
Microglial-Targeted nSMase2 Inhibitor Fails to Reduce Tau Propagation in PS19 Mice
 , ,
, ,  ,
,  ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Studies and D-DPTIP Dosing
2.2. Magnetic Resonance Imaging (MRI)
2.3. Behavioral Testing
2.3.1. Y-Maze Spatial Recognition
2.3.2. Novel Object Recognition Test (NORT)
2.3.3. Rotarod
2.4. Sample Collection
2.5. Immunofluorescence Staining
2.6. Immunoblotting
2.7. Microglial Isolation
2.8. Fluorescence-Activated Cell Sorting (FACS)
2.9. nSMase2 Activity Assay
2.10. EVs Isolation
2.11. Nanoparticle Tracking Analysis (NTA)
2.12. Surface Plasmon Resonance Imaging (SPRi) Analysis
2.13. Statistical Analysis
3. Results
3.1. D-DPTIP Had No Effect on Recognition or Spatial Memory Deficits in PS19 Mice
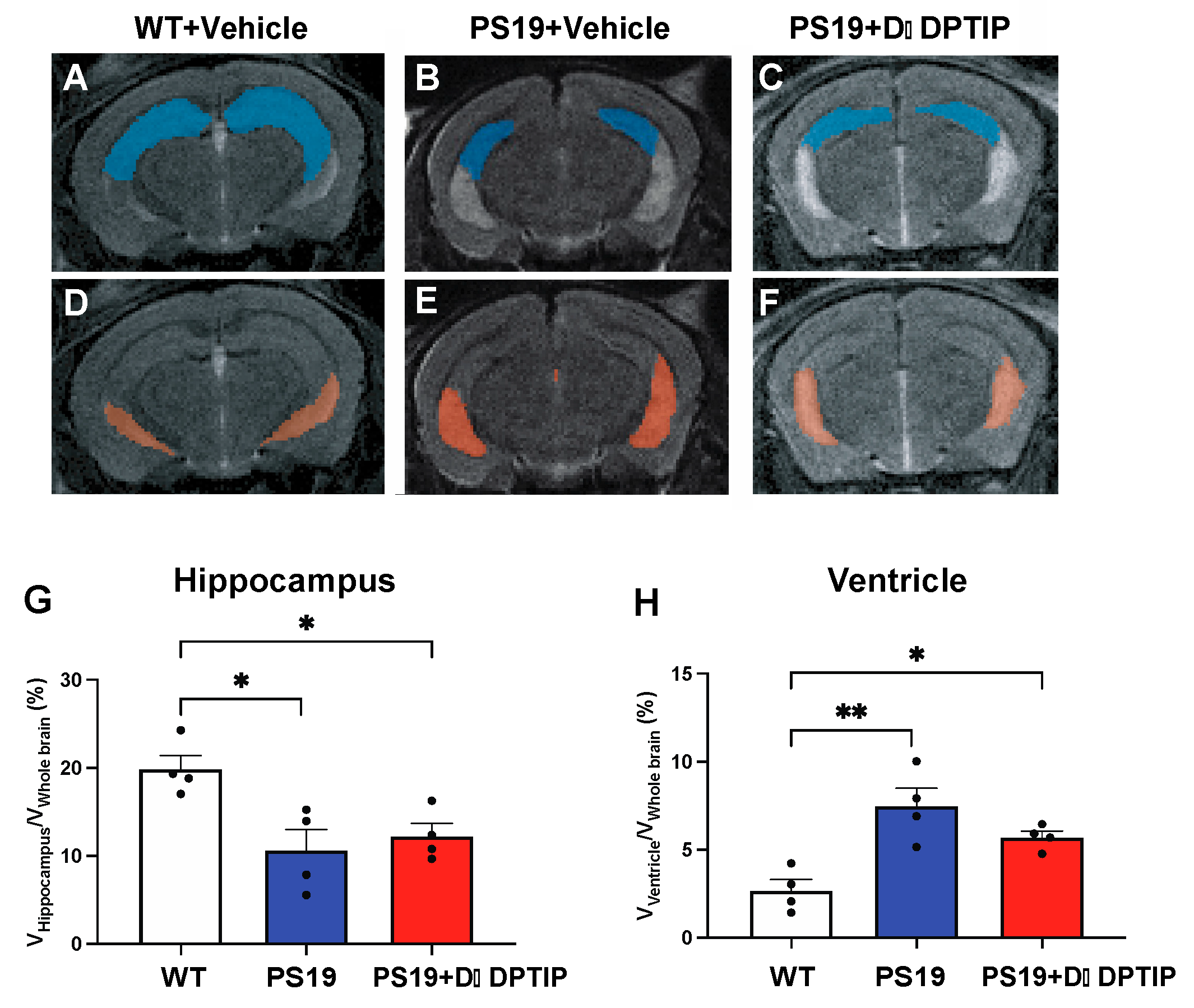
3.2. D-DPTIP Had No Effect on Hippocampal Atrophy or Ventricular Enlargement in PS19 Mice
3.3. D-DPTIP Had No Effect on Hippocampal Tau or pTau Levels in PS19 Mice
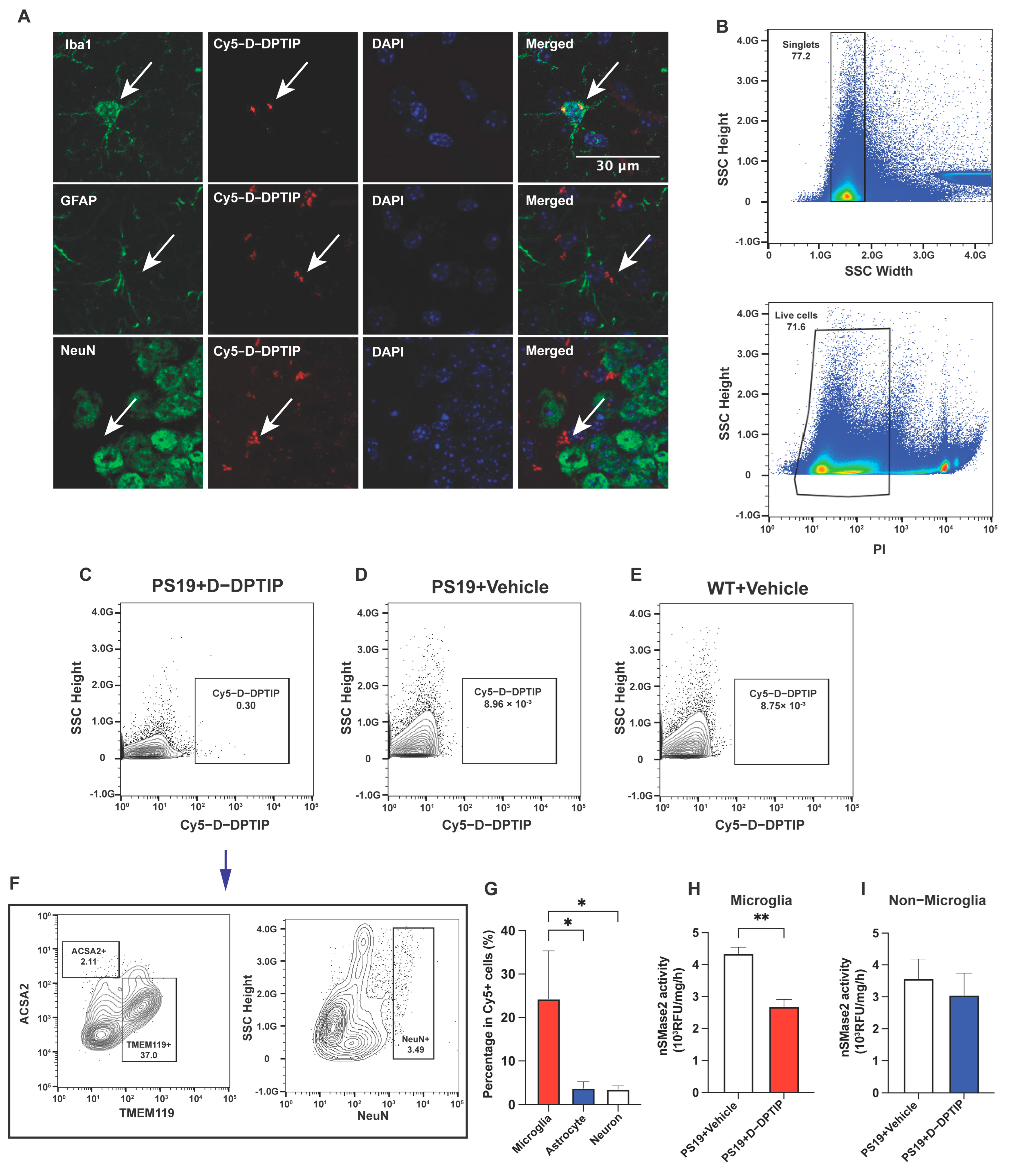
3.4. D-DPTIP Colocalizes with Microglia and Selectively Inhibits Microglial nSMase2 Activity in PS19 Mice
3.5. D-DPTIP Selectively Reduced the Number of Microglia-Derived EVs in the Plasma of PS19 Mice, with No Effect on EVs from Other Brain-Cell Types
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Siemers, E.R.; Mawuenyega, K.G.; Wen, G.; Browning, K.R.; Sigurdson, W.C.; Yarasheski, K.E.; Friedrich, S.W.; Demattos, R.B.; May, P.C.; et al. A gamma-secretase inhibitor decreases amyloid-beta production in the central nervous system. Ann. Neurol. 2009, 66, 48–54. [Google Scholar] [CrossRef]
- Fleisher, A.S.; Raman, R.; Siemers, E.R.; Becerra, L.; Clark, C.M.; Dean, R.A.; Farlow, M.R.; Galvin, J.E.; Peskind, E.R.; Quinn, J.F.; et al. Phase 2 Safety Trial Targeting Amyloid β Production With a γ-Secretase Inhibitor in Alzheimer Disease. Arch. Neurol. 2008, 65, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Henley, D.B.; May, P.C.; Dean, R.A.; Siemers, E.R. Development of semagacestat (LY450139), a functional γ-secretase inhibitor, for the treatment of Alzheimer’s disease. Expert Opin. Pharmacother. 2009, 10, 1657–1664. [Google Scholar] [CrossRef]
- Wolfe, M.S. γ-Secretase as a target for Alzheimer’s disease. Adv. Pharmacol. 2012, 64, 127–153. [Google Scholar]
- Bazzari, F.H.; Bazzari, A.H. BACE1 Inhibitors for Alzheimer’s Disease: The Past, Present and Any Future? Molecules 2022, 27, 8823. [Google Scholar] [PubMed]
- Panza, F.; Solfrizzi, V.; Imbimbo, B.P.; Tortelli, R.; Santamato, A.; Logroscino, G. Amyloid-based immunotherapy for Alzheimer’s disease in the time of prevention trials: The way forward. Expert Rev. Clin. Immunol. 2014, 10, 405–419. [Google Scholar]
- Schenk, D.; Hagen, M.; Seubert, P. Current progress in beta-amyloid immunotherapy. Curr. Opin. Immunol. 2004, 16, 599–606. [Google Scholar]
- Andreasen, N.; Sjögren, M.; Blennow, K. CSF markers for Alzheimer’s disease: Total tau, phospho-tau and Aβ42. World J. Biol. Psychiatry 2003, 4, 147–155. [Google Scholar] [CrossRef]
- Karikari, T.K.; Ashton, N.J.; Brinkmalm, G.; Brum, W.S.; Benedet, A.L.; Montoliu-Gaya, L.; Lantero-Rodriguez, J.; Pascoal, T.A.; Suárez-Calvet, M.; Rosa-Neto, P.; et al. Blood phospho-tau in Alzheimer disease: Analysis, interpretation, and clinical utility. Nat. Rev. Neurol. 2022, 18, 400–418. [Google Scholar]
- Gibbons, G.S.; Lee, V.M.Y.; Trojanowski, J.Q. Mechanisms of Cell-to-Cell Transmission of Pathological Tau: A Review. JAMA Neurol. 2019, 76, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Simón, D.; García-García, E.; Gómez-Ramos, A.; Falcón-Pérez, J.M.; Díaz-Hernández, M.; Hernández, F.; Avila, J. Tau overexpression results in its secretion via membrane vesicles. Neurodegener. Dis. 2012, 10, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Juan, T.; Fürthauer, M. Biogenesis and function of ESCRT-dependent extracellular vesicles. Semin. Cell Dev. Biol. 2018, 74, 66–77. [Google Scholar] [CrossRef]
- Urbanelli, L.; Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Porcellati, S.; Emiliani, C. The Role of Extracellular Vesicles in Viral Infection and Transmission. Vaccines 2019, 7, 102. [Google Scholar] [CrossRef] [PubMed]
- Tallon, C.; Bell, B.J.; Sharma, A.; Pal, A.; Malvankar, M.M.; Thomas, A.G.; Yoo, S.W.; Hollinger, K.R.; Coleman, K.; Wilkinson, E.L.; et al. Dendrimer-Conjugated nSMase2 Inhibitor Reduces Tau Propagation in Mice. Pharmaceutics 2022, 14, 2066. [Google Scholar] [CrossRef]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kugler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- DeLeo, A.M.; Ikezu, T. Extracellular Vesicle Biology in Alzheimer’s Disease and Related Tauopathy. J. Neuroimmune. Pharmacol. 2018, 13, 292–308. [Google Scholar] [CrossRef]
- Bell, B.J.; Malvankar, M.M.; Tallon, C.; Slusher, B.S. Sowing the Seeds of Discovery: Tau-Propagation Models of Alzheimer’s Disease. ACS Chem. Neurosci. 2020, 11, 3499–3509. [Google Scholar] [CrossRef]
- Rojas, C.; Barnaeva, E.; Thomas, A.G.; Hu, X.; Southall, N.; Marugan, J.; Chaudhuri, A.D.; Yoo, S.-W.; Hin, N.; Stepanek, O.; et al. DPTIP, a newly identified potent brain penetrant neutral sphingomyelinase 2 inhibitor, regulates astrocyte-peripheral immune communication following brain inflammation. Sci. Rep. 2018, 8, 17715. [Google Scholar] [CrossRef]
- Hayder, M.; Poupot, M.; Baron, M.; Nigon, D.; Turrin, C.O.; Caminade, A.M.; Majoral, J.P.; Eisenberg, R.A.; Fournie, J.J.; Cantagrel, A.; et al. A phosphorus-based dendrimer targets inflammation and osteoclastogenesis in experimental arthritis. Sci. Transl. Med. 2011, 3, 81ra35. [Google Scholar] [CrossRef]
- Hayder, M.; Varilh, M.; Turrin, C.-O.; Saoudi, A.; Caminade, A.-M.; Poupot, R.; Liblau, R.S. Phosphorus-Based Dendrimer ABP Treats Neuroinflammation by Promoting IL-10-Producing CD4+ T Cells. Biomacromolecules 2015, 16, 3425–3433. [Google Scholar] [CrossRef]
- Arbez-Gindre, C.; Steele, B.R.; Micha-Screttas, M. Dendrimers in Alzheimer’s Disease: Recent Approaches in Multi-Targeting Strategies. Pharmaceutics 2023, 15, 898. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Johnson, M.A.; Mehrabian, Z.; Mishra, M.K.; Kannan, R.; Miller, N.R.; Bernstein, S.L. Dendrimers Target the Ischemic Lesion in Rodent and Primate Models of Nonarteritic Anterior Ischemic Optic Neuropathy. PLoS ONE 2016, 11, e0154437. [Google Scholar] [CrossRef] [PubMed]
- Niño, D.F.; Zhou, Q.; Yamaguchi, Y.; Martin, L.Y.; Wang, S.; Fulton, W.B.; Jia, H.; Lu, P.; Prindle, T., Jr.; Zhang, F.; et al. Cognitive impairments induced by necrotizing enterocolitis can be prevented by inhibiting microglial activation in mouse brain. Sci. Transl. Med. 2018, 10, eaan0237. [Google Scholar] [CrossRef]
- Nance, E.; Kambhampati, S.P.; Smith, E.S.; Zhang, Z.; Zhang, F.; Singh, S.; Johnston, M.V.; Kannan, R.M.; Blue, M.E.; Kannan, S. Dendrimer-mediated delivery of N-acetyl cysteine to microglia in a mouse model of Rett syndrome. J. Neuroinflammation 2017, 14, 252. [Google Scholar] [CrossRef] [PubMed]
- Nance, E.; Zhang, F.; Mishra, M.K.; Zhang, Z.; Kambhampati, S.P.; Kannan, R.M.; Kannan, S. Nanoscale effects in dendrimer-mediated targeting of neuroinflammation. Biomaterials 2016, 101, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.R.; Nemeth, C.L.; Marx, J.S.; Tiffany, C.; Jones, R.; Theisen, B.; Kambhampati, S.; Ramireddy, R.; Singh, S.; Rosen, M.; et al. Dendrimer-N-acetyl-L-cysteine modulates monophagocytic response in adrenoleukodystrophy. Ann. Neurol. 2018, 84, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Nance, E.; Alnasser, Y.; Kannan, R.; Kannan, S. Microglial migration and interactions with dendrimer nanoparticles are altered in the presence of neuroinflammation. J. Neuroinflammation 2016, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.K.; Beaty, C.A.; Lesniak, W.G.; Kambhampati, S.P.; Zhang, F.; Wilson, M.A.; Blue, M.E.; Troncoso, J.C.; Kannan, S.; Johnston, M.V.; et al. Dendrimer brain uptake and targeted therapy for brain injury in a large animal model of hypothermic circulatory arrest. ACS Nano 2014, 8, 2134–2147. [Google Scholar] [CrossRef]
- Sharma, A.; Porterfield, J.E.; Smith, E.; Sharma, R.; Kannan, S.; Kannan, R.M. Effect of mannose targeting of hydroxyl PAMAM dendrimers on cellular and organ biodistribution in a neonatal brain injury model. J. Control. Release 2018, 283, 175–189. [Google Scholar] [CrossRef]
- Gusdon, A.M.; Faraday, N.; Aita, J.S.; Kumar, S.; Mehta, I.; Choi, H.A.; Cleland, J.L.; Robinson, K.; McCullough, L.D.; Ng, D.K.; et al. Dendrimer nanotherapy for severe COVID-19 attenuates inflammation and neurological injury markers and improves outcomes in a phase2a clinical trial. Sci. Transl. Med. 2022, 14, eabo2652. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, T.; Dewachter, I.; Lechat, B.; Croes, S.; Termont, A.; Demedts, D.; Borghgraef, P.; Devijver, H.; Filipkowski, R.K.; Kaczmarek, L.; et al. AAV-tau mediates pyramidal neurodegeneration by cell-cycle re-entry without neurofibrillary tangle formation in wild-type mice. PLoS ONE 2009, 4, e7280. [Google Scholar] [CrossRef]
- Wegmann, S.; Bennett, R.E.; Delorme, L.; Robbins, A.B.; Hu, M.; McKenzie, D.; Kirk, M.J.; Schiantarelli, J.; Tunio, N.; Amaral, A.C.; et al. Experimental evidence for the age dependence of tau protein spread in the brain. Sci. Adv. 2019, 5, eaaw6404. [Google Scholar] [CrossRef]
- You, Y.; Botros, M.B.; Enoo, A.A.V.; Bockmiller, A.; Herron, S.; Delpech, J.C.; Ikezu, T. Cre-inducible Adeno Associated Virus-mediated Expression of P301L Mutant Tau Causes Motor Deficits and Neuronal Degeneration in the Substantia Nigra. Neuroscience 2019, 422, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Siman, R.; Lin, Y.G.; Malthankar-Phatak, G.; Dong, Y. A rapid gene delivery-based mouse model for early-stage Alzheimer disease-type tauopathy. J. Neuropathol. Exp. Neurol. 2013, 72, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Luengo, E.; Buendia, I.; Fernandez-Mendivil, C.; Trigo-Alonso, P.; Negredo, P.; Michalska, P.; Hernandez-Garcia, B.; Sanchez-Ramos, C.; Bernal, J.A.; Ikezu, T.; et al. Pharmacological doses of melatonin impede cognitive decline in tau-related Alzheimer models, once tauopathy is initiated, by restoring the autophagic flux. J. Pineal Res. 2019, 67, e12578. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zeng, K.; Li, M.; Wang, Q.; Liu, R.; Zhang, B.; Wang, J.Z.; Shu, X.; Wang, X. Expression of P301L-hTau in mouse MEC induces hippocampus-dependent memory deficit. Sci. Rep. 2017, 7, 3914. [Google Scholar] [CrossRef]
- Clayton, K.A.; Delpech, J.C.; Herron, S.; Iwahara, N.; Saito, T.; Saido, T.C.; Ikezu, S.; Ikezu, T. Amyloid plaque deposition accelerates tau propagation via activation of microglia in a humanized APP mouse model. bioRxiv 2020, arXiv:2020.09.22.308015. [Google Scholar]
- Clayton, K.; Delpech, J.C.; Herron, S.; Iwahara, N.; Ericsson, M.; Saito, T.; Saido, T.C.; Ikezu, S.; Ikezu, T. Plaque associated microglia hyper-secrete extracellular vesicles and accelerate tau propagation in a humanized APP mouse model. Mol. Neurodegener. 2021, 16, 18. [Google Scholar] [CrossRef]
- Španić, E.; Langer Horvat, L.; Hof, P.R.; Šimić, G. Role of Microglial Cells in Alzheimer’s Disease Tau Propagation. Front. Aging Neurosci. 2019, 11, 271. [Google Scholar] [CrossRef]
- Duwat, C.; Léal, P.; Vautheny, A.; Aurégan, G.; Joséphine, C.; Gaillard, M.-C.; Hérard, A.-S.; Jan, C.; Gipchtein, P.; Mitja, J.; et al. Development of an AAV-based model of tauopathy targeting retinal ganglion cells and the mouse visual pathway to study the role of microglia in Tau pathology. Neurobiol. Dis. 2023, 181, 106116. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.J.; Yu, P.; Miller, B.B.; Shcherbinin, S.; Dickson, J.; Navitsky, M.; Joshi, A.D.; Devous, M.D., Sr.; Mintun, M.S. Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 2016, 139 Pt 5, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Calafate, S.; Buist, A.; Miskiewicz, K.; Vijayan, V.; Daneels, G.; de Strooper, B.; de Wit, J.; Verstreken, P.; Moechars, D. Synaptic Contacts Enhance Cell-to-Cell Tau Pathology Propagation. Cell Rep. 2015, 11, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Demaegd, K.; Schymkowitz, J.; Rousseau, F. Transcellular Spreading of Tau in Tauopathies. Chembiochem 2018, 19, 2424–2432. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Kruger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.; Stack, C.; Elipenahli, C.; Jainuddin, S.; Gerges, M.; Starkova, N.; Calingasan, N.Y.; Yang, L.; Tampellini, D.; Starkov, A.A.; et al. Bezafibrate administration improves behavioral deficits and tau pathology in P301S mice. Hum. Mol. Genet. 2012, 21, 5091–5105. [Google Scholar] [CrossRef]
- Sarver, D.C.; Xu, C.; Cheng, Y.; Terrillion, C.E.; Wong, G.W. CTRP4 ablation impairs associative learning and memory. Faseb. J. 2021, 35, e21910. [Google Scholar] [CrossRef]
- Lueptow, L.M. Novel Object Recognition Test for the Investigation of Learning and Memory in Mice. J. Vis. Exp. 2017, 126, e55718. [Google Scholar]
- Su, Y.; Claflin, D.R.; Huang, M.; Davis, C.S.; Macpherson, P.C.D.; Richardson, A.; Van Remmen, H.; Brooks, S.V. Deletion of Neuronal CuZnSOD Accelerates Age-Associated Muscle Mitochondria and Calcium Handling Dysfunction That Is Independent of Denervation and Precedes Sarcopenia. Int. J. Mol. Sci. 2021, 22, 10735. [Google Scholar] [CrossRef]
- Sarkar, S.; Dammer, E.B.; Malovic, E.; Olsen, A.L.; Raza, S.A.; Gao, T.; Xiao, H.; Oliver, D.L.; Duong, D.; Joers, V.; et al. Molecular Signatures of Neuroinflammation Induced by alphaSynuclein Aggregates in Microglial Cells. Front. Immunol. 2020, 11, 33. [Google Scholar] [CrossRef]
- Huang, M.; Modeste, E.; Dammer, E.; Merino, P.; Taylor, G.; Duong, D.M.; Deng, Q.; Holler, C.J.; Gearing, M.; Dickson, D.; et al. Network analysis of the progranulin-deficient mouse brain proteome reveals pathogenic mechanisms shared in human frontotemporal dementia caused by GRN mutations. Acta Neuropathol. Commun. 2020, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Nedelcovych, M.T.; Kim, B.H.; Zhu, X.; Lovell, L.E.; Manning, A.A.; Kelschenbach, J.; Hadas, E.; Chao, W.; Prchalova, E.; Dash, R.P.; et al. Glutamine Antagonist JHU083 Normalizes Aberrant Glutamate Production and Cognitive Deficits in the EcoHIV Murine Model of HIV-Associated Neurocognitive Disorders. J. Neuroimmune Pharmacol. 2019, 14, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Peyron, L.; Henry, M.; Sohier, R. Biological diagnosis of influenza B. Observations made during a recent influenza epidemic. Ann. Biol. Clin. 1966, 24, 1135–1145. [Google Scholar]
- Tallon, C.; Picciolini, S.; Yoo, S.W.; Thomas, A.G.; Pal, A.; Alt, J.; Carlomagno, C.; Gualerzi, A.; Rais, R.; Haughey, N.J.; et al. Inhibition of neutral sphingomyelinase 2 reduces extracellular vesicle release from neurons, oligodendrocytes, and activated microglial cells following acute brain injury. Biochem. Pharmacol. 2021, 194, 114796. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Iba, M.; McBride, J.D.; Guo, J.L.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol. 2015, 130, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Tallon, C.; Bell, B.J.; Malvankar, M.M.; Deme, P.; Nogueras-Ortiz, C.; Eren, E.; Thomas, A.G.; Hollinger, K.R.; Pal, A.; Mustapic, M.; et al. Inhibiting tau-induced elevated nSMase2 activity and ceramides is therapeutic in murine Alzheimer’s disease. Res. Sq. 2023, preprint. [Google Scholar]
- Pascoal, T.A.; Benedet, A.L.; Ashton, N.J.; Kang, M.S.; Therriault, J.; Chamoun, M.; Savard, M.; Lussier, F.Z.; Tissot, C.; Karikari, T.K.; et al. Microglial activation and tau propagate jointly across Braak stages. Nat. Med. 2021, 27, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xie, C. Microglia activation linking amyloid-beta drive tau spatial propagation in Alzheimer’s disease. Front. Neurosci. 2022, 16, 951128. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Fan, L.; Khawaja, R.R.; Liu, B.; Zhan, L.; Kodama, L.; Chin, M.; Li, Y.; Le, D.; Zhou, Y.; et al. Microglial NF-kappaB drives tau spreading and toxicity in a mouse model of tauopathy. Nat. Commun. 2022, 13, 1969. [Google Scholar] [CrossRef] [PubMed]
- Stancu, I.C.; Lodder, C.; Botella Lucena, P.; Vanherle, S.; Gutierrez de Rave, M.; Terwel, D.; Bottelbergs, A.; Dewachter, I. The NLRP3 inflammasome modulates tau pathology and neurodegeneration in a tauopathy model. Glia 2022, 70, 1117–1132. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, M.; Tallon, C.; Zhu, X.; Huizar, K.D.J.; Picciolini, S.; Thomas, A.G.; Tenora, L.; Liyanage, W.; Rodà, F.; Gualerzi, A.; et al. Microglial-Targeted nSMase2 Inhibitor Fails to Reduce Tau Propagation in PS19 Mice. Pharmaceutics 2023, 15, 2364. https://doi.org/10.3390/pharmaceutics15092364
Huang M, Tallon C, Zhu X, Huizar KDJ, Picciolini S, Thomas AG, Tenora L, Liyanage W, Rodà F, Gualerzi A, et al. Microglial-Targeted nSMase2 Inhibitor Fails to Reduce Tau Propagation in PS19 Mice. Pharmaceutics. 2023; 15(9):2364. https://doi.org/10.3390/pharmaceutics15092364
Chicago/Turabian StyleHuang, Meixiang, Carolyn Tallon, Xiaolei Zhu, Kaitlyn D. J. Huizar, Silvia Picciolini, Ajit G. Thomas, Lukas Tenora, Wathsala Liyanage, Francesca Rodà, Alice Gualerzi, and et al. 2023. "Microglial-Targeted nSMase2 Inhibitor Fails to Reduce Tau Propagation in PS19 Mice" Pharmaceutics 15, no. 9: 2364. https://doi.org/10.3390/pharmaceutics15092364
APA StyleHuang, M., Tallon, C., Zhu, X., Huizar, K. D. J., Picciolini, S., Thomas, A. G., Tenora, L., Liyanage, W., Rodà, F., Gualerzi, A., Kannan, R. M., Bedoni, M., Rais, R., & Slusher, B. S. (2023). Microglial-Targeted nSMase2 Inhibitor Fails to Reduce Tau Propagation in PS19 Mice. Pharmaceutics, 15(9), 2364. https://doi.org/10.3390/pharmaceutics15092364