



Synthesis and Preliminary Evaluation of an ASGPr-Targeted Polycationic β-Cyclodextrin Carrier for Nucleosides and Nucleotides



Abstract
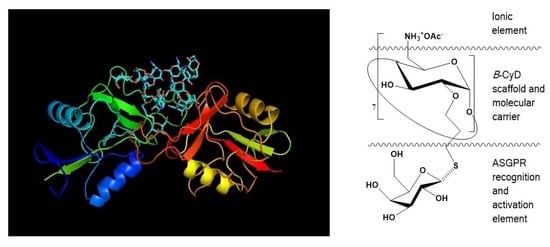
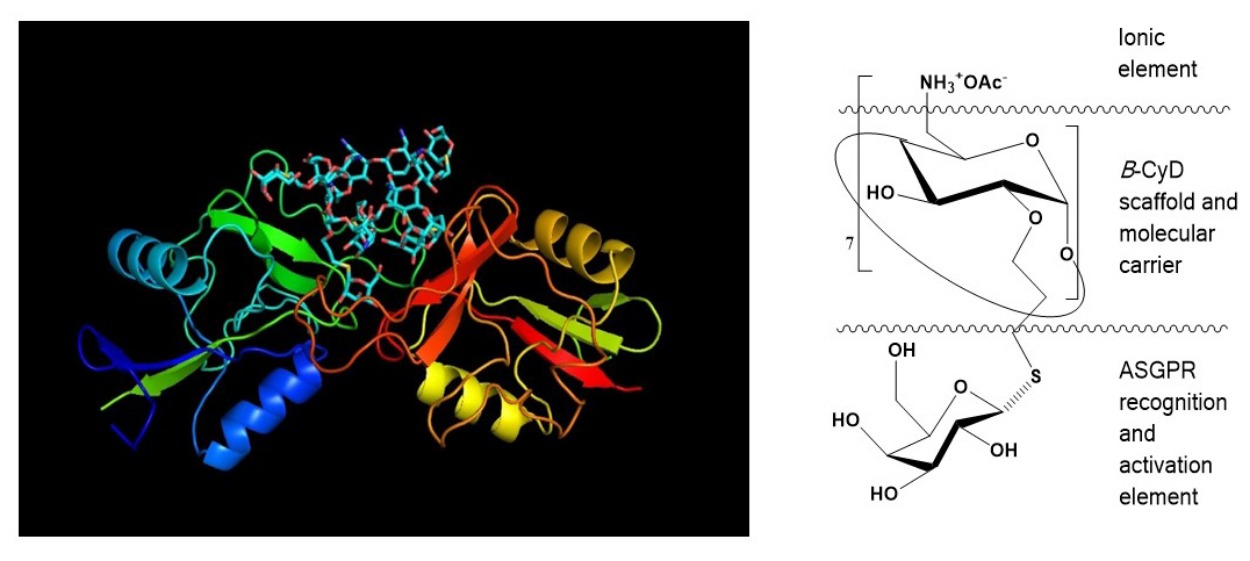
:
1. Introduction
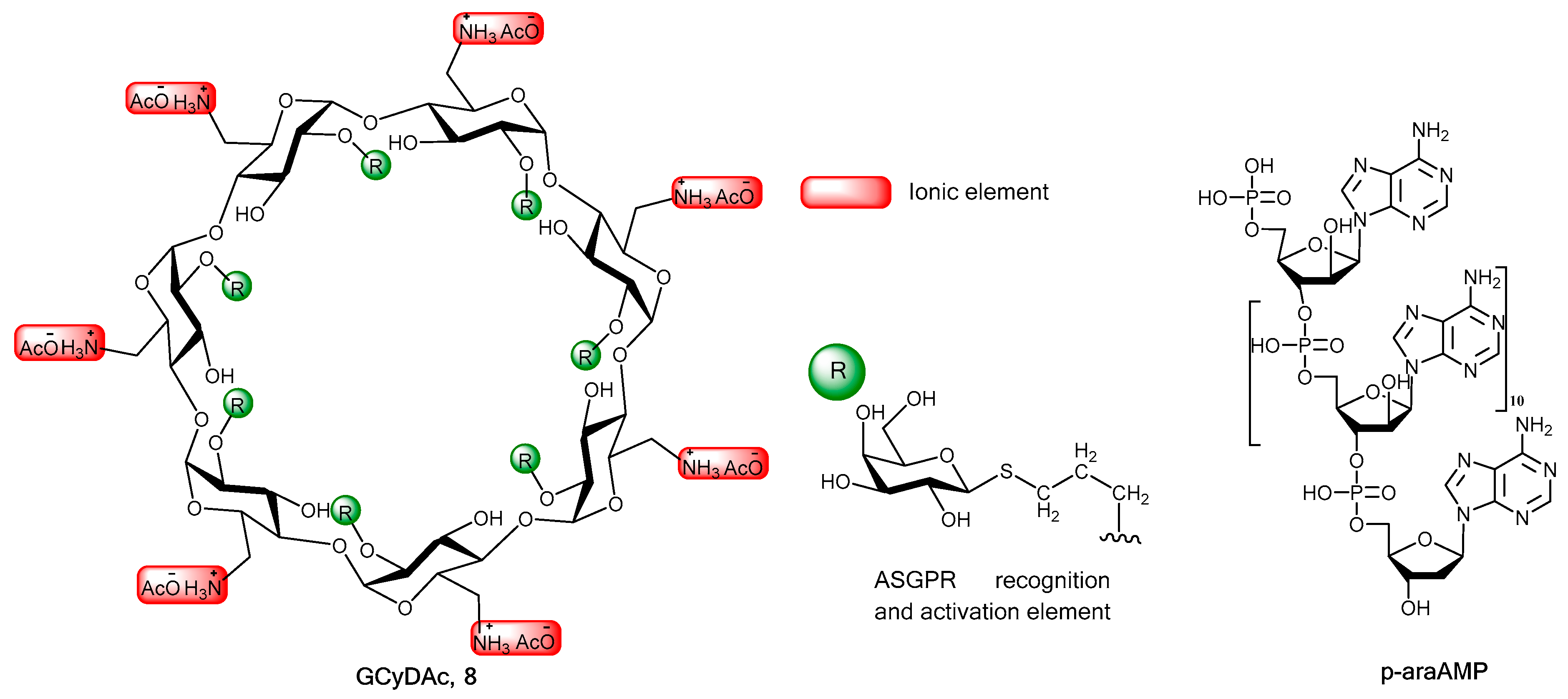
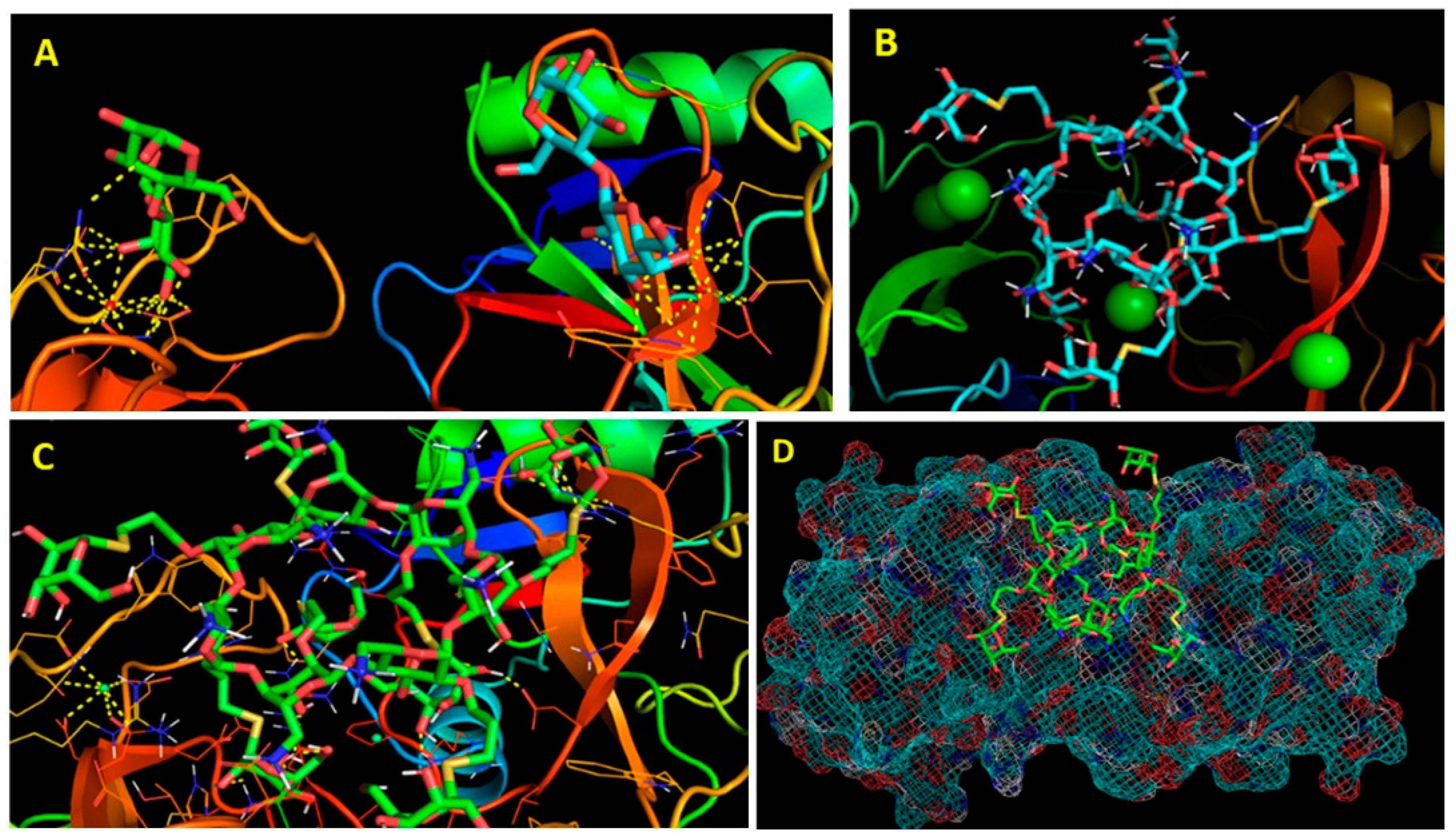
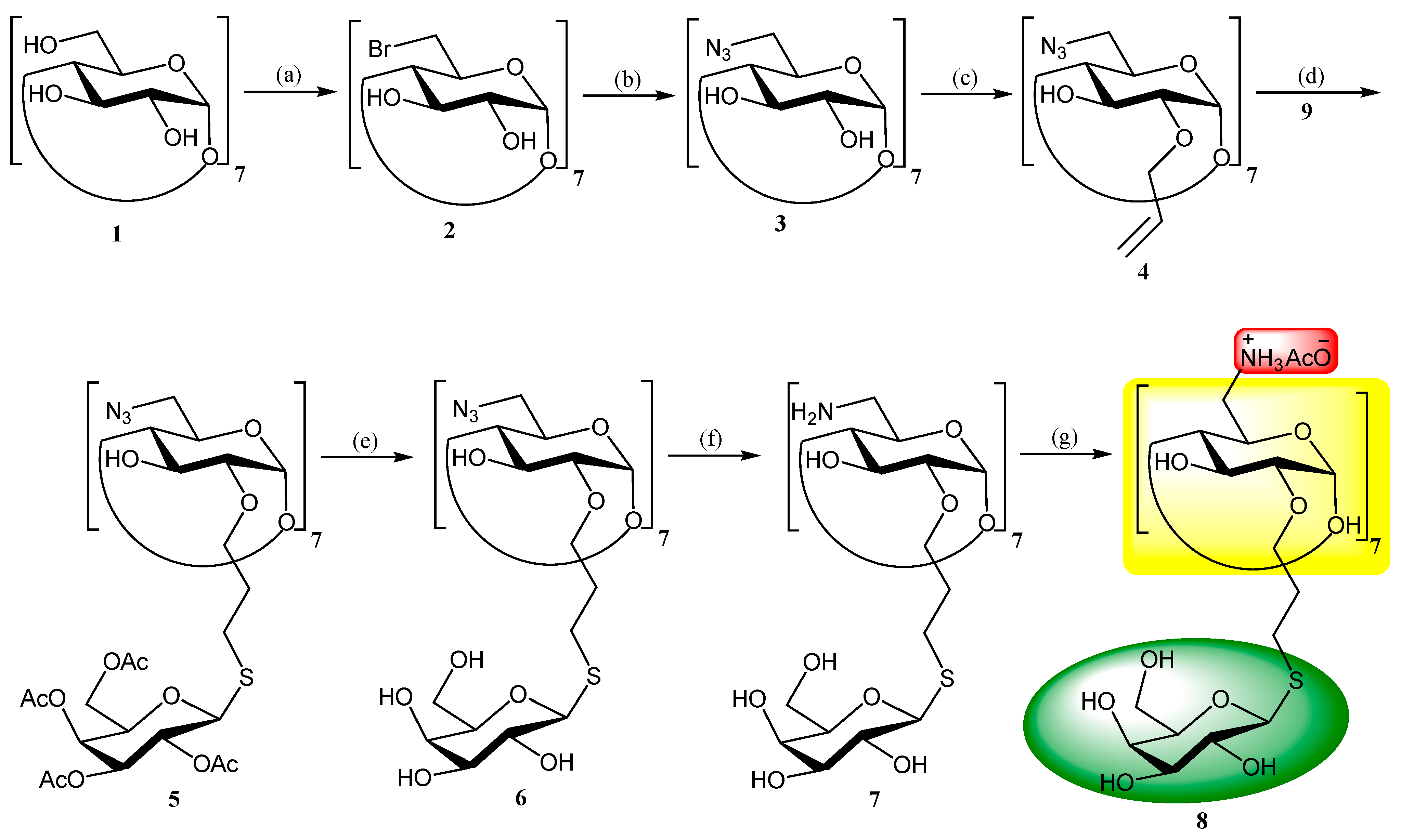
2. Chemistry, Formulation and Molecular Docking
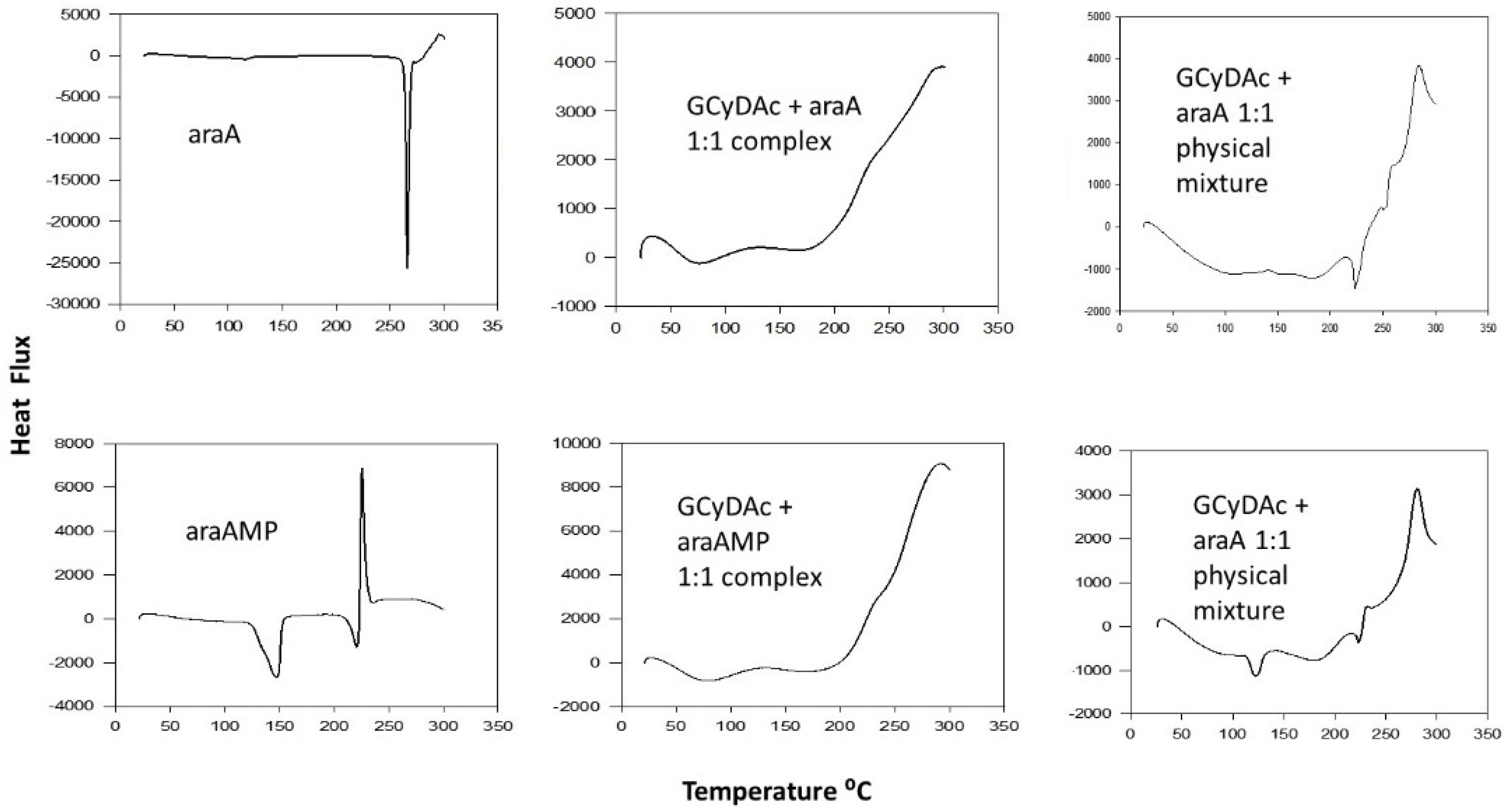
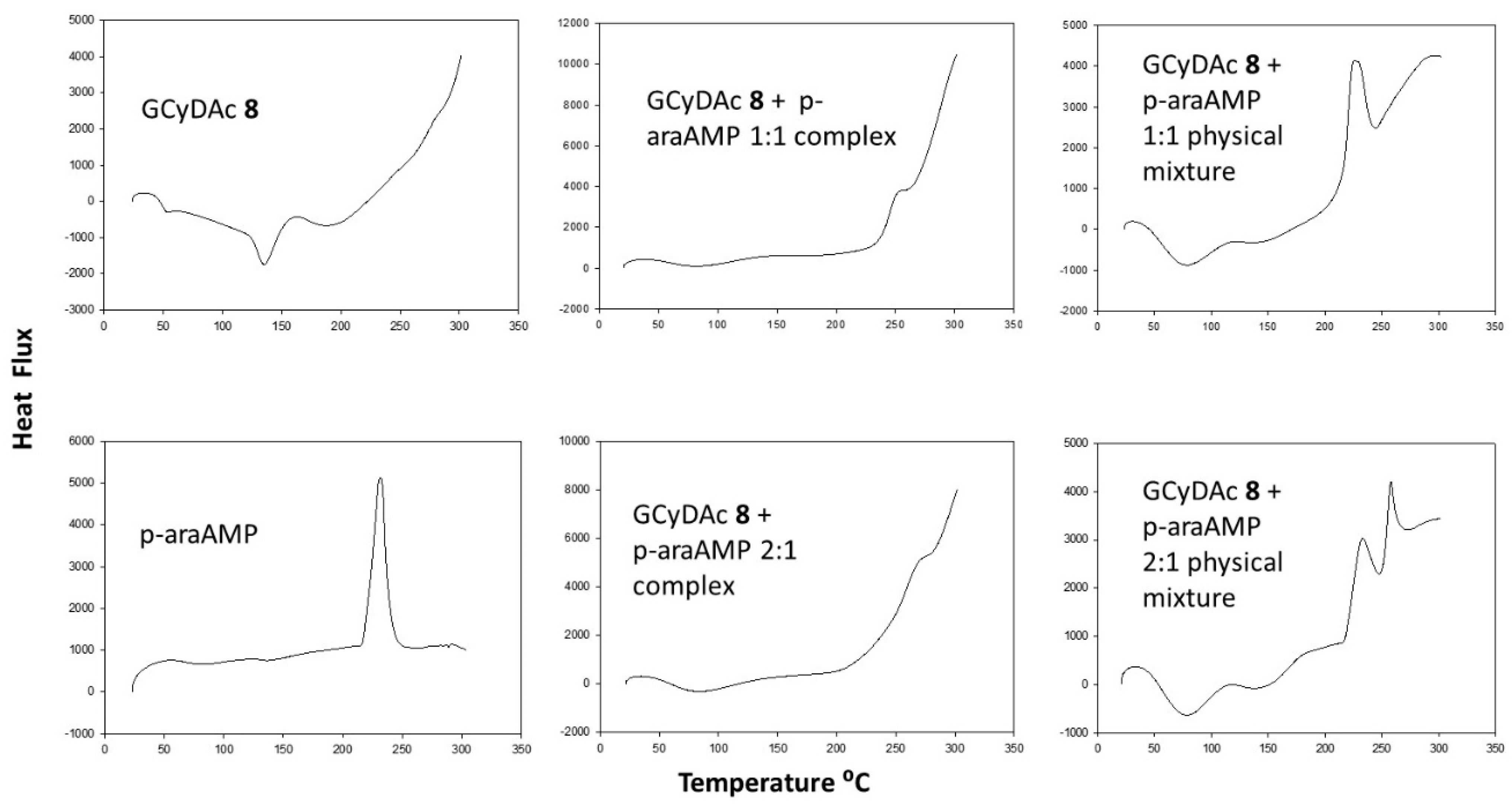
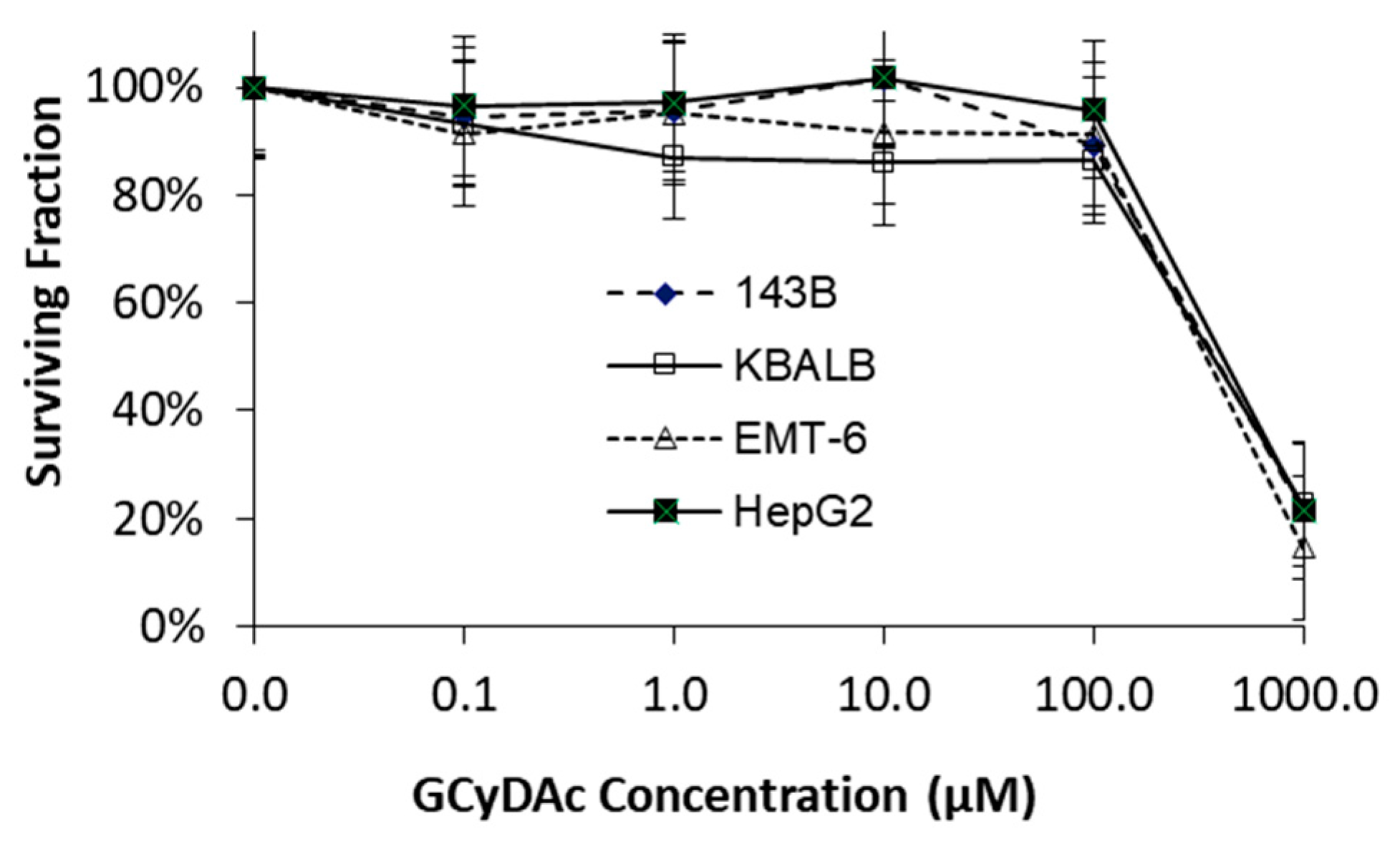
3. Results
4. Discussion
5. Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- LePage, G.A.; Naik, S.R.; Katakkar, S.B.; Khaliq, A. 9-Beta-D-arabinofuranosyladenine 5’-Phosphate Metabolism and Excretion in Humans. Cancer Res. 1975, 35, 3036–3040. [Google Scholar] [PubMed]
- Holzer, S.; Rzechorzek, N.J.; Short, I.R.; Jenkyn-Bedford, M.; Pellegrini, L.; Kilkenny, M.L. Structural Basis for Inhibition of Human Primase by Arabinofuranosyl Nucleoside Analogues Fludarabine and Vidarabine. ACS Chem. Biol. 2019, 14, 1904–1912. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R.J.; Ch’ien, L.T.; Dolin, R.; Galasso, G.J.; Alford, C.A., Jr. Adenine Arabinoside Therapy of Herpes Zoster in the Immunosuppressed—NIAID Collaborative Antiviral Study. N. Engl. J. Med. 1976, 294, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Sagar, S.; Kaur, M.; Minneman, K.P. Antiviral Lead Compounds from Marine Sponges. Mar. Drugs 2010, 8, 2619–2638. [Google Scholar] [CrossRef] [PubMed]
- Balzarini, J.; De Clercq, E. 9-Beta-D-arabinofuranosyladenine 5′-monophosphate (araAMP) is Converted Directly to its Antivirally Active 5′-triphosphate Form by 5-Phosphoribosyl-1-pyrophosphate (PRPP) Synthtase. Biochem. Biophys. Res. Commun. 1990, 173, 781–787. [Google Scholar] [CrossRef] [PubMed]
- York, J.L.; LePage, G.A. A Proposed Mechanism for the Action of 9-Beta-D-arabinofuranosyladenine as an Inhibitor of the Growth of Some Ascites Cells. Can. J. Biochem. Physiol. 1966, 44, 19–28. [Google Scholar] [CrossRef]
- Furth, J.J.; Cohen, S.S. Inhibition of Mammalian DNA Polymerase by the 5′-Triphosphate of 1-Beta-D-Arabinofuranosylcytosine and the 5′-Triphosphate of 9-Beta-D-arabinofuranosyladenineoxyladenine. Cancer Res. 1968, 60, 2061–2067. [Google Scholar]
- Derse, D.; Cheng, Y.C. Herpes Simplex Virus Type I DNA Polymerase. Kinetic Properties of the Associated 3′-5′ Exonuclease Activity and its Role in araAMP incorporation. J. Biol. Chem. 1981, 256, 8525–8530. [Google Scholar] [CrossRef]
- Bijsterbosch, M.K.; Ying, C.; de Vrueh, R.L.A.; de Clercq, E.; Biessen, E.A.L.; Neyts, J.; van Berkel, T.J.C. Carrier-mediated Delivery Improves the Efficacy of 9-(2-Phosphonylmethoxyethyl)adenine against Hepatitis B Virus. Mol. Pharmac. 2001, 60, 521–527. [Google Scholar]
- Cass, C.E. Nucleoside transport. In Drug Transport in Antimicrobial and Anticancer Chemotherapy; Georgopapadakou, N.H., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 1995; pp. 408–451. ISBN 10978-0824793999. [Google Scholar]
- Young, J.D.; Yao, S.Y.M.; Sun, L.; Cass, C.E.; Baldwin, S.A. Human Equilibrative Nucleoside Transporter (ENT) Family of Nucleoside and Nucleobase Transporter Proteins. Xenobiotica 2008, 38, 995–1021. [Google Scholar] [CrossRef]
- LePage, G.A.; Khaliq, A.; Gottlieb, J.A. Studies of 9-Beta-D-arabinofuranosyladenine in Man. Drug Metab. Disp. 1973, 1, 756–759. [Google Scholar]
- Van Rompay, A.; Johansson, M.; Karlsson, A. Phosphorylation of Nucleosides and Nucleoside Analogs by Mammalian Nucleoside Monophosphate Kinases. Pharmacol. Therapeut. 2000, 87, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Pavan-Langston, D.; North, R.D.; Geary, P.A. Ara AMP- a Highly Soluble New Antiviral Drug. Ann. Ophthalmol. 1976, 8, 571–579. [Google Scholar] [PubMed]
- LePage, G.A.; Lin, Y.T.; Orth, R.E.; Gottlieb, J.A. 5′-Nucleotides as Potential Formulations for Administering Nucleoside Analogs in Man. Cancer Res. 1972, 32, 2441–2444. [Google Scholar] [PubMed]
- Sawada, K.; Echigo, N.; Juge, N.; Miyaji, T.; Otsuka, M.; Omote, H.; Yamamoto, A.; Moriyama, Y. Identification of a Vesicular Nucleotide Transporter. Proc. Natl. Acad. Sci. USA 2008, 105, 5683–5686. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.H.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; et al. The Concise Guide to PHARMACOLOGY 2019/20: Transporters. Br. J. Pharmacol. 2019, 176, S397–S493. [Google Scholar] [CrossRef]
- Kim, J.; Chou, T.-F.; Griesgraber, G.W.; Wagner, C.R. Direct Measurement of Nucleoside Monophosphate Delivery from a Phosphoramidate Pronucleotide by Stable Isotope Labeling and LC−ESI(-)-MS/MS. Mol. Pharmaceut. 2004, 1, 102–111. [Google Scholar] [CrossRef]
- Ahuja, S.; Whorton, M.R. Structural Basis for Mammalian Nucleotide Sugar Transport. eLife 2019, 8, e45221. [Google Scholar] [CrossRef]
- Markov, O.V.; Filatov, A.V.; Kupryushkin, M.S.; Chernikov, I.V.; Patutina, O.A.; Strunov, A.A.; Chernolovskaya, E.L.; Vlassov, V.V.; Pyshnyi, D.V.; Zenkova, M.A. Transport Oligonucleotides—A Novel System for Intracellular Delivery of Antisense Therapeutics. Molecules 2020, 25, 3663. [Google Scholar] [CrossRef]
- Hiasa, M.; Togawa, N.; Moriyama, Y. Vesicular Nucleotide Transport: A Brief History and the Vesicular Nucleotide Transporter as a Target for Drug Development. Curr. Pharm. Des. 2014, 20, 2745–2749. [Google Scholar] [CrossRef]
- Bender, M.L.; Komiyama, M. Cyclodextrin Chemistry. Reactivity and Structure: Concepts in Organic Chemistry; Springer: Berlin/Heidelberg, Germany, 1978; Volume 6. [Google Scholar] [CrossRef]
- Szejtli, J.; Osa, T. Cyclodextrins. In Comprehensive Supramolecular Chemistry; Lehn, J.M., Atwood, J.L., Davies, J.E.D., Eds.; Pergamon: New York, NY, USA, 1999; Volume 3, ISBN 9780080427157. [Google Scholar]
- Muankaew, C.; Loftsson, T. Cyclodextrin-based Formulations: A Non-invasive Platform for Targeted Drug Delivery. Basic Clin. Pharmacol. Toxicol. 2018, 122, 46–55. [Google Scholar] [CrossRef]
- Hoffman, J.L.; Bock, R.M. The Interaction of Cyclodextrins with Nucleic Acids. A Study of Secondary Structure in Three Transfer Ribonucleic Acids. Biochemistry 1970, 9, 3542–3550. [Google Scholar] [CrossRef]
- Xiang, T.-X.; Anderson, B.D. Inclusion Complexes of Purine Nucleosides with Cyclodextrins. II. Investigation of Inclusion Complex Geometry and Cavity Microenvironment. Int. J. Pharmaceut. 1990, 59, 45–55. [Google Scholar] [CrossRef]
- Juliano, R.L. The Delivery of Therapeutic Oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in Oligonucleotide Drug Delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Hermann, R.; Krajcsi, P.; Fluck, M.; Seithel-Keuth, A.; Bytyqi, A.; Galazka, A.; Munafo, A. Review of Transporter Substrate, Inhibitor, and Inducer Characteristics of Cladribine. Clin. Pharmacokinet. 2021, 60, 1509–1535. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, J.W.; Ryu, J.-H.; Le, X.C.; Wiebe, L.I. Characterization of a Cyclodextrin-Oligonucleotide Complex by Capillary Electrophoresis using Laser-Induced Fluorescence. J. Pharm. Pharmaceut. Sci. 2007, 10, 246–255. [Google Scholar]
- Suzuki, I.; Miura, T.; Anzai, J.-I. Superiority of Secondary Hydroxy Side Modification in Cyclodextrin Complexation for Highly Hydrophilic Adenine Nucleotides. J. Supramol. Chem. 2001, 1, 283–288. [Google Scholar] [CrossRef]
- Eliseev, A.V.; Schneider, H.-J. Molecular Recognition of Nucleotides, Nucleosides, and Sugars by Aminocyclodextrins. J. Am. Chem. Soc. 1994, 116, 6081–6088. [Google Scholar] [CrossRef]
- Ladner, S.K.; Otto, M.J.; Barker, C.S.; Zaifert, K.; Wang, G.H.; Guo, J.T.; Seeger, C.; King, R.W. Inducible Expression of Human Hepatitis B Virus (HBV) in Stably Transfected Hepatoblastoma Cells: A Novel System for Screening Potential Inhibitors of HBV Replication. Antimicrob. Agents Chemother. 1997, 41, 1715–1720. [Google Scholar] [CrossRef]
- King, R.W.; Ladner, S.K. Hep AD38 Assay: A High-Throughput, Cell-Based Screen for the Evaluation of Compounds against Hepatitis B Virus. In Antiviral Methods and Protocols. Methods in Molecular Medicine™; Kinchington, D., Schinazi, R.F., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2001; Volume 24, pp. 43–50. [Google Scholar] [CrossRef]
- Szurmai, Z.; Liptak, A.; Szejtli, J. Halogen Azide Displacement to Prepare Some Symmetrically Substituted β-Cyclodextrin Derivatives. Starch 1990, 42, 447–449. [Google Scholar] [CrossRef]
- Parrot-Lopez, H.; Ling, C.C.; Zhang, P.; Baszkin, A.; Albrecht, G.; de Rango, C.; Coleman, A.W. Self-Assembling Systems of the Amphiphilic Cationic Per-6-amino-β-cyclodextrin 2,3-di-O-alkyl ethers. J. Am. Chem. Soc. 1992, 114, 5479–5480. [Google Scholar] [CrossRef]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.B.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P.; et al. Advances in Methods and Algorithms in a Modern Quantum Chemistry Program Package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.B.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in Molecular Quantum Chemistry Contained in the Q-Chem 4 Program Package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef]
- Mamidyala, S.K.; Dutta, S.; Chrunyk, B.A.; Preville, C.; Wang, H.; Withka, J.M.; McColl, A.; Subashi, T.A.; Hawrylik, S.J.; Griffor, M.C.; et al. Glycomimetic Ligands for the Human Asialoglycoprotein Receptor. J. Am. Chem. Soc. 2012, 134, 1978–1981. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Sanhueza, C.A.; Baksh, M.M.; Thuma, B.; Roy, M.D.; Dutta, S.; Preville, C.; Chrunyk, B.A.; Beaumont, K.; Dullea, R.; Ammirati, M.; et al. Efficient Liver Targeting by Polyvalent Display of a Compact Ligand for the Asialoglycoprotein Receptor. J. Am. Chem. Soc. 2017, 139, 3528–3536. [Google Scholar] [CrossRef]
- Huang, J.-R.; Zhuang, H.-N.; Jin, Z.-Y. Chapter 1: Introduction. In Cyclodextrin Chemistry Preparation and Application; Jin, Z.-Y., Ed.; World Scientific Publishing Co. Pte Ltd.: Singapore, 2013; pp. 1–18. ISBN 978-981-4436-79-3. [Google Scholar] [CrossRef]
- Sugihara, J.M. Relative reactivities of hydroxyl groups of carbohydrates. In Advances in Carbohydrate Chemistry; Hudson, C.S., Wolfrom, M.L., Eds.; Academic Press: Cambridge, MA, USA, 1953; Volume 8, pp. 1–44. [Google Scholar] [CrossRef]
- Khan, A.R.; Forgo, P.; Stine, K.J.; D’Souza, V.T. Methods for Selective Modifications of Cyclodextrins. Chem. Rev. 1998, 98, 1977–1996. [Google Scholar] [CrossRef]
- Przybyla, M.A.; Yilmaz, G.; Becer, C.R. Natural Cyclodextrins and their Derivatives for Polymer Synthesis. Polym. Chem. 2020, 11, 7582–7602. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, P.X. Cyclodextrin-based Supramolecular Systems for Drug Delivery: Recent Progress and Future Perspective. Adv. Drug Deliv. Rev. 2013, 65, 1215–1233. [Google Scholar] [CrossRef]
- Haley, R.M.; Gottardi, R.; Langer, R.; Mitchell, M.J. Cyclodextrins in Drug Delivery: Applications in Gene and Combination Therapy. Drug Deliv. Transl. Res. 2020, 10, 661–677. [Google Scholar] [CrossRef]
- Mazzaglia, A.; Forde, D.; Garozzo, D.; Malvagna, P.; Ravoo, B.J.; Darcy, R. Multivalent Binding of Galactosylated Cyclodextrin Vesicles to Lectin. Org. Biomol. Chem. 2004, 2, 957–960. [Google Scholar] [CrossRef]
- Malhotra, M.; Gooding, M.; Evans, J.C.; O’Driscoll, D.; Darcy, R.; O’Driscoll, C.M. Cyclodextrin-siRNA Conjugates as Versatile Gene Silencing Agents. Eur. J. Pharmaceut. Sci. 2018, 114, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Rekharsky, M.V.; Inoue, Y. Complexation and Chiral Recognition Thermodynamics of 6-Amino-6-deoxy-β-cyclodextrin with Anionic, Cationic, and Neutral Chiral Guests: Counterbalance Between van der Waals and Coulombic Interactions. J. Am. Chem. Soc. 2002, 124, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Aggelidou, C.; Mavridis, I.M.; Yannakopoulou, K. Binding of Nucleotides and Nucleosides to Per(6-guanidino-6-deoxy)-cyclodextrins in Solution. Eur. J. Org. Chem. 2009, 14, 2299–2305. [Google Scholar] [CrossRef]
- Rodriguez-Ruiz, V.; Maksimenko, A.; Salzano, G.; Lampropoulou, M.; Lazarou, Y.G.; Agostoni, V.; Couvreur, P.; Gref, R.; Yannakopoulou, K. Positively Charged Cyclodextrins as Effective Molecular Transporters of Active Phosphorylated Forms of Gemcitabine into Cancer Cells. Sci. Rep. 2017, 7, 8353. [Google Scholar] [CrossRef] [PubMed]
- Jicsinszky, L.; Caporaso, M.; Martina, K.; Calcio Gaudino, E.; Cravotto, G. Efficient Mechanochemical Synthesis of Regioselective Persubstituted Cyclodextrins. Beilstein J. Org. Chem. 2016, 12, 2364–2371. [Google Scholar] [CrossRef] [PubMed]
- Zultanski, S.L.; Kuhl, N.; Zhong, W.; Cohen, R.D.; Reibarkh, M.; Jurica, J.; Kim, J.; Weisel, L.; Ekkati, A.R.; Klapars, A.; et al. Mechanistic Understanding of a Robust and Scalable Synthesis of Per(6-deoxy-6-halo)cyclodextrins, Versatile Intermediates for Cyclodextrin Modification. Org. Process Res. Dev. 2020, 25, 597–607. [Google Scholar] [CrossRef]
- Motoyama, K.; Nishiyama, R.; Maeda, Y.; Higashi, T.; Ishitsuka, Y.; Kondo, Y.; Irie, T.; Era, T.; Arima, H. Synthesis of Multi-lactose-appended β-Cyclodextrin and its Cholesterol-lowering Effects in Niemann–Pick Type C Disease-like HepG2 Cells. Beilstein J. Org. Chem. 2017, 13, 10–18. [Google Scholar] [CrossRef]
- Alali, U.; Vallin, A.; Bil, A.; Khanchouche, T.; Mathiron, D.; Przybylski, C.; Beaulieu, R.; Kovensky, J.; Benazza, M.; Bonnet, V. The Uncommon Strong Inhibition of α-Glucosidase by Multivalent Glycoclusters Based on Cyclodextrin Scaffolds. Org. Biomol. Chem. 2019, 17, 7228–7237. [Google Scholar] [CrossRef]
- Eliseev, A.V.; Schneider, H.-J. Aminocyclodextrins as Selective Hosts with Several Binding Sites for Nucleotides. Angew. Chem. Int. Ed. Engl. 1993, 32, 1331–1333. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, J.; Wang, T.; Sun, L. Why Does β-Cyclodextrin Prefer to Bind Nucleotides with an Adenine Base Rather than Other 2′-Deoxyribonucleoside 5′-Monophosphates? J. Mol. Model. 2017, 23, 149. [Google Scholar] [CrossRef] [PubMed]
- Fallon, R.J.; Schwartz, A.L. Receptor–Mediated Endocytosis and Targeted Drug Delivery. Hepatology 1985, 5, 899–901. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.J.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in Humans from Systemically Administered siRNA via Targeted Nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Bon, C.; Hofer, T.; Bousquet-Mélou, A.; Davies, M.R.; Krippendorff, B.-F. Capacity Limits of Asialoglycoprotein Receptor-mediated Liver Targeting. mAbs 2017, 9, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, G.; Diakur, J.; Wiebe, L.I. Targeted Delivery of Macromolecular Drugs: Asialoglycoprotein Receptor (ASGPr) Expression by Selected Hepatoma Cell Lines Used in Antiviral Drug Development. Curr. Drug Deliv. 2008, 5, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.L.; Fridovich, S.E.; Knowles, B.B.; Lodish, H.F. Characterization of the Asialoglycoprotein Receptor in a Continuous Hepatoma Line. J. Biol. Chem. 1981, 256, 8878–8881. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.L.; Geuze, H.J.; Lodish, H.F. Recycling of the Asialoglycoprotein Receptor: Biochemical and Immunocytochemical Evidence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1982, 300, 229–235. [Google Scholar] [CrossRef]
- Pei, D.; Buyanova, M. Overcoming Endosomal Entrapment in Drug Delivery. Bioconjugate Chem. 2019, 30, 273–283. [Google Scholar] [CrossRef]
- Li, C.; Cao, X.-W.; Zhao, J.; Wang, F.-J. Effective Therapeutic Drug Delivery by GALA3, an Endosomal Escape Peptide with Reduced Hydrophobicity. J. Membr. Biol. 2020, 253, 139–152. [Google Scholar] [CrossRef]
- Du Rietz, H.; Hedlund, H.; Wilhelmson, S.; Nordenfelt, P.; Wittrup, A. Imaging Small Molecule-induced Endosomal Escape of siRNA. Nat. Commun. 2020, 11, 1809. [Google Scholar] [CrossRef] [PubMed]
- Huotari, J.; Helenius, A. Endosome Maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Wada, K.; Kabuta, T. Lysosomal Degradation of Intracellular Nucleic Acids—Multiple Autophagic Pathways. J. Biochem. 2017, 161, 145–154. [Google Scholar] [CrossRef]
- Ghosh, M.; Zhang, R.; Lawler, R.G.; Seto, C.T. The Effects of Buffers on the Thermodynamics and Kinetics of Binding between Positively-Charged Cyclodextrins and Phosphate Ester Guests. J. Org. Chem. 2000, 65, 735–741. [Google Scholar] [CrossRef]
- Awumey, E.M.K.; Somayaji, V.V.; Wiebe, L.I.; Tyrrell, D.L.; Paterson, A.R.P. Synthesis, Hepatocyte Uptake and In Vivo Biodistribution of Lactosyl-9-β-D-arabinofuranosyl Adenine (Lactosyl-araA), a Proposed Prodrug for Targeting the Delivery of 9-β-D-Arabinofuranosyl Adenine (araA) to Liver. Pharmaceut. Sci. Comm. 1993, 4, 59–67. [Google Scholar]
- Mercer, J.R.; Mannan, R.H.; Somayaji, V.V.; Lee, J.; Chapman, J.D.; Wiebe, L.I. Sugar-coupled 2-Nitroimidazoles: Novel In Vivo Markers for Hypoxic Tissue. In Advances in Radiopharmacology; Maddalena, D.J., Snowden, G.M., Boniface, G.R., Eds.; Wollongong University Printery Services: Wollongong, Australia, 1990; pp. 104–113. [Google Scholar]
- Yokoyama, R.; Taharabaru, T.; Nishida, T.; Ohno, Y.; Maeda, Y.; Sato, M.; Ishikura, K.; Yanagihara, K.; Takagi, H.; Nakamura, T.; et al. Lactose-appended β-Cyclodextrin as an Effective Nanocarrier for Brain Delivery. J. Control Release 2020, 328, 722–735. [Google Scholar] [CrossRef]
- Yang, W. Nucleases: Diversity of Structure, Function and Mechanism. Q. Rev. Biophys. 2011, 44, 1–93. [Google Scholar] [CrossRef]
- Mikkola, S.; Lönnberg, T.; Lönnberg, H. Phosphodiester Models for Cleavage of Nucleic Acids. Beilstein J. Org. Chem. 2018, 14, 803–837. [Google Scholar] [CrossRef]
- Smith, L.D.; Kizer, D.E. Purification and Properties of Rat Liver AMP Deaminase. Biochim. Biophys. Acta 1969, 191, 415–424. [Google Scholar] [CrossRef]
- Connor, J.D.; Sweetman, L.; Carey, S.; Stuckey, M.A.; Buchanan, R. Effect of Adenosine Deaminase upon the Antiviral Activity In Vitro of Adenine Arabinoside for Vaccinia Virus. Antimicrob. Agents Chemother. 1974, 6, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Jicsinszky, L.; Martina, K.; Cravotto, G. Cyclodextrins in the Antiviral Therapy. J. Drug. Deliv. Sci. Technol. 2021, 64, 102589. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, S.B.; Costa Duarte, F.Í.; Heimfarth, L.; Siqueira Quintans, J.d.S.; Quintans-Júnior, L.J.; Veiga Júnior, V.F.d.; Neves de Lima, Á.A. Cyclodextrin-Drug Inclusion Complexes: In Vivo and In Vitro Approaches. Int. J. Mol. Sci. 2019, 20, 642. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound/Complex | Test Conc. (µg/mL) | EC50 (µg/mL) (araA Equivalent) | Potency Relative to araA |
|---|---|---|---|
| GcyDAc (ASGPr targeted carrier) | 0.8–100 | >100 | N/A |
| Lamivudine * (LV,3TC) (reference antiviral) | 0.8–100 | <0.8 | 43.7 |
| GcyDAc:LV (reference antiviral complex) | 0.8–100 | 1.5 | 23.3 |
| PMPA ** (reference antiviral) | 0.08–10 | 0.7 | 50 |
| araA | 0.8–100 | 35 | 1.0 |
| GcyDAc:araA (1:1) | 0.8–100 | 14 | 2.5 |
| araAMP | 0.8–100 | 33 | 1.1 |
| GcyDAc:araAMP (1:1) | 0.8–100 | 26 | 1.3 |
| p-araAMP | 0.8–100 | 29 | 1.2 |
| GcyDA:p-araAMP (2:1) | 0.8–100 | 29 | 1.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryu, J.-H.; Zheng, W.; Yang, X.-H.; Elsaidi, H.; Diakur, J.; Wiebe, L.I. Synthesis and Preliminary Evaluation of an ASGPr-Targeted Polycationic β-Cyclodextrin Carrier for Nucleosides and Nucleotides. Pharmaceutics 2024, 16, 323. https://doi.org/10.3390/pharmaceutics16030323
Ryu J-H, Zheng W, Yang X-H, Elsaidi H, Diakur J, Wiebe LI. Synthesis and Preliminary Evaluation of an ASGPr-Targeted Polycationic β-Cyclodextrin Carrier for Nucleosides and Nucleotides. Pharmaceutics. 2024; 16(3):323. https://doi.org/10.3390/pharmaceutics16030323
Chicago/Turabian StyleRyu, Jang-Ha, Weizhong Zheng, Xiao-Hong Yang, Hassan Elsaidi, Jim Diakur, and Leonard I. Wiebe. 2024. "Synthesis and Preliminary Evaluation of an ASGPr-Targeted Polycationic β-Cyclodextrin Carrier for Nucleosides and Nucleotides" Pharmaceutics 16, no. 3: 323. https://doi.org/10.3390/pharmaceutics16030323
APA StyleRyu, J. -H., Zheng, W., Yang, X. -H., Elsaidi, H., Diakur, J., & Wiebe, L. I. (2024). Synthesis and Preliminary Evaluation of an ASGPr-Targeted Polycationic β-Cyclodextrin Carrier for Nucleosides and Nucleotides. Pharmaceutics, 16(3), 323. https://doi.org/10.3390/pharmaceutics16030323