

Drug Repurposing for Targeting Myeloid-Derived Suppressor-Cell-Generated Immunosuppression in Ovarian Cancer: A Literature Review of Potential Candidates

 , ,
, ,  ,
,  and
and 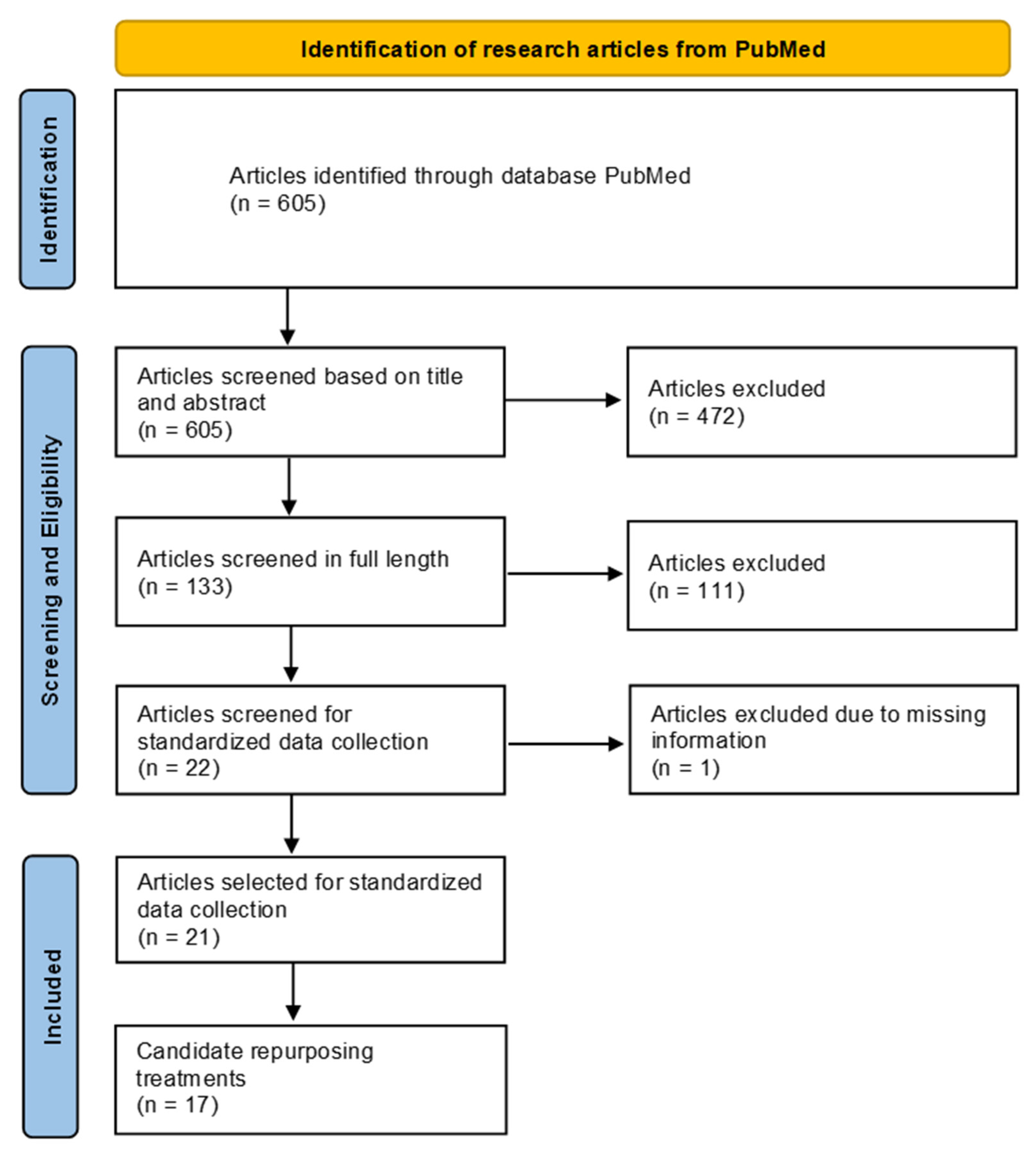{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
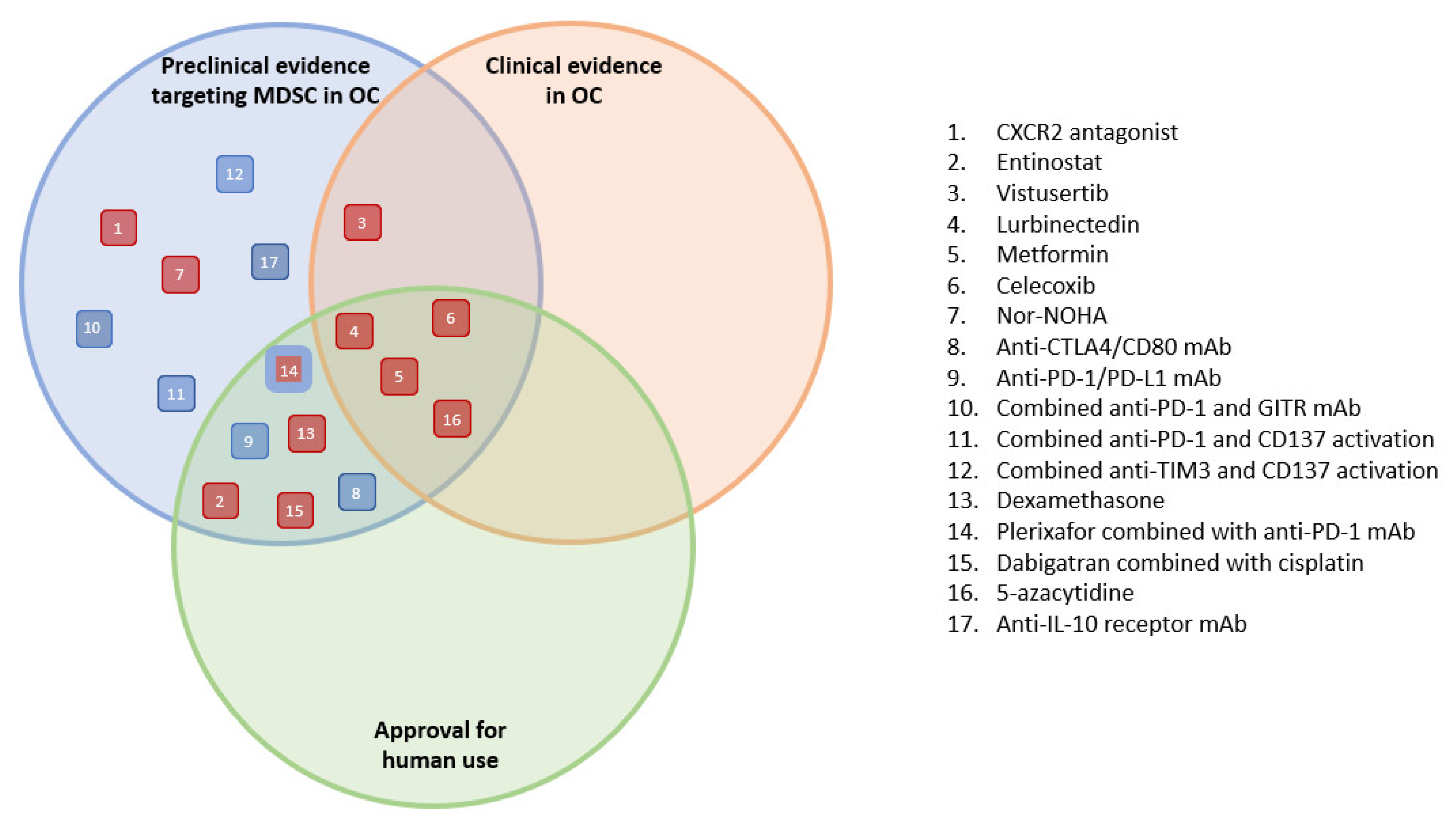
3. Results
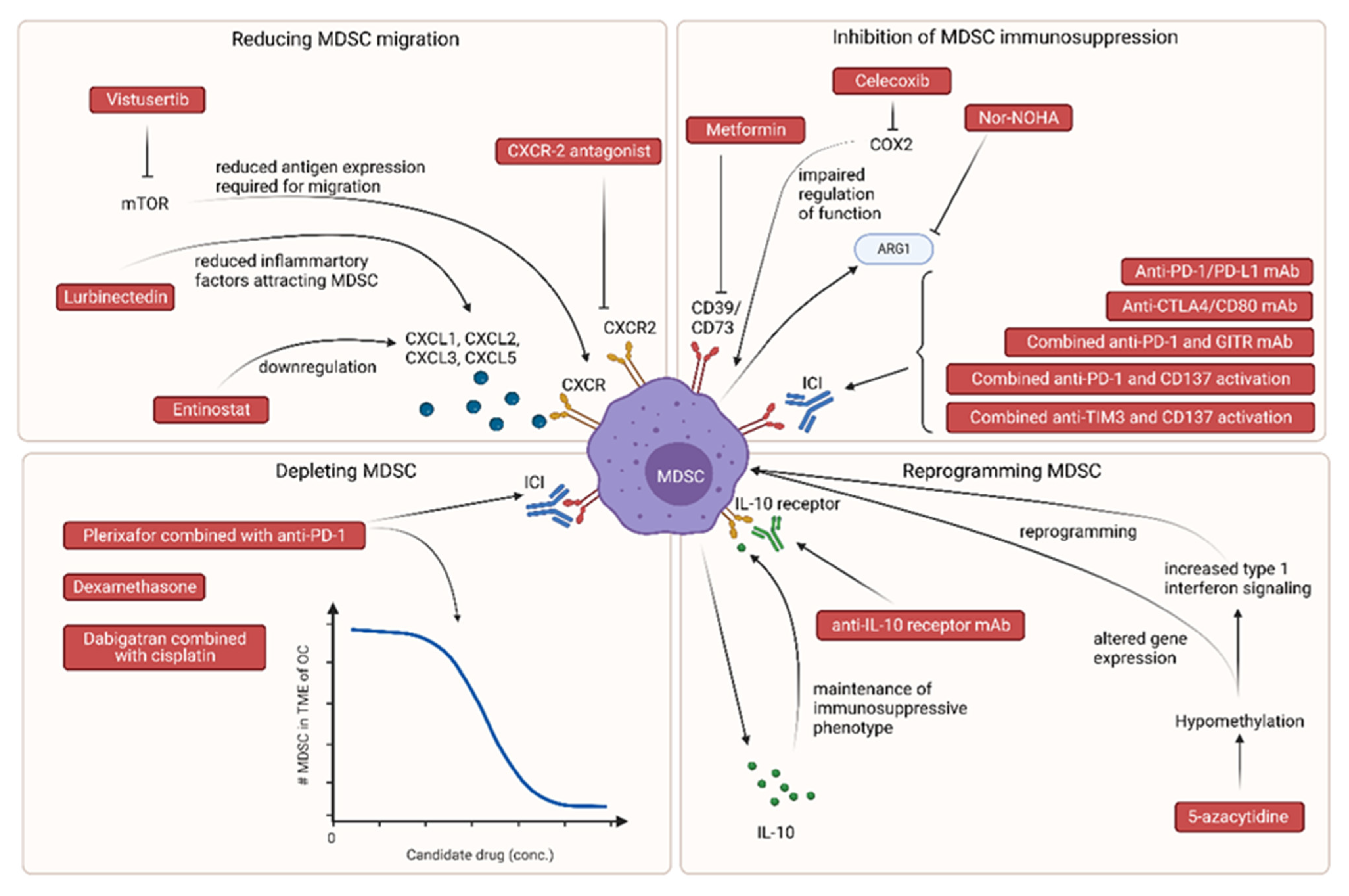
3.1. Candidate Treatments Reducing MDSC Migration to the Tumor Microenvironment
3.1.1. CXCR-2 Antagonists
3.1.2. Entinostat
3.1.3. Vistusertib
3.1.4. Lurbinectedin
3.2. Candidate Treatments Inhibiting MDSC Immunosuppressive Functions
3.2.1. Metformin
3.2.2. Celecoxib
3.2.3. Nor-NOHA
3.2.4. Anti-CTLA4/CD80 mAb
3.2.5. Anti-PD-1/PD-L1 mAb
Combined Anti-PD-1 and GITR mAb
Combined Anti-PD-1 and CD137 Activation
3.2.6. Combined Anti-TIM3 and CD137 Activation
3.3. Candidate Treatments Depleting MDSC
3.3.1. Dexamethasone
3.3.2. CXCR4 Antagonist, Plerixafor, Combined with Anti-PD-1
3.3.3. Dabigatran Combined with Cisplatin
3.4. Candidate Treatments Able to Reprogram MDSC
3.4.1. 5-Azacytidine
3.4.2. Anti-IL-10 Receptor mAb
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Brett, M.R.; Jennifer, B.P.; Thomas, A.S. Epidemiology of Ovarian Cancer: A Review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, J. Staging Classification for Cancer of the Ovary, Fallopian Tube, and Peritoneum. Int. J. Gynecol. Obstet. 2014, 124, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Blair, A.R.; Casas, C.M. Gynecologic Cancers. Prim. Care Clin. Off. Pract. 2009, 36, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Jelovac, D.; Armstrong, D.K. Recent Progress in the Diagnosis and Treatment of Ovarian Cancer. CA Cancer J. Clin. 2011, 61, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian Cancer. Nat. Rev. Dis. Prim. 2016, 2, 16061. [Google Scholar] [CrossRef]
- Lisio, M.-A.; Fu, L.; Goyeneche, A.; Gao, Z.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef] [Green Version]
- Doherty, J.A.; Jensen, A.; Kelemen, L.E.; Pearce, C.L.; Poole, E.; Schildkraut, J.M.; Terry, K.L.; Tworoger, S.S.; Epidemiology Working Group Steering Committee; Ovarian Cancer Association Consortium Members of the EWG SC, in Alphabetical Order; et al. Current Gaps in Ovarian Cancer Epidemiology: The Need for New Population-Based Research. JNCI J. Natl. Cancer Inst. 2017, 109, djx144. [Google Scholar] [CrossRef]
- Baert, T.; Ferrero, A.; Sehouli, J.; O’Donnell, D.M.; González-Martín, A.; Joly, F.; van der Velden, J.; Blecharz, P.; Tan, D.S.P.; Querleu, D.; et al. The Systemic Treatment of Recurrent Ovarian Cancer Revisited. Ann. Oncol. 2021, 32, 710–725. [Google Scholar] [CrossRef]
- Maiorano, B.A.; Maiorano, M.F.P.; Lorusso, D.; Maiello, E. Ovarian Cancer in the Era of Immune Checkpoint Inhibitors: State of the Art and Future Perspectives. Cancers 2021, 13, 4438. [Google Scholar] [CrossRef]
- Gaillard, S.L.; Secord, A.A.; Monk, B. The Role of Immune Checkpoint Inhibition in the Treatment of Ovarian Cancer. Gynecol. Oncol. Res. Pract. 2016, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, L.; Zitvogel, L.; Eggermont, A.; Marabelle, A. PD-Loma: A Cancer Entity with a Shared Sensitivity to the PD-1/PD-L1 Pathway Blockade. Br. J. Cancer 2019, 120, 3–5. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.N.; Bookman, M.; Sehouli, J.; Miller, A.; Anderson, C.; Scambia, G.; Myers, T.; Taskiran, C.; Robison, K.; Mäenpää, J.; et al. Atezolizumab, Bevacizumab, and Chemotherapy for Newly Diagnosed Stage III or IV Ovarian Cancer: Placebo-Controlled Randomized Phase III Trial (IMagyn050/GOG 3015/ENGOT-OV39). J. Clin. Oncol. 2021, 39, 1842–1855. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.B.; Buchsbaum, D.J.; Straughn, J.M.; Randall, T.D.; Arend, R.C. Ovarian Cancer and the Immune System—The Role of Targeted Therapies. Gynecol. Oncol. 2016, 142, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Coosemans, A.; Vankerckhoven, A.; Baert, T.; Boon, L.; Ruts, H.; Riva, M.; Blagden, S.; Delforge, M.; Concin, N.; Mirza, M.R.; et al. Combining Conventional Therapy with Immunotherapy: A Risky Business? Eur. J. Cancer 2019, 113, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Muenst, S.; Läubli, H.; Soysal, S.D.; Zippelius, A.; Tzankov, A.; Hoeller, S. The Immune System and Cancer Evasion Strategies: Therapeutic Concepts. J. Intern. Med. 2016, 279, 541–562. [Google Scholar] [CrossRef]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A High M1/M2 Ratio of Tumor-Associated Macrophages Is Associated with Extended Survival in Ovarian Cancer Patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Vankerckhoven, A.; Wouters, R.; Mathivet, T.; Ceusters, J.; Baert, T.; Van Hoylandt, A.; Gerhardt, H.; Vergote, I.; Coosemans, A. Opposite Macrophage Polarization in Different Subsets of Ovarian Cancer: Observation from a Pilot Study. Cells 2020, 9, 305. [Google Scholar] [CrossRef] [Green Version]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ Tumor-Infiltrating Lymphocytes and a High CD8+/Regulatory T Cell Ratio Are Associated with Favorable Prognosis in Ovarian Cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Deng, Z.; Peng, Y.; Han, L.; Liu, J.; Wang, L.; Li, B.; Zhao, J.; Jiao, S.; Wei, H. Ascites-Derived IL-6 and IL-10 Synergistically Expand CD14+HLA-DR-/Low Myeloid-Derived Suppressor Cells in Ovarian Cancer Patients. Oncotarget 2017, 8, 76843–76856. [Google Scholar] [CrossRef] [Green Version]
- Cui, T.X.; Kryczek, I.; Zhao, L.; Zhao, E.; Kuick, R.; Roh, M.H.; Vatan, L.; Szeliga, W.; Mao, Y.; Thomas, D.G.; et al. Myeloid-Derived Suppressor Cells Enhance Stemness of Cancer Cells by Inducing MicroRNA101 and Suppressing the Corepressor CtBP2. Immunity 2013, 39, 611–621. [Google Scholar] [CrossRef] [Green Version]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines 2016, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Bronte, V.; Serafini, P.; Apolloni, E.; Zanovello, P. Tumor-Induced Immune Dysfunctions Caused by Myeloid Suppressor Cells. J. Immunother. 2001, 24, 431–446. [Google Scholar] [CrossRef]
- Hanson, E.M.; Clements, V.K.; Sinha, P.; Ilkovitch, D.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells Down-Regulate L-Selectin Expression on CD4+ and CD8+ T Cells. J. Immunol. 2009, 183, 937–944. [Google Scholar] [CrossRef] [Green Version]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front. Immunol. 2018, 9, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trovato, R.; Canè, S.; Petrova, V.; Sartoris, S.; Ugel, S.; De Sanctis, F. The Engagement Between MDSCs and Metastases: Partners in Crime. Front. Oncol. 2020, 10, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaraj, S.; Gabrilovich, D.I. Tumor Escape Mechanism Governed by Myeloid-Derived Suppressor Cells. Cancer Res. 2008, 68, 2561–2563. [Google Scholar] [CrossRef] [Green Version]
- Ohl, K.; Tenbrock, K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 2018, 9, 2499. [Google Scholar] [CrossRef] [Green Version]
- Yaseen, M.M.; Abuharfeil, N.M.; Darmani, H.; Daoud, A. Mechanisms of Immune Suppression by Myeloid-Derived Suppressor Cells: The Role of Interleukin-10 as a Key Immunoregulatory Cytokine. Open Biol. 2020, 10, 200111. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Fierro, A.; Dueñas-González, A. Drug Repurposing for Cancer Therapy, Easier Said than Done. Semin. Cancer Biol. 2021, 68, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Parvathaneni, V.; Kulkarni, N.S.; Muth, A.; Gupta, V. Drug Repurposing: A Promising Tool to Accelerate the Drug Discovery Process. Drug Discov. Today 2019, 24, 2076–2085. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.; Duarte, D.; Vale, N. Drug Repurposing in Cancer Therapy: Influence of Patient’s Genetic Background in Breast Cancer Treatment. Int. J. Mol. Sci. 2022, 23, 4280. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Verbaanderd, C.; Sukhatme, V.; Rica Capistrano, I.; Crispino, S.; Gyawali, B.; Rooman, I.; Van Nuffel, A.M.T.; Meheus, L.; Sukhatme, V.P.; et al. Redo_DB: The Repurposing Drugs in Oncology Database. Ecancermedicalscience 2018, 12, 886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.K.B.; Tiong, K.H.; Chang, J.K.; Liew, C.S.; Abdul Rahman, Z.A.; Tan, A.C.; Khang, T.F.; Cheong, S.C. DeSigN: Connecting Gene Expression with Therapeutics for Drug Repurposing and Development. BMC Genom. 2017, 18, 934. [Google Scholar] [CrossRef] [Green Version]
- Hintzsche, J.D.; Yoo, M.; Kim, J.; Amato, C.M.; Robinson, W.A.; Tan, A.C. IMPACT Web Portal: Oncology Database Integrating Molecular Profiles with Actionable Therapeutics. BMC Med. Genom. 2018, 11, 26. [Google Scholar] [CrossRef] [Green Version]
- Amelio, I.; Gostev, M.; Knight, R.A.; Willis, A.E.; Melino, G.; Antonov, A. V DRUGSURV: A Resource for Repositioning of Approved and Experimental Drugs in Oncology Based on Patient Survival Information. Cell Death Dis. 2014, 5, e1051. [Google Scholar] [CrossRef] [Green Version]
- Dodds, M.; Xiong, Y.; Mouksassi, S.; Kirkpatrick, C.M.; Hui, K.; Doyle, E.; Patel, K.; Cox, E.; Wesche, D.; Brown, F.; et al. Model-informed Drug Repurposing: A Pharmacometric Approach to Novel Pathogen Preparedness, Response and Retrospection. Br. J. Clin. Pharmacol. 2021, 87, 3388–3397. [Google Scholar] [CrossRef]
- Romano, F.; D’Agate, S.; Pasqua, O. Model-Informed Repurposing of Medicines for SARS-CoV-2: Extrapolation of Antiviral Activity and Dose Rationale for Paediatric Patients. Pharmaceutics 2021, 13, 1299. [Google Scholar] [CrossRef]
- Ouzzani, M.; Hammady, H.; Fedorowicz, Z.; Elmagarmid, A. Rayyan—A Web and Mobile App for Systematic Reviews. Syst. Rev. 2016, 5, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesolowski, R.; Markowitz, J.; Carson, W.E. Myeloid Derived Suppressor Cells—A New Therapeutic Target in the Treatment of Cancer. J. Immunother. Cancer 2013, 1, 10. [Google Scholar] [CrossRef] [Green Version]
- Bradley, M.; Bond, M.; Manini, J.; Brown, Z.; Charlton, S. SB265610 Is an Allosteric, Inverse Agonist at the Human CXCR2 Receptor. Br. J. Pharmacol. 2009, 158, 328–338. [Google Scholar] [CrossRef] [Green Version]
- O’Byrne, P.M.; Metev, H.; Puu, M.; Richter, K.; Keen, C.; Uddin, M.; Larsson, B.; Cullberg, M.; Nair, P. Efficacy and Safety of a CXCR2 Antagonist, AZD5069, in Patients with Uncontrolled Persistent Asthma: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Respir. Med. 2016, 4, 797–806. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. European Medicines Agency Decision P/0103/2014. Available online: https://www.ema.europa.eu/en/documents/pip-decision/p/0103/2014-ema-decision-2-may-2014-agreement-paediatric-investigation-plan-granting-deferral-granting_en.pdf (accessed on 11 April 2023).
- Guo, C.; Sharp, A.; Vogl, U.; Colombo, I.; Stathis, A. 454O—A Phase (Ph) I/II Trial of the CXCR2 Antagonist AZD5069 in Combination with Enzalutamide (ENZA) in Patients (Pts) with Metastatic Castration Resistant Prostate Cancer (MCRPC). Ann. Oncol. 2022, 33, S745. [Google Scholar] [CrossRef]
- Taki, M.; Abiko, K.; Baba, T.; Hamanishi, J.; Yamaguchi, K.; Murakami, R.; Yamanoi, K.; Horikawa, N.; Hosoe, Y.; Nakamura, E.; et al. Snail Promotes Ovarian Cancer Progression by Recruiting Myeloid-Derived Suppressor Cells via CXCR2 Ligand Upregulation. Nat. Commun. 2018, 9, 1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriques, T.B.; dos Santos, D.Z.; dos Santos Guimarães, I.; Tessarollo, N.G.; Lyra-Junior, P.C.M.; Mesquita, P.; Pádua, D.; Amaral, A.L.; Cavadas, B.; Pereira, L.; et al. Inhibition of CXCR2 Plays a Pivotal Role in Re-Sensitizing Ovarian Cancer to Cisplatin Treatment. Aging 2021, 13, 13405–13420. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Public Summary of Opinion on Orphan Designation Entinostat for the Treatment of Hodgkin’s Lymphoma; European Medicines Agency: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Garmpis, N.; Damaskos, C.; Garmpi, A.; Valsami, S.; Dimitroulis, D. Pharmacoepigenetics of Histone Deacetylase Inhibitors in Cancer. In Pharmacoepigenetics; Elsevier: Amsterdam, The Netherlands, 2019; pp. 501–521. [Google Scholar]
- Connolly, R.M.; Rudek, M.A.; Piekarz, R. Entinostat: A Promising Treatment Option for Patients with Advanced Breast Cancer. Future Oncol. 2017, 13, 1137–1148. [Google Scholar] [CrossRef]
- McCaw, T.R.; Goel, N.; Brooke, D.J.; Katre, A.A.; Londoño, A.I.; Smith, H.J.; Randall, T.D.; Arend, R.C. Class I Histone Deacetylase Inhibition Promotes CD8 T Cell Activation in Ovarian Cancer. Cancer Med. 2021, 10, 709–717. [Google Scholar] [CrossRef]
- Cadoo, K.A.; Meyers, M.L.; Burger, R.A.; Armstrong, D.K.; Penson, R.T.; Gordon, M.S.; Fleming, G.F.; Moroney, J.W.; Hamilton, E.P.; Duska, L.R.; et al. A Phase II Randomized Study of Avelumab plus Entinostat versus Avelumab plus Placebo in Patients (Pts) with Advanced Epithelial Ovarian Cancer (EOC). J. Clin. Oncol. 2019, 37, 5511. [Google Scholar] [CrossRef]
- Lapointe, S.; Mason, W.; MacNeil, M.; Harlos, C.; Tsang, R.; Sederias, J.; Luchman, H.A.; Weiss, S.; Rossiter, J.P.; Tu, D.; et al. A Phase I Study of Vistusertib (Dual MTORC1/2 Inhibitor) in Patients with Previously Treated Glioblastoma Multiforme: A CCTG Study. Investig. New Drugs 2020, 38, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Pi, R.; Yang, Y.; Hu, X.; Li, H.; Shi, H.; Liu, Y.; Wang, X.; Tong, A.; Lu, T.; Wei, Y.; et al. Dual MTORC1/2 Inhibitor AZD2014 Diminishes Myeloid-Derived Suppressor Cells Accumulation in Ovarian Cancer and Delays Tumor Growth. Cancer Lett. 2021, 523, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Bouyahya, A.; El Allam, A.; Aboulaghras, S.; Bakrim, S.; El Menyiy, N.; Alshahrani, M.M.; Al Awadh, A.A.; Benali, T.; Lee, L.-H.; El Omari, N.; et al. Targeting MTOR as a Cancer Therapy: Recent Advances in Natural Bioactive Compounds and Immunotherapy. Cancers 2022, 14, 5520. [Google Scholar] [CrossRef] [PubMed]
- Wong Te Fong, A.-C.; Thavasu, P.; Gagrica, S.; Swales, K.E.; Leach, M.O.; Cosulich, S.C.; Chung, Y.-L.; Banerji, U. Evaluation of the Combination of the Dual M-TORC1/2 Inhibitor Vistusertib (AZD2014) and Paclitaxel in Ovarian Cancer Models. Oncotarget 2017, 8, 113874–113884. [Google Scholar] [CrossRef] [Green Version]
- Pancholi, S.; Leal, M.F.; Ribas, R.; Simigdala, N.; Schuster, E.; Chateau-Joubert, S.; Zabaglo, L.; Hills, M.; Dodson, A.; Gao, Q.; et al. Combination of MTORC1/2 Inhibitor Vistusertib plus Fulvestrant in Vitro and in Vivo Targets Oestrogen Receptor-Positive Endocrine-Resistant Breast Cancer. Breast Cancer Res. 2019, 21, 135. [Google Scholar] [CrossRef]
- Banerjee, S.; Giannone, G.; Clamp, A.R.; Ennis, D.P.; Glasspool, R.M.; Herbertson, R.; Krell, J.; Riisnaes, R.; Mirza, H.B.; Cheng, Z.; et al. Efficacy and Safety of Weekly Paclitaxel Plus Vistusertib vs. Paclitaxel Alone in Patients With Platinum-Resistant Ovarian High-Grade Serous Carcinoma: The OCTOPUS Multicenter, Phase 2, Randomized Clinical Trial. JAMA Oncol. 2023, 9, 675–682. [Google Scholar] [CrossRef]
- Musacchio, L.; Cicala, C.M.; Salutari, V.; Camarda, F.; Carbone, M.V.; Ghizzoni, V.; Giudice, E.; Nero, C.; Perri, M.T.; Ricci, C.; et al. Preclinical and Clinical Evidence of Lurbinectedin in Ovarian Cancer: Current Status and Future Perspectives. Front. Oncol. 2022, 12, 585. [Google Scholar] [CrossRef]
- Singh, S.; Jaigirdar, A.A.; Mulkey, F.; Cheng, J.; Hamed, S.S.; Li, Y.; Liu, J.; Zhao, H.; Goheer, A.; Helms, W.S.; et al. FDA Approval Summary: Lurbinectedin for the Treatment of Metastatic Small Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 2378–2382. [Google Scholar] [CrossRef]
- Dumoulin, D.W.; Cantini, L.; Cornelissen, R.; Vink, M.; Klaase, L.; Slooff, K.; Tebayna, N.; Mankor, J.M.; Baart, S.J.; Hendriks, R.; et al. Lurbinectedin Shows Clinical Activity and Immune-Modulatory Functions in Patients with Pre-Treated Small Cell Lung Cancer and Malignant Pleural Mesothelioma. Eur. J. Cancer 2022, 172, 357–366. [Google Scholar] [CrossRef]
- Belgiovine, C.; Bello, E.; Liguori, M.; Craparotta, I.; Mannarino, L.; Paracchini, L.; Beltrame, L.; Marchini, S.; Galmarini, C.M.; Mantovani, A.; et al. Lurbinectedin Reduces Tumour-Associated Macrophages and the Inflammatory Tumour Microenvironment in Preclinical Models. Br. J. Cancer 2017, 117, 628–638. [Google Scholar] [CrossRef]
- Gaillard, S.; Oaknin, A.; Ray-Coquard, I.; Vergote, I.; Scambia, G.; Colombo, N.; Fernandez, C.; Alfaro, V.; Kahatt, C.; Nieto, A.; et al. Lurbinectedin versus Pegylated Liposomal Doxorubicin or Topotecan in Patients with Platinum-Resistant Ovarian Cancer: A Multicenter, Randomized, Controlled, Open-Label Phase 3 Study (CORAIL). Gynecol. Oncol. 2021, 163, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Garralda, E.; Stathis, A.; Szyldergemajn, S.; Hyman, D.; Boni, V.; Griguolo, G.; Martinez, E.J.; Makker, V.; Canziani, L.; et al. Lurbinectedin (PM01183) plus Paclitaxel (P), Recommended Dose (RD) Expansion Results with or without the Addition of Bevacizumab (Bev) in Patients (Pts) with Selected Solid Tumors. Ann. Oncol. 2016, 27, vi125. [Google Scholar] [CrossRef]
- Gong, L.; Goswami, S.; Giacomini, K.M.; Altman, R.B.; Klein, T.E. Metformin Pathways pharmacokinetics and pharmacodynamics. Pharm. Genom. 2012, 22, 820–827. [Google Scholar] [CrossRef] [Green Version]
- Nasri, H.; Rafieian-Kopaei, M. Metformin: Current Knowledge. J. Res. Med. Sci. 2014, 19, 658–664. [Google Scholar]
- Kuan, I.H.S.; Wright, D.F.B.; Duffull, S.B.; Zhu, X. Understanding the Association between Metformin Plasma Concentrations and Lactate. Br. J. Clin. Pharmacol. 2021, 87, 700–701. [Google Scholar] [CrossRef] [PubMed]
- Johansson, S.; Read, J.; Oliver, S.; Steinberg, M.; Li, Y.; Lisbon, E.; Mathews, D.; Leese, P.T.; Martin, P. Pharmacokinetic Evaluations of the Co-Administrations of Vandetanib and Metformin, Digoxin, Midazolam, Omeprazole or Ranitidine. Clin. Pharmacokinet. 2014, 53, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, C.; Jacobs, T.F. Metformin; StatPearls: Tampa, FL, USA, 2022. [Google Scholar]
- Li, L.; Wang, L.; Li, J.; Fan, Z.; Yang, L.; Zhang, Z.; Zhang, C.; Yue, D.; Qin, G.; Zhang, T.; et al. Metformin-Induced Reduction of CD39 and CD73 Blocks Myeloid-Derived Suppressor Cell Activity in Patients with Ovarian Cancer. Cancer Res. 2018, 78, 1779–1791. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-Y.; Lim, M.C.; Baek, M.-H.; Park, Y.-H.; Kim, S. Impact of Metformin on Survival Outcome in Ovarian Cancer: A Nationwide Population-Based Cohort Study. J. Gynecol. Oncol. 2021, 32, e65. [Google Scholar] [CrossRef]
- European Medicines Agency. Celecoxib Article 31 Referral Annex I II III. Available online: https://www.ema.europa.eu/en/documents/referral/celecoxib-article-31-referral-annex-i-ii-iii_en.pdf (accessed on 5 April 2023).
- Goldenberg, M.M. Celecoxib, a Selective Cyclooxygenase-2 Inhibitor for the Treatment of Rheumatoid Arthritis and Osteoarthritis. Clin. Ther. 1999, 21, 1497–1513. [Google Scholar] [CrossRef]
- Gong, L.; Thorn, C.F.; Bertagnolli, M.M.; Grosser, T.; Altman, R.B.; Klein, T.E. Celecoxib Pathways. Pharm. Genom. 2012, 22, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.L.; Obermajer, N.; Odunsi, K.; Edwards, R.P.; Kalinski, P. Synergistic COX2 Induction by IFNγ and TNFα Self-Limits Type-1 Immunity in the Human Tumor Microenvironment. Cancer Immunol. Res. 2016, 4, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legge, F.; Paglia, A.; D’Asta, M.; Fuoco, G.; Scambia, G.; Ferrandina, G. Phase II Study of the Combination Carboplatin plus Celecoxib in Heavily Pre-Treated Recurrent Ovarian Cancer Patients. BMC Cancer 2011, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Kövamees, O.; Shemyakin, A.; Pernow, J. Effect of Arginase Inhibition on Ischemia-Reperfusion Injury in Patients with Coronary Artery Disease with and without Diabetes Mellitus. PLoS ONE 2014, 9, e103260. [Google Scholar] [CrossRef]
- Kövamees, O.; Shemyakin, A.; Checa, A.; Wheelock, C.E.; Lundberg, J.O.; Östenson, C.-G.; Pernow, J. Arginase Inhibition Improves Microvascular Endothelial Function in Patients With Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2016, 101, 3952–3958. [Google Scholar] [CrossRef]
- Bak, S.P.; Alonso, A.; Turk, M.J.; Berwin, B. Murine Ovarian Cancer Vascular Leukocytes Require Arginase-1 Activity for T Cell Suppression. Mol. Immunol. 2008, 46, 258–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javle, M.M.; Bridgewater, J.A.; Gbolahan, O.B.; Jungels, C.; Cho, M.T.; Papadopoulos, K.P.; Thistlethwaite, F.C.; Canon, J.-L.R.; Cheng, L.; Ioannidis, S.; et al. A Phase I/II Study of Safety and Efficacy of the Arginase Inhibitor INCB001158 plus Chemotherapy in Patients (Pts) with Advanced Biliary Tract Cancers. J. Clin. Oncol. 2021, 39, 311. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- European Medicines Agency. Yervoy EPAR Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/yervoy-epar-product-information_en.pdf (accessed on 6 April 2023).
- Yang, R.; Cai, Z.; Zhang, Y.; Yutzy, W.H.; Roby, K.F.; Roden, R.B.S. CD80 in Immune Suppression by Mouse Ovarian Carcinoma–Associated Gr-1+CD11b+ Myeloid Cells. Cancer Res. 2006, 66, 6807–6815. [Google Scholar] [CrossRef] [Green Version]
- Zamarin, D.; Burger, R.A.; Sill, M.W.; Powell, D.J.; Lankes, H.A.; Feldman, M.D.; Zivanovic, O.; Gunderson, C.; Ko, E.; Mathews, C.; et al. Randomized Phase II Trial of Nivolumab Versus Nivolumab and Ipilimumab for Recurrent or Persistent Ovarian Cancer: An NRG Oncology Study. J. Clin. Oncol. 2020, 38, 1814–1823. [Google Scholar] [CrossRef]
- Ai, L.; Chen, J.; Yan, H.; He, Q.; Luo, P.; Xu, Z.; Yang, X. Research Status and Outlook of PD-1/PD-L1 Inhibitors for Cancer Therapy. Drug Des. Dev. Ther. 2020, 14, 3625–3649. [Google Scholar] [CrossRef]
- Nuñez, N.G.; Berner, F.; Friebel, E.; Unger, S.; Wyss, N.; Gomez, J.M.; Purde, M.-T.; Niederer, R.; Porsch, M.; Lichtensteiger, C.; et al. Immune Signatures Predict Development of Autoimmune Toxicity in Patients with Cancer Treated with Immune Checkpoint Inhibitors. Med 2023, 4, 113–129.e7. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, J. Immune Checkpoint Inhibitors and Their Side Effects. Pathology 2019, 51, S17. [Google Scholar] [CrossRef]
- Liu, Y.; Zeng, B.; Zhang, Z.; Zhang, Y.; Yang, R. B7-H1 on Myeloid-Derived Suppressor Cells in Immune Suppression by a Mouse Model of Ovarian Cancer. Clin. Immunol. 2008, 129, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Paijens, S.T.; Leffers, N.; Daemen, T.; Helfrich, W.; Boezen, H.M.; Cohlen, B.J.; Melief, C.J.; de Bruyn, M.; Nijman, H.W. Antigen-Specific Active Immunotherapy for Ovarian Cancer. Cochrane Database Syst. Rev. 2018, 9, CD007287. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Xu, X.; Zhang, B.; Zhang, R.; Ji, H.; Wang, X. Combined PD-1 Blockade and GITR Triggering Induce a Potent Antitumor Immunity in Murine Cancer Models and Synergizes with Chemotherapeutic Drugs. J. Transl. Med. 2014, 12, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappasodi, R.; Sirard, C.; Li, Y.; Budhu, S.; Abu-Akeel, M.; Liu, C.; Yang, X.; Zhong, H.; Newman, W.; Qi, J.; et al. Rational Design of Anti-GITR-Based Combination Immunotherapy. Nat. Med. 2019, 25, 759–766. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, X.; Cheng, D.; Xia, Z.; Luan, M.; Zhang, S. PD-1 Blockade and OX40 Triggering Synergistically Protects against Tumor Growth in a Murine Model of Ovarian Cancer. PLoS ONE 2014, 9, e89350. [Google Scholar] [CrossRef]
- Chu, D.-T.; Bac, N.D.; Nguyen, K.-H.; Tien, N.L.B.; Thanh, V.V.; Nga, V.T.; Ngoc, V.T.N.; Anh Dao, D.T.; Hoan, L.N.; Hung, N.P.; et al. An Update on Anti-CD137 Antibodies in Immunotherapies for Cancer. Int. J. Mol. Sci. 2019, 20, 1822. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H. Update of Early Phase Clinical Trials in Cancer Immunotherapy. BMB Rep. 2021, 54, 70–88. [Google Scholar] [CrossRef]
- Massarelli, E. Clinical Safety and Efficacy Assessment of the CD137 Agonist Urelumab Alone and in Combination with Nivolumab in Patients with Hematologic and Solid Tumor Malignancies. In Proceedings of the 31st Annual Meeting & Associated Programs of the Society for Immunotherapy of Cancer (SITC)’s 2016, National Harbor, MD, USA, 11–16 November 2016. [Google Scholar]
- Segal, N.H.; He, A.R.; Doi, T.; Levy, R.; Bhatia, S.; Pishvaian, M.J.; Cesari, R.; Chen, Y.; Davis, C.B.; Huang, B.; et al. Phase I Study of Single-Agent Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 1816–1823. [Google Scholar] [CrossRef] [Green Version]
- Dubrot, J.; Azpilikueta, A.; Alfaro, C.; Murillo, O.; Arina, A.; Berraondo, P.; Hervás-Stubbs, S.; Melero, I. Absence of Surface Expression of CD137 (4-1BB) on Myeloid-Derived Suppressor Cells. Inmunología 2007, 26, 121–126. [Google Scholar] [CrossRef]
- Guo, Z.; Cheng, D.; Xia, Z.; Luan, M.; Wu, L.; Wang, G.; Zhang, S. Combined TIM-3 Blockade and CD137 Activation Affords the Long-Term Protection in a Murine Model of Ovarian Cancer. J. Transl. Med. 2013, 11, 215. [Google Scholar] [CrossRef] [Green Version]
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 Finds Its Place in the Cancer Immunotherapy Landscape. J. Immunother. Cancer 2020, 8, e000911. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.; Tang, S.; Powderly, J.; Balmanoukian, A.; Janik, J.; Hoyle, P.; Wei, W.; Gong, X.; Hamid, O. 730MO—First-in-Human Phase I Study of INCAGN02390, a TIM-3 Monoclonal Antibody Antagonist in Patients with Advanced Malignancies. Ann. Oncol. 2022, 33, S331–S355. [Google Scholar] [CrossRef]
- Curigliano, G.; Gelderblom, H.; Mach, N.; Doi, T.; Tai, D.; Forde, P.M.; Sarantopoulos, J.; Bedard, P.L.; Lin, C.-C.; Hodi, F.S.; et al. Phase I/Ib Clinical Trial of Sabatolimab, an Anti–TIM-3 Antibody, Alone and in Combination with Spartalizumab, an Anti–PD-1 Antibody, in Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3620–3629. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.M.; Lawrence, T.; Kleiman, A.; Warden, P.; Medghalchi, M.; Tuckermann, J.; Saklatvala, J.; Clark, A.R. Antiinflammatory Effects of Dexamethasone Are Partly Dependent on Induction of Dual Specificity Phosphatase 1. J. Exp. Med. 2006, 203, 1883–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.H.; Hassan, A. Dexamethasone for the Treatment of Coronavirus Disease (COVID-19): A Review. SN Compr. Clin. Med. 2020, 2, 2637–2646. [Google Scholar] [CrossRef]
- Tomazini, B.M.; Maia, I.S.; Cavalcanti, A.B.; Berwanger, O.; Rosa, R.G.; Veiga, V.C.; Avezum, A.; Lopes, R.D.; Bueno, F.R.; Silva, M.V.A.O.; et al. Effect of Dexamethasone on Days Alive and Ventilator-Free in Patients With Moderate or Severe Acute Respiratory Distress Syndrome and COVID-19. JAMA 2020, 324, 1307. [Google Scholar] [CrossRef]
- Pinzón, M.A.; Ortiz, S.; Holguín, H.; Betancur, J.F.; Cardona Arango, D.; Laniado, H.; Arias Arias, C.; Muñoz, B.; Quiceno, J.; Jaramillo, D.; et al. Dexamethasone vs Methylprednisolone High Dose for Covid-19 Pneumonia. PLoS ONE 2021, 16, e0252057. [Google Scholar] [CrossRef]
- Bouadma, L.; Mekontso-Dessap, A.; Burdet, C.; Merdji, H.; Poissy, J.; Dupuis, C.; Guitton, C.; Schwebel, C.; Cohen, Y.; Bruel, C.; et al. High-Dose Dexamethasone and Oxygen Support Strategies in Intensive Care Unit Patients With Severe COVID-19 Acute Hypoxemic Respiratory Failure. JAMA Intern. Med. 2022, 182, 906. [Google Scholar] [CrossRef]
- European Medicines Agency. Neofordex EPAR Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/neofordex-epar-product-information_en.pdf (accessed on 5 April 2023).
- Buchman, A.L. Side Effects of Corticosteroid Therapy. J. Clin. Gastroenterol. 2001, 33, 289–294. [Google Scholar] [CrossRef]
- Ferro, A.; Graikioti, D.; Gezer, E.; Athanassopoulos, C.M.; Cuendet, M. Entinostat-Bortezomib Hybrids against Multiple Myeloma. Molecules 2023, 28, 1456. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.-T.; Sun, S.-P.; Wu, J.-I.; Wang, L.-H. Low-Dose Glucocorticoids Suppresses Ovarian Tumor Growth and Metastasis in an Immunocompetent Syngeneic Mouse Model. PLoS ONE 2017, 12, e0178937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Shen, X.-F.; Cao, K.; Ding, J.; Kang, X.; Guan, W.; Ding, Y.; Liu, B.; Du, J.-F. Dexamethasone-Induced Myeloid-Derived Suppressor Cells Prolong Allo Cardiac Graft Survival through INOS- and Glucocorticoid Receptor-Dependent Mechanism. Front. Immunol. 2018, 9, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency. Mozobil EPAR Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/mozobil-epar-product-information_en.pdf (accessed on 6 April 2023).
- Slater, S. Plerixafor. J. Adv. Pract. Oncol. 2012, 3, 49–54. [Google Scholar] [PubMed]
- Jiang, Y.; Wu, X.; Shi, B.; Wu, W.; Yin, G. Expression of Chemokine CXCL12 and Its Receptor CXCR4 in Human Epithelial Ovarian Cancer: An Independent Prognostic Factor for Tumor Progression. Gynecol. Oncol. 2006, 103, 226–233. [Google Scholar] [CrossRef]
- Zeng, Y.; Li, B.; Liang, Y.; Reeves, P.M.; Qu, X.; Ran, C.; Liu, Q.; Callahan, M.V.; Sluder, A.E.; Gelfand, J.A.; et al. Dual Blockade of CXCL12-CXCR4 and PD-1–PD-L1 Pathways Prolongs Survival of Ovarian Tumor–Bearing Mice by Prevention of Immunosuppression in the Tumor Microenvironment. FASEB J. 2019, 33, 6596–6608. [Google Scholar] [CrossRef] [PubMed]
- Biasci, D.; Smoragiewicz, M.; Connell, C.M.; Wang, Z.; Gao, Y.; Thaventhiran, J.E.D.; Basu, B.; Magiera, L.; Johnson, T.I.; Bax, L.; et al. CXCR4 Inhibition in Human Pancreatic and Colorectal Cancers Induces an Integrated Immune Response. Proc. Natl. Acad. Sci. USA 2020, 117, 28960–28970. [Google Scholar] [CrossRef]
- Stangier, J.; Clemens, A. Pharmacology, Pharmacokinetics, and Pharmacodynamics of Dabigatran Etexilate, an Oral Direct Thrombin Inhibitor. Clin. Appl. Thromb. Hemost. 2009, 15, 9S–16S. [Google Scholar] [CrossRef]
- European Medicines Agency. Pradaxa EPAR Medicine Overview. Available online: https://www.ema.europa.eu/en/documents/overview/pradaxa-epar-medicine-overview_en.pdf (accessed on 11 April 2023).
- Zhou, Y.; Yao, Z.; Zhu, L.; Tang, Y.; Chen, J.; Wu, J. Safety of Dabigatran as an Anticoagulant: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2021, 12, 626063. [Google Scholar] [CrossRef]
- Khorana, A.A. Venous Thromboembolism and Prognosis in Cancer. Thromb. Res. 2010, 125, 490–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasthuri, R.S.; Taubman, M.B.; Mackman, N. Role of Tissue Factor in Cancer. J. Clin. Oncol. 2009, 27, 4834–4838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.Y.; Landen, C.N.; Kamat, A.A.; Lopez, A.; Bender, D.P.; Mueller, P.; Schmandt, R.; Gershenson, D.M.; Sood, A.K. Preoperative Serum Tissue Factor Levels Are an Independent Prognostic Factor in Patients with Ovarian Carcinoma. J. Clin. Oncol. 2006, 24, 755–761. [Google Scholar] [CrossRef]
- Alexander, E.T.; Minton, A.R.; Peters, M.C.; van Ryn, J.; Gilmour, S.K. Thrombin Inhibition and Cisplatin Block Tumor Progression in Ovarian Cancer by Alleviating the Immunosuppressive Microenvironment. Oncotarget 2016, 7, 85291–85305. [Google Scholar] [CrossRef] [Green Version]
- Alexander, E.; Minton, A.; Peters, M.; van Ryn, J.; Gilmour, S. Dabigatran and Cisplatin Co-Treatment Enhances the Antitumor Efficacy of Immune Checkpoint Blockade in A Murine Model of Resistant Ovarian Cancer. J. Cancer Res. Therap. Oncol. 2020, 8, 1–12. [Google Scholar]
- Müller, A.; Florek, M. 5-Azacytidine/Azacitidine. In Small Molecules in Oncology; Springer: Berlin/Heidelberg, Germany, 2010; pp. 159–170. [Google Scholar]
- European Medicines Agency. Azacitidine Accord EPAR Medicine Overview. Available online: https://www.ema.europa.eu/en/documents/overview/azacitidine-accord-epar-medicine-overview_en.pdf (accessed on 6 April 2023).
- European Medicines Agency. Azacitidine Accord EPAR Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/azacitidine-accord-epar-product-information_en.pdf (accessed on 6 April 2023).
- Stone, M.L.; Chiappinelli, K.B.; Li, H.; Murphy, L.M.; Travers, M.E.; Topper, M.J.; Mathios, D.; Lim, M.; Shih, I.-M.; Wang, T.-L.; et al. Epigenetic Therapy Activates Type I Interferon Signaling in Murine Ovarian Cancer to Reduce Immunosuppression and Tumor Burden. Proc. Natl. Acad. Sci. USA 2017, 114, E10981–E10990. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Han, X.; Shang, C.; Wang, Y.; Xu, B.; Jiang, S.; Mo, Y.; Wang, D.; Ke, Y.; Zeng, X. The Downregulation of Type I IFN Signaling in G-MDSCs under Tumor Conditions Promotes Their Development towards an Immunosuppressive Phenotype. Cell Death Dis. 2022, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Fu, S.; Naing, A.; Hong, D.S.; Hu, W.; Moulder, S.; Wheler, J.J.; Sood, A.K.; Bustinza-Linares, E.; Parkhurst, K.L.; et al. Methylation and Histone Deacetylase Inhibition in Combination with Platinum Treatment in Patients with Advanced Malignancies. Investig. New Drugs 2013, 31, 1192–1200. [Google Scholar] [CrossRef] [Green Version]
- Saraiva, M.; Vieira, P.; O’Garra, A. Biology and Therapeutic Potential of Interleukin-10. J. Exp. Med. 2020, 217, e20190418. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wong, K.; Ouyang, W.; Rutz, S. Targeting IL-10 Family Cytokines for the Treatment of Human Diseases. Cold Spring Harb. Perspect. Biol. 2019, 11, a028548. [Google Scholar] [CrossRef]
- Llorente, L.; Richaud-Patin, Y.; Garcia-Padilla, C.; Claret, E.; Jakez-Ocampo, J.; Cardiel, M.; Alcocer-Varela, J.; Grangeot-Keros, L.; Alarcon-Sergovia, D.; Wijdenes, J.; et al. Clinical and Biologic Effects of Anti-Interleukin-10 Monoclonal Antibody Administration in Systemic Lupus Erythematosus. Arthritis Rheum 2000, 43, 1790–1800. [Google Scholar] [CrossRef] [PubMed]
- Hart, K.M.; Byrne, K.T.; Molloy, M.J.; Usherwood, E.M.; Berwin, B. IL-10 Immunomodulation of Myeloid Cells Regulates a Murine Model of Ovarian Cancer. Front. Immunol. 2011, 2, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, S.; Bak, G.; Hart, K.; Berwin, B. Ovarian Tumor-Induced T Cell Suppression Is Alleviated by Vascular Leukocyte Depletion. Transl. Oncol. 2009, 2, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risnik, D.; Colado, A.; Podaza, E.; Almejún, M.B.; Elías, E.E.; Bezares, R.F.; Fernández-Grecco, H.; Seija, N.; Oppezzo, P.; Borge, M.; et al. Immunoregulatory Effects of Lurbinectedin in Chronic Lymphocytic Leukemia. Cancer Immunol. Immunother. 2020, 69, 813–824. [Google Scholar] [CrossRef]
- Veltman, J.D.; Lambers, M.E.; van Nimwegen, M.; Hendriks, R.W.; Hoogsteden, H.C.; Aerts, J.G.; Hegmans, J.P. COX-2 Inhibition Improves Immunotherapy and Is Associated with Decreased Numbers of Myeloid-Derived Suppressor Cells in Mesothelioma. Celecoxib Influences MDSC Function. BMC Cancer 2010, 10, 464. [Google Scholar] [CrossRef] [Green Version]
- Mikyšková, R.; Indrová, M.; Vlková, V.; Bieblová, J.; Šímová, J.; Paračková, Z.; Pajtasz-Piasecka, E.; Rossowska, J.; Reiniš, M. DNA Demethylating Agent 5-Azacytidine Inhibits Myeloid-Derived Suppressor Cells Induced by Tumor Growth and Cyclophosphamide Treatment. J. Leukoc. Biol. 2014, 95, 743–753. [Google Scholar] [CrossRef]
- Rook, L. Dose–Exposure-Response Relationship. Available online: https://www.ema.europa.eu/en/documents/presentation/presentation-dose-exposure-response-relationship-break-out-session-2-theme-1_en.pdf (accessed on 12 June 2023).
- Maloney, A. A New Paradigm. “Learn—Learn More”; Dose-Exposure-Response at the Center of Drug Development and Regulatory Approval. Clin. Pharmacol. Ther. 2017, 102, 942–950. [Google Scholar] [CrossRef]
- Jiang, S.; Yang, Y.; Zhang, Y.; Ye, Q.; Song, J.; Zheng, M.; Li, X. Overexpression of CAPG Is Associated with Poor Prognosis and Immunosuppressive Cell Infiltration in Ovarian Cancer. Dis. Markers 2022, 2022, 9719671. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berckmans, Y.; Hoffert, Y.; Vankerckhoven, A.; Dreesen, E.; Coosemans, A. Drug Repurposing for Targeting Myeloid-Derived Suppressor-Cell-Generated Immunosuppression in Ovarian Cancer: A Literature Review of Potential Candidates. Pharmaceutics 2023, 15, 1792. https://doi.org/10.3390/pharmaceutics15071792
Berckmans Y, Hoffert Y, Vankerckhoven A, Dreesen E, Coosemans A. Drug Repurposing for Targeting Myeloid-Derived Suppressor-Cell-Generated Immunosuppression in Ovarian Cancer: A Literature Review of Potential Candidates. Pharmaceutics. 2023; 15(7):1792. https://doi.org/10.3390/pharmaceutics15071792
Chicago/Turabian StyleBerckmans, Yani, Yannick Hoffert, Ann Vankerckhoven, Erwin Dreesen, and An Coosemans. 2023. "Drug Repurposing for Targeting Myeloid-Derived Suppressor-Cell-Generated Immunosuppression in Ovarian Cancer: A Literature Review of Potential Candidates" Pharmaceutics 15, no. 7: 1792. https://doi.org/10.3390/pharmaceutics15071792
APA StyleBerckmans, Y., Hoffert, Y., Vankerckhoven, A., Dreesen, E., & Coosemans, A. (2023). Drug Repurposing for Targeting Myeloid-Derived Suppressor-Cell-Generated Immunosuppression in Ovarian Cancer: A Literature Review of Potential Candidates. Pharmaceutics, 15(7), 1792. https://doi.org/10.3390/pharmaceutics15071792