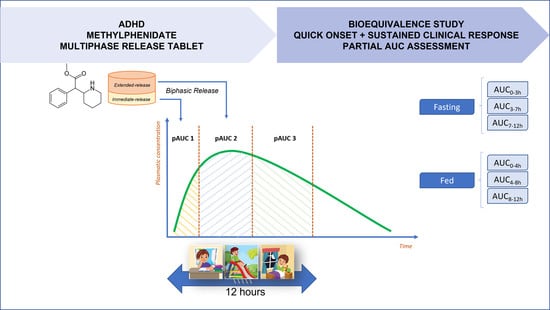
6]. Once administered in the morning, the early peak concentration is needed to control morning hyperactivity, while the later peak is needed to control hyperactivity during school hours. Thus, the ideal scenario is for plasma levels to decrease 10 h after administration, so that the stimulation is minimal, avoiding cases of insomnia. The presence of multiple peaks of maximum concentration is directly related to the efficacy of the drug but makes it difficult to establish bioequivalence [
9]. According to the Concerta
® label, the MPH extended-release formulation has an action onset of 1 to 2 h after administration. The plasma concentrations continue to gradually increase, maintaining the clinical effect until 12 h. The Tmax is typically in the range of 6 to 8 h and the drug could be administered with or without food, with no bioavailability impact [
6]. Nonetheless, a delay in MPH absorption is observed when it is taken with high-fat meals, which could alter the clinical response profile [
10].
Generic medicines are considered to be therapeutically equivalent to the corresponding reference products if they have the same efficacy and safety and are, therefore, interchangeable. The comparison between test and reference formulations is performed through pharmacokinetic profiles and bioequivalence assessment to guarantee that both drug formulations present the same rate and extent of absorption [
11,
12].
For the design of bioequivalence studies with MPH extended-release tablets, the FDA Product Specific Guidance recommends the use of the partial area under the curve (pAUC) metrics in bioequivalence assessment, in addition to the traditional primary pharmacokinetic parameters (Cmax and AUC
0–t). These metrics relate the drug’s release profile to its pharmacodynamic (PD) properties [
13,
14]. The PK/PD relationships of MPH were elucidated by Swanson (1978), comparing the clinical response time to drug plasma concentration, showing clinical superiority to the formulation with the higher concentration [
15]. Therefore, pAUC metrics seem fundamental in developing generic and branded-generic formulations of MPH extended-release tablets.
The present study aimed to assess the bioequivalence and tolerability of two formulations of MPH 54 mg extended-release tablets administered under fasting and fed conditions to attend regulatory requirements for generic drug product registration in Brazil [
11,
12].
2. Materials and Methods
2.1. Ethical Aspects and Good Clinical Practices
Both fasting and fed study protocols were approved by the Research Ethics Committee of the Instituto de Ciências Farmacêuticas de Estudos e Pesquisas (Aparecida de Goiânia, Goiás, Brazil) with protocol numbers 4,108,995 and 3,548,871, respectively. The clinical, analytical, and statistical phases were performed at the Instituto de Ciências Farmacêuticas de Estudos e Pesquisas (Goiânia, Goiás, Brazil), a Brazilian clinical research center certified by ANVISA to conduct bioequivalence studies.
Both studies were conducted in compliance with the ethical principles of the Good Clinical Practices Guidelines [
16], the Declaration of Helsinki [
17], the local laws [
18], and requirements for bioequivalence studies [
11,
12]. All subjects gave their informed consent for inclusion before the initiation of study procedures.
2.2. Study Design and Subjects
Two independent studies were performed, one under fasting and the other under fed conditions. Both studies were conducted as a single-center, open-label, randomized, balanced, single-dose, 2-period, 2-treatment, and 2-sequence crossover design.
Ninety-six (96) adult healthy subjects of both genders (48 male and 48 nonpregnant female subjects) were enrolled in the fasting study. Fifty-two (52) adult healthy subjects of both genders (26 male and 26 nonpregnant female subjects) were enrolled in the fed study. The sample size for both studies was calculated considering a within-subject variability (CVws = 30%) obtained from pilot studies performed by the sponsor (data unpublished).
Regarding the inclusion and exclusion criteria, for both studies, subjects had not previously participated in another clinical trial nor donated blood during the preceding six months and had no history of alcohol or drug abuse. They were aged between 18 and 50 years with a body mass index (BMI) between 18.5 and 28.6 kg/m2.
All subjects showed good health conditions or the absence of significant diseases after assessing their medical history, verifying vital signs, and conducting physical examinations, electrocardiograms, and routine laboratory tests. In addition, subjects must not have had lactose intolerance, a positive or indeterminate result for the RT-qPCR test for SARS-CoV-2, be smokers, be vegetarians, or have dietary habits that would prevent the intake of the provided fed study diet. They also should not have a clinical history or episodes of gastrointestinal disorders or have taken medications that would interfere with the pharmacokinetics of methylphenidate.
2.3. Formulations Studied
The test formulation was Consiv®, a methylphenidate hydrochloride 54 mg extended-release tablet (batch No. 84497, expiry date: February 2021) manufactured by Monte Verde S.A. (San Juan, Argentina) and imported to Brazil by Adium S.A. (Pindamonhangaba, São Paulo, Brazil). The reference formulation was Concerta® 54 mg extended-release tablet (batch No. 8LE798, expiry date: September 2020) manufactured by Janssen Cilag Manufacturing LLC (Rockford, IL, USA) and imported to Brazil by Janssen-Cilag Farmacêutica Ltd. (São Paulo, Brazil). The same products and batches were administered in both the fasting and fed studies.
2.4. Drug Administration and Sampling Times
Fasting study: Each period began with a minimum overnight fasting period of eight hours. The subjects received a single dose of 54 mg MPH extended-release tablets from one of the two formulations, along with 200 mL of water. Following drug administration, the subjects remained fasted for four hours, with restrictions on water intake from seven hours before to two hours after drug administration. The diet, including food and drink, was standardized for all subjects in both periods. Alcoholic beverages, as well as food or beverages containing caffeine or xanthine (such as coffee, tea, chocolate, and cola- or guarana-based soft drinks), were not allowed in the 24 h before study admission. A total of 25 blood samples were collected at 0 h (before drug administration) and 0.33, 0.67, 1.00, 1.33, 1.67, 2.00, 2.50, 3.00, 3.50, 4.00, 4.50, 5.00, 5.50, 6.00, 6.50, 7.00, 7.50, 8.00, 8.50, 9.00, 10.0, 12.0, 16.0, and 24.0 h after drug administration, in tubes containing K3EDTA as the anticoagulant.
Fed study: In each period, the subjects were given a hypercaloric meal consisting of approximately 1000 kcal, 30 min before drug administration. Then, they receive a single dose of 54 mg of MPH extended-release tablets from one of the two formulations along with 200 mL of water. Following drug administration, the subjects remained fasted for four hours, with restrictions on water intake from seven hours before to two hours after drug administration. The diet, including food and drink, was standardized for all subjects in both periods, to maintain the standardization of treatment groups. Alcoholic beverages, and food or beverages containing caffeine or xanthine (such as coffee, tea, chocolate, and cola- or guarana-based soft drinks), were not allowed in the 24 h before study admission. A total of 26 blood samples were collected at 0 h (before drug administration) and 0.33, 0.67, 1.00, 1.50, 2.00, 2.50, 3.00, 3.50, 4.00, 4.50, 5.00, 5.50, 6.00, 6.50, 7.00, 7.50, 8.00, 8.50, 9.00, 9.50, 10.0, 11.0, 12.0, 16.0, and 24.0 h after drug administration, in tubes containing K3EDTA as the anticoagulant.
The blood samples from both studies were centrifuged at 3000 rpm for 5 min at 4 °C; the plasma was separated (2 mL) and transferred into cryogenic tubes. Then, 100 µL of citric acid 10% was immediately added to 2 mL of plasma and homogenized. Finally, the samples were stored at −80 °C with appropriate labeling until sample analysis.
2.5. Bioanalytical Method
The plasma samples were analyzed using validated high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS), to obtain MPH concentrations. The system includes an Agilent 1200 Series (Agilent Technologies Inc., Santa Clara, CA, USA) and an API 5000 MS/MS (Applied Biosystems/Sciex, Framingham, MA, USA). The analytes were extracted from the plasma using a protein precipitation method and methylphenidate-d9 was used as an internal standard (IS). To avoid interassay variations, all samples from the same participant were assessed in the same analytical run.
An amount of 3.0 µL of each sample was injected onto a Zorbax Eclipse XDB-Phenyl (4.6 × 150 mm; 3.5 µm) column, maintained at 20 °C. The mobile phase for MPH consisted of a mixture (70:30) of (A) acetonitrile and (B) ammonium acetate 5 mM solution (v/v, with 0.025% formic acid). The flow rate was 1 mL/min, in an isocratic performance. The detection of MPH was carried out in the mass spectrometer with the positive electrospray ionization multiple-reaction monitoring mode set to transmit at m/z 234.1 → 84.1 for MPH and m/z 243.3 → 93.2 for methylphenidate-d9 (IS).
The analyte concentrations were calculated through interpolation on the calibration curve, and the linearity range used was from 25 to 30,000 pg/mL. The bioanalytical method was validated in compliance with ANVISA guidance for bioanalytical method validation [
19] including the evaluation of selectivity, concomitant medication interference, matrix effect, carry-over, calibration curve, precision, accuracy, reinjection reproducibility, and the stabilities of MPH under different conditions.
2.6. Pharmacokinetic and Statistical Analysis
The pharmacokinetic parameters were obtained from the curves of MPH plasma concentration versus time and statistically compared for the determination of bioequivalence in both the fasting and fed studies using Phoenix WinNonlin™ version 6.4 (Princeton, NJ, USA). The calculation of the area under the curve from zero to the last quantifiable concentration (AUC0–t) was performed using the trapezoidal method, and the area under the curve from zero to infinity (AUC0–inf) was calculated using the formula AUC0–t + (Cn/kel), where Cn was the last quantifiable plasma concentration. The elimination constant (kel) was determined by analyzing the elimination phase of the graph depicting the log plasma concentration versus time. t1/2 was defined using the equation t1/2 = Ln(2)/kel and the maximum plasma drug concentration (Cmax) was obtained directly from the experimental data, as well as the time of the occurrence of Cmax (Tmax).
The Concerta
® tablet is an extended-release formulation with a bimodal release profile (designed to release a bolus of MPH followed by slower delivery later in the day) [
6]. Thus, as per FDA product-specific guidance for generic drug development of MPH, the following three partial AUC (pAUC) metrics were necessary in addition to the traditional (Cmax and AUC
0–t) metrics for each study:
Fasting Study: log-transformed AUC0–3, AUC3–7, and AUC7–12, where AUC0–3 is the area under the plasma concentration vs. time curve from 0 to 3 h, AUC3–7 is the area under the curve from 3 to 7 h, and AUC7–12 is the area under the curve from 7 to 12 h.
Fed Study: log-transformed AUC0–4, AUC4–8, and AUC8–12, where AUC0–4 is the area under the plasma concentration vs. time curve from 0 to 4 h, AUC4–8 is the area under the curve from 4 to 8 h, and AUC8–12 is the area under the curve from 8 to 12 h.
The selected pAUCs have been identified as the most appropriate parameters for drug bioavailability evaluation, which are responsible for ensuring a quick onset and sustained maintenance of the clinical response throughout the drug effect duration.
To assess the bioequivalence, predefined acceptance criteria were applied to the 90% confidence interval for the ratio of the test and reference (T/R) formulations for the log-transformed data of Cmax and AUCs (AUC
0–t, AUC
0–inf, and pAUCs), where the acceptance range was set at 80.00–125.00% [
11,
12]. An analysis of variance (ANOVA) test was conducted to evaluate the effects of sequence, treatment, and period on these parameters.
2.7. Safety
All participants were continuously and carefully monitored in both the fasting and fed studies. Safety was assessed by monitoring baseline and ongoing vital signs including temperature, blood pressure, heart rate, and respiratory rate throughout the study. Additionally, laboratory tests (such as hematology, urinalysis, and blood biochemistry), physical examinations, and electrocardiograms (ECGs) were conducted at the beginning and conclusion of the study. Adverse events were assessed by the nursing and medical staff throughout the entire study. Subjects were instructed regarding the need to immediately report any undesirable symptoms or medical conditions during the study or after the hospitalization period. Adverse events were graded as mild, moderate, or severe, and their causality to the drug was determined by the medical staff as suspected or not suspected.
3. Results
3.1. Study Subjects
After the medical history assessment, verification of vital signs, physical examination, electrocardiogram, and routine laboratory tests, all subjects showed good health conditions and the absence of significant diseases.
Table 1 shows the demographic subjects characteristics in both fasting and fed studies.
3.2. Sample Bioanalysis
The validated method covered all required tests, including the evaluation of the selectivity, concomitant medication interference, matrix effect, carry-over, calibration curve, precision, accuracy, reinjection reproducibility, and stabilities.
The method was linear in a concentration range of 25.0 to 30,000.0 pg/mL, and the lower limit of quantification (LLOQ) was 25.0 pg/mL. The method selectivity was demonstrated to be suitable by confirming that substances in the blank plasma samples did not affect MPH and IS retention times. In terms of precision and accuracy, the method was deemed appropriate for both within-assay (intra-run) and between-assay (inter-run) samples.
The stability assessments demonstrated that samples remained stable, with a variation less than 15% from the nominal value up to 6 h at room temperature (15 °C to 25 °C), and could remain stable for up to 52 h after extraction when stored in the auto-sampler at 10 °C. In terms of freeze–thaw stability, samples maintained their stability after undergoing four cycles of freezing in a standard freezer (−20 °C) and an ultrafreezer (−80 °C) followed by thawing at room temperature. Furthermore, the samples were proven to be stable and could be kept frozen for up to 168 days in an ultrafreezer (−80 °C). These stability tests are crucial to ensure proper sample storage prior to analysis, guaranteeing the accurate determination of drug concentrations.
The absence of carryover effects was confirmed since no predose samples from any participant showed the presence of MPH in plasma, confirming the appropriate washout period. Finally, all validation parameters met the predetermined acceptance criteria following ANVISA guidelines for bioanalytical method validation [
19].
3.3. Pharmacokinetic Analysis
3.3.1. Fasting Study
Ninety-six (96) healthy subjects were enrolled in the fasting study and eighty subjects (39 women and 41 men) completed the two study periods, being included in the pharmacokinetic and statistical analysis. A total of six subjects were withdrawn due to personal reasons, eight subjects were excluded due to adverse events, and two subjects were excluded due to drug abuse detection before the second drug administration period.
Figure 1 shows the mean plasma concentration versus time curves of MPH (reference and test formulations) when administered under fasting conditions. It is possible to observe the similarity of both test and reference pharmacokinetic profiles. Moreover, the sampling time can be considered adequate since it was possible to correctly describe the drug absorption and elimination phases. The pharmacokinetic parameters of MPH for both formulations are described in
Table 2. The use of pAUC metrics was applied to ensure that, in fasting conditions, the products are therapeutic equivalents in different parts of the daily dosing interval.
3.3.2. Fed Study
Fifty-two (52) healthy subjects were enrolled in the fed study, in which forty-six (24 women and 22 men) completed the two study periods and, therefore, were included in the pharmacokinetic and statistical analysis. A total of three subjects were withdrawn due to personal reasons, and three subjects were excluded due to adverse events before the second drug administration period.
Figure 2 shows the mean plasma concentration versus time curves of MPH (reference and test formulations) when administered under fed conditions. It is possible to observe that the pharmacokinetic profiles of both test and reference formulations are very similar when administered under fed conditions. The pharmacokinetic parameters of MPH for both formulations are described in
Table 3. The use of pAUC metrics was applied to ensure that, in the fed condition, the products are therapeutic equivalents in different parts of the daily dosing interval.
3.4. Bioequivalence Assessment
Table 4 and
Table 5 present the test/reference geometric mean ratio for pharmacokinetic parameters Cmax, AUC
0–t, AUC
0–inf, and pAUCs, and the 90% CIs for the bioequivalence analysis for the fasting and fed studies, respectively.
Regarding the fasting study, the result obtained for Cmax is very similar to those obtained for AUC3–7. This happened mainly because the mean values of Tmax were between 3 and 7 h, and the median was estimated at 6.50 h for both formulations. The ratios were shown to be displaced upwards (above 112%); the CVws were considered to be low, presenting a result below 16%; and the power estimates (TOST method) were above 94%. For AUC0–3 the geometric mean ratio was around 94%, and the power estimate was higher than 99%, with similar low CVws values. The results obtained for AUC7–12, AUC0–t, and AUC0–inf were also similar presenting ratios centered on the CI, CVws ranging between 8 and 11%, and all power estimates reaching 100%.
Concerning the fed study, the geometric mean ratios for Cmax and AUC0–4 showed a slight shift, presenting values around 111% and 88%, respectively. The CVws for both parameters could be considered low, however, the power (TOST method) estimates did not reach 80%. So, the CVws equality test was performed, and the results indicate that both AUC0–4 and Cmax present variability equivalence in terms of variance. For AUC4–8, despite the observed geometric mean ratio having been slightly displaced upwards, the power estimate was higher than 93%. For AUC8–12, AUC0–t, and AUC0–inf the results were very similar. The ratios presented values around 96 and 99%, estimated CVws lower than 17%, and power estimates that reached 100%.
The ANOVA
p-values did not demonstrate statistically relevant differences for all evaluated parameters for sequence and period fixed effects at 10% and 5% significance levels, respectively. All 90% CIs of test/reference geometric mean ratios for both studies (fasting and fed) fell within the bioequivalence acceptance range of 80.00–125.00%, established by ANVISA [
11,
12]. So, the two MPH extended-release formulations (test and reference) are bioequivalent in terms of the rate and extent of absorption.
In addition to the bioequivalence data analysis, the present study did not show significant differences (
p < 0.05) in the pharmacokinetic parameters regarding subject gender.
Table 6 summarizes the obtained pharmacokinetic data for both test and reference formulations, according to gender for the fasting and fed studies, respectively. Our results agree with the statement in the Concerta
® label that, in healthy adults, the mean dose-adjusted AUC
(0–inf) data were 36.7 ng/mL.h in men and 37.1 ng/mL.h in women, with no significant differences between the two groups [
6].
3.5. Safety
In the fasting study, a total of 90 adverse events were reported by 53 of the 96 participants for both test and reference formulations; the most common were headache (30.0%) and leukocyturia (17.8%) (
Table 7). In turn, in the fed study a total of 55 adverse events were reported by 30 of the 52 participants for both test and reference formulations. The most common adverse events were leukocyturia (18.2%) and headache (14.5%) (
Table 8). Regarding the fasting study, eight adverse events were classified as causality suspected related to the drug and twelve of them were considered causality not suspected to the drug, whilst in the fed study five adverse events were classified as causality suspected related to the drug and thirteen of them were considered causality not suspected to the drug.
4. Discussion
The development of generic products for multiphasic release formulations is a challenge, mainly in terms of galenic and clinical studies. In this work, we have studied two formulations of MPH multiphasic release tablets (test and reference) in order to assess the bioequivalence for generic product registration. Both test (Consiv
®) and reference (Concerta
®) formulations demonstrated an ascending pharmacokinetic profile with plateau concentrations around 4 to 6 h; a similar behavior was observed by Markowitz and collaborators (2003) for Concerta
® [
20].
Schapperer and collaborators (2014) [
21] performed bioequivalence studies comparing Concerta
® with an MPH osmotic-controlled release (OCR) tablet and presented data for fasting (N = 24) and fed (N = 21) conditions [
17]. For the MPH OCR (54 mg) in the fasting study, a Cmax of 10.89 ± 2.09 ng/mL, AUC
0–t of 116.70 ± 26.52 ng.h/mL, AUC
0–inf of 121.98 ± 28.07 ng.h/mL, Tmax of 5.81 ± 1.01 h, and t
1/2 of 4.36 ± 0.55 h were found. On the other hand, for Concerta
® (54 mg) a Cmax of 12.11 ± 2.95 ng/mL, AUC
0–t of 121.43 ± 27.00 ng.h/mL, AUC
0–inf of 125.36 ± 28.12 ng.h/mL, Tmax of 6.96 ± 1.56 h, and t
1/2 of 3.88 ± 0.46 h were found. In the fed study for the MPH OCR (54 mg), a Cmax of 12.55 ± 3.36 ng/mL, AUC
0–t of 141.02 ± 43.11 ng.h/mL, AUC
0–inf of 148.92 ± 47.46 ng.h/mL, Tmax of 5.17 ± 0.64 h, and t
1/2 of 4.52 ± 0.92 h were found. Moreover, for Concerta
® (54 mg) a Cmax of 13.35 ± 4.04 ng/mL, AUC
0–t of 148.57 ± 47.73 ng.h/mL, AUC
0–inf of 154.38 ± 51.61 ng.h/mL, Tmax of 8.19 ± 2.64 h, and t
1/2 of 3.79 ± 0.52 h were found. The pharmacokinetic parameters shown in the work by Schapperer and collaborators (2014) [
21] are similar to our results (
Table 2 and
Table 3). Unfortunately, it was not possible to compare the pAUC metric values because the authors calculated different intervals for the fasting study and did not present these metrics for the fed study.
A comparative bioavailability study was performed by Reiz and collaborators (2008) [
22] evaluating a multilayer-based bead and an osmotic system formulation [
18]. The study was conducted with twenty-one healthy subjects, the formulations were administered under fed conditions, and the authors evaluated the pAUC metrics using 20 mg of MPH. Despite the differences in pharmacokinetic parameters caused by the different drug doses, the author’s plasma concentration versus time profile was very similar to our results. Notably, the formulations were designed to provide a multiphasic behavior, including a rapid initial release and a second sustained one.
Considering the pharmacokinetic data described in the Concerta
® label [
6], MPH concentrations increase rapidly reaching an initial maximum at about 1 h, followed by gradual ascending concentrations over the next 5 to 9 h after which a gradual decrease begins. Similar behavior was presented in our study (
Figure 1 and
Figure 2) for both formulations. Additionally, the Concerta
® label describes that Tmax occurs between 6 to 10 h and T
1/2 of 3.5 h. In our fasting study, Tmax was 6.44 (±1.33) h for the test formulation and 6.97 (±1.23) h for the reference formulation. For the fed study, we found a Tmax of 6.35 (±2.35) h for the test formulation and 6.44 (±2.59) h for the reference formulation. Regarding the T
1/2, our results (3.6–4.6 h) were similar to those described in the Concerta
® label [
6]. These comparisons are important to guarantee that the study design, sample size, and blood sampling adopted in our work were adequate to correctly describe the pharmacokinetic profile of each formulation and, consequently, to assess the bioequivalence. Moreover, it is important to mention that the adopted study design was adequate to calculate pAUC metrics (following the FDA requirements [
14]) and necessary measures to compare the formulations in different parts of the daily dosing interval.
This study compared the bioavailability of test and reference MPH 54 mg extended-release tablets in different intervals, and also monitored safety and tolerability under fasting and fed conditions. Here, we provide important information about pAUC metrics that could help the understanding of the MPH PK/PD relationship, and the oral intake of test or reference MPH tablets showed similar favorable safety profiles. However, it is important to exercise caution when evaluating adverse events resulting from a bioequivalence study, as this was conducted with healthy subjects and the drug was administered as a single dose. Continuous pharmacovigilance and a post-marketing surveillance plan are responsible for any safety concern monitoring.


 ,
, 



{kind=link}
{kind=link}
{kind=link}